Heterotrimeric GAIT Complex Drives Transcript-Selective Translation Inhibition in Murine Macrophages (original) (raw)
Abstract
The gamma interferon (IFN-γ)-activated inhibitor of translation (GAIT) complex in human myeloid cells is heterotetrameric, consisting of glutamyl-prolyl-tRNA synthetase (EPRS), NS1-associated protein 1 (NSAP1), ribosomal protein L13a, and glyceraldehyde-3-phosphate dehydrogenase (GAPDH). The complex binds a structural GAIT element in the 3′ untranslated region of VEGF-A and other inflammation-related transcripts and inhibits their translation. EPRS is dually phosphorylated by cyclin-dependent kinase 5 (Cdk5) at Ser886 and then by a Cdk5-dependent-AGC kinase at Ser999; L13a is phosphorylated at Ser77 by death-associated protein kinases DAPK and ZIPK. Because profound differences in inflammatory responses between mice and humans are known, we investigated the GAIT system in mouse macrophages. The murine GAIT complex is heterotrimeric, lacking NSAP1. As in humans, IFN-γ activates the mouse macrophage GAIT system via induced phosphorylation of EPRS and L13a. Murine L13a is phosphorylated at Ser77 by the DAPK-ZIPK cascade, but EPRS is phosphorylated only at Ser999. Loss of EPRS Ser886 phosphorylation prevents NSAP1 incorporation into the GAIT complex. However, the triad of Ser999-phosphorylated EPRS, Ser77-phosphorylated L13a, and GAPDH forms a functional GAIT complex that inhibits translation of GAIT target mRNAs. Thus, translational control by the heterotrimeric GAIT complex in mice exemplifies the distinctive species-specific responses of myeloid cells to inflammatory stimuli.
INTRODUCTION
Transcript-selective translation inhibition is an important posttranscriptional mechanism to regulate gene expression. It is generally mediated by the binding of a protein or complex (or noncoding RNA) to specific sequence or structural elements in the 5′ or 3′ untranslated region (UTR) (8, 44). 3′ UTR-specific posttranscriptional mechanisms are particularly critical in regulating the expression of genes associated with inflammation (22, 26). In human myeloid cells, the inflammatory cytokine gamma interferon (IFN-γ) induces the formation of a heterotetrameric, IFN-γ-activated inhibitor of translation (GAIT) complex that binds a defined, split stem-loop GAIT element in the 3′ UTR of multiple inflammation-related mRNAs and inhibits their translation (26). Among the targets are vascular endothelial growth factor A (VEGF-A), ceruloplasmin (Cp), death-associated protein kinases DAPK and ZIPK, and several chemokines and their receptors (26, 43). The human GAIT complex is heterotetrameric, consisting of glutamyl-prolyl-tRNA synthetase (EPRS), NS1-associated protein 1 (NSAP1), ribosomal protein L13a, and glyceraldehyde-3-phosphate dehydrogenase (GAPDH) (37).
Recruitment of the four GAIT proteins and their assembly into GAIT complex is regulated by a network of IFN-γ-activated kinases. The human GAIT complex is assembled in two distinct stages. After about 2 to 4 h of IFN-γ treatment, EPRS is phosphorylated at two sites, first at Ser886 by cyclin-dependent kinase 5 (Cdk5) and subsequently at Ser999 by a Cdk5-dependent AGC kinase (2, 3). Both phosphorylation events occur in the noncatalytic linker domain that joins the functional ERS and PRS synthetases. Two-site Ser phosphorylation induces the release of EPRS from its residence in the tRNA multisynthetase complex (MSC). Free, phosphorylated EPRS binds NSAP1 to form the nonfunctional pre-GAIT complex that does not bind GAIT element RNA; EPRS Ser886 phosphorylation is essential for NSAP1 interaction. About 12 to 16 h later, ribosomal protein L13a is phosphorylated at Ser77 by a kinase cascade of DAPK and ZIPK (27). L13a phosphorylation induces its translocation from the 60S ribosomal subunit to join GAPDH and then the pre-GAIT complex to form the heterotetrameric GAIT complex that binds GAIT element RNA. Phosphorylation of EPRS at Ser999 is critical for translational repression activity, as it facilitates the interaction of L13a in the RNA-bound GAIT complex with a eukaryotic translation initiation factor, eIF4G, of the translation initiation complex at or near the eIF3-binding site (3, 15). This interaction blocks the recruitment of 40S ribosome-containing preinitiation complex, thereby inhibiting translation of GAIT element-bearing mRNA.
To date, the GAIT system has been investigated in detail in human cells only. To begin to understand how widespread the GAIT system is in vertebrates, we have investigated its presence and function in mouse myeloid cells. Mice have small genetic differences compared to humans, with only about 300 unique genes (23). Moreover, the primary sequences of the four human GAIT proteins are highly conserved in mice. Mice can be genetically manipulated and thus are oft-used models of human disease. Not surprisingly, mouse models are imperfect and differ from humans in several fundamental processes, including inflammation. For example, several inflammation-responsive genes in humans, such as the genes for interleukin-8 (IL-8) and monocyte chemoattractant protein 4 (MCP4), are absent in mice (23). Marked differences between mouse and human intestinal immunity have also been observed (9). Differences between humans and mice in both innate and acquired immunity are not surprising because the two species diverged about 65 million to 75 million years ago and inhabit very different environments with markedly dissimilar pathogenic challenges (23, 33).
Previous work showed that cytosolic extracts from IFN-γ-activated mouse macrophages inhibit in vitro translation of a GAIT element-containing reporter, suggesting a functional GAIT pathway in murine macrophages (34). To unambiguously establish the activity of the GAIT pathway, its regulation, and the identity of its constituents, we investigated the GAIT system in murine macrophages. Here we report that IFN-γ activates GAIT-mediated translational silencing in mouse macrophages, with important similarities to and unexpected differences from activation in human macrophages. Unlike that in humans, the GAIT complex in mice is heterotrimeric and contains EPRS phosphorylated at Ser999 but not at Ser886, Ser77-phosphorylated L13a, and GAPDH, but it is devoid of NSAP1.
MATERIALS AND METHODS
Reagents.
Affinity-purified antibodies against human peptides containing EPRS Ser886 and Ser999 phosphorylation sites, EPRS, and ribosomal protein L13a were obtained as described previously (3, 21, 33). Small interfering RNAs (siRNAs) targeting DAPK, ZIPK, and Cdk5 were from Santa Cruz. Flag-tagged human and mouse EPRS linker (Pro683 to Asn1023) and full-length L13a cDNA constructs were generated by cloning into pcDNA3 expression vector (3, 14). Point mutations at Ser886 and Ser999 in EPRS and Ser77 in L13a to either Ala or Asp were introduced using primers bearing the desired mutation and a site-directed mutagenesis system from Invitrogen. Purified recombinant human NSAP1 and phosphorylated L13a were as described previously (13). Human GAPDH was purchased from Sigma (St. Louis, MO). Recombinant, His-tagged human and mouse EPRS linker proteins (wild type and mutants) were cloned in pET 30 expression vector and purified (3, 14). Kinase inhibitors were purchased from EMD (Darmstadt, Germany).
Cell culture.
Human U937 monocytic cells, HEK293 embryonic kidney cells, and mouse J774A.1 and RAW 264.7 macrophages were from the ATCC (Manassas, VA). U937 cells were cultured in RPMI 1640 medium containing 10% fetal bovine serum (FBS). Mouse J774A.1, RAW 264.7, and HEK293 cells were cultured in Dulbecco's modified Eagle's medium (DMEM) with 10% FBS. Bone marrow-derived macrophages (BMM) were isolated from 8-week-old C57BL6/J mice by flushing the marrows of femurs and tibias and cultured for 6 days in RPMI 1640 medium containing 10% FBS and 20% L929 cell-conditioned medium. Cells (1 × 107) were treated with human or mouse IFN-γ (500 U/ml; R&D Systems) for up to 24 h (21). Cell lysates were prepared in Phosphosafe extraction buffer (Novagen) supplemented with protease inhibitor cocktail.
Cell transfection.
Cells were transiently transfected with plasmid DNAs or siRNAs) using Amaxa Nucleofector kit V (Lonza, Cologne, Germany). Transfected cells were immediately transferred to prewarmed Opti-MEM medium for 6 h and then to RPMI 1640 medium containing 10% FBS for 18 to 24 h.
RNA electrophoretic mobility shift assay (EMSA).
α-32P-labeled Cp GAIT element probe was prepared using the oligonucleotide-directed MEGAshortscript transcription system (Ambion) and custom-synthesized RNA probe templates (Invitrogen) (36). Labeled probe was incubated with cytosolic lysates from U937 and RAW 264.7 cells. RNA-protein complexes were resolved by native gel electrophoresis and visualized by autoradiography. For RNA-protein complex supershift, antibodies against EPRS, NSAP1, L13a, and GAPDH were preincubated with cytosolic extracts for 30 min at 4°C before incubation with labeled probe.
In vitro translation and reconstitution assays.
T7 gene 10 and luciferase-Cp 3′ UTR GAIT element (Luc-Cp GAIT) reporter mRNAs were prepared using the mMESSAGE mMACHINE kit (Ambion) as described previously (13). Gel-purified, capped poly(A) template RNAs were translated in a rabbit reticulocyte lysate (RRL) system (Promega) in the presence of [35S]Met. Translated reactions were resolved by SDS-PAGE and autoradiography. For in vitro reconstitution of GAIT system activity, the complex was reconstituted by incubating 5 pmol each of purified GAIT constituent proteins NSAP1, GAPDH, phosphorylated L13a, and EPRS linker proteins. The effect of reconstituted GAIT complex on in vitro translation of reporter mRNAs was determined using wheat germ extract and [35S]Met.
In vitro phosphorylation and Cdk5 activity assay.
Purified His-tagged EPRS linker protein permitting Ser886 and Ser999 phosphorylation was phosphorylated in vitro by incubation with cell lysate and 5 μCi of [γ-32P]ATP (Perkin-Elmer) in kinase assay buffer (50 mM Tris-HCl [pH 7.6], 1 mM dithiothreitol, 10 mM MgCl2, 1 mM CaCl2, and phosphatase inhibitor cocktail) (2, 3). For Cdk5 activity assay, immunoprecipitates obtained by incubation with anti-Cdk5 antibody were used to phosphorylate QRRDR886SPTRNREPA derived from human EPRS linker peptide as described above. 32P incorporation into peptide was determined by spotting equal volumes on phosphocellulose P-81 paper and scintillation counting.
32P-labeling experiments.
U937 and RAW 264.7 cells were cultured in phosphate-free RPMI 1640 medium and incubated with IFN-γ for 2 h and then with [32P]orthophosphate (500 μCi; Perkin-Elmer, Boston, MA) for 24 h. The 32P-labeled cells were lysed in Phosphosafe extraction buffer (Novagen, Darmstadt, Germany) and immunoprecipitated with anti-EPRS antibody conjugated to protein A-Sepharose (Sigma, St. Louis, MO).
Protein-protein interaction by coimmunoprecipitation.
Cell lysates (1 mg) were immunoprecipitated by incubation with antibody cross-linked to protein A-Sepharose or agarose in detergent-free immunoprecipitation buffer (50 mM Tris-HCl [pH 7.6], 150 mM NaCl, EDTA-free protease, and phosphatase inhibitor cocktail) as described previously (3). The beads were washed three times in the same buffer, eluted, and analyzed by SDS-PAGE and immunoblotting.
RESULTS
GAIT system silences VEGF-A expression in mouse macrophages.
We determined the IFN-γ-regulated expression of VEGF-A, which is a human GAIT target, in RAW 264.7 cells, and in bone marrow-derived macrophages (BMM) isolated from femurs and tibias of C57BL6/J mice. VEGF-A protein in lysates was markedly induced in both cell types after 8 h of IFN-γ treatment (Fig. 1A). After 24 h, VEGF-A returned to a near-basal level despite abundant VEGF-A mRNA, consistent with translational silencing by the GAIT system (33, 34). To verify the role of the GAIT pathway, two key activation events were inhibited using specific siRNAs. Inhibition of either ZIPK or Cdk5, responsible for L13a and EPRS phosphorylation, respectively, caused sustained expression of VEGF-A (Fig. 1B). To establish the role of the 3′ UTR GAIT element in the observed translational silencing, we determined binding of the mouse GAIT complex to the GAIT element by RNA electrophoretic mobility shift assay using 32P-labeled Cp GAIT RNA element as a probe. As reported previously (26), lysates from human U937 cells treated with IFN-γ for 24 h shifted the GAIT element probe (Fig. 1C). Unexpectedly, both 8- and 24-h lysates from mouse RAW 264.7 cells bound the GAIT element. Finally, we determined whether lysates from IFN-γ-treated cells could inhibit in vitro translation of a luciferase reporter transcript containing the human ceruloplasmin (Cp) 3′ UTR GAIT element (Luc-Cp GAIT). As previously observed, 24-h lysates from human U937 monocytic cells repressed translation of the Luc-Cp GAIT transcript without affecting the T7 gene 10 control transcript lacking the GAIT element (Fig. 1D). Likewise, 24-h lysates from mouse BMM, RAW 264.7, and J774A.1 macrophages, but not from the nonmacrophage HEK293 cells, were inhibitory. These results provide compelling evidence for GAIT-mediated regulation of gene expression in mouse macrophages. However, the binding of the mouse GAIT complex to target elements after 8 h of IFN-γ treatment suggests that the GAIT system is not identical to that in human cells.
Fig 1.

IFN-γ activates the GAIT system in murine macrophages. (A) RAW 264.7 cells and BMM from wild-type C57BL6/J mice were treated with IFN-γ for 8 and 24 h. Cell lysates were immunoblotted (IB) using anti-VEGF-A and anti-β-actin antibodies. VEGF-A and β-actin mRNA was determined by reverse transcription-PCR (RT-PCR) of total cellular RNA. (B) RAW 264.7 cells were transfected with siRNAs against ZIPK and Cdk5, and control scrambled siRNAs, and incubated with IFN-γ for up to 24 h. Lysates were immunoblotted as shown. (C) Analysis of protein binding to the Cp GAIT element. Cytosolic lysates from IFN-γ-treated U937 and RAW 264.7 cells were incubated with a 32P-labeled Cp GAIT element RNA probe, and binding was determined by EMSA. (D) In vitro translation. Cells were incubated with IFN-γ and lysates added to rabbit reticulocyte lysates in the presence of capped, polyadenylated luciferase (Luc) reporter RNA bearing the 3′ UTR Cp GAIT element and T7 gene 10 control RNA and [35S]Met. Luc translation was quantified by densitometry, normalized by T7 gene 10, and expressed as a percentage of the control (mean ± SEM; n = 3 experiments).
DAPK and ZIPK mediate L13a phosphorylation at Ser77 in mouse macrophages.
IFN-γ-induced phosphorylation of L13a at Ser77 is critical for GAIT pathway activation in human monocytes (3, 27). The human Ser77 phosphorylation site in L13a is conserved in other mammalian species, including mice (Fig. 2A). To investigate temporal phosphorylation of L13a in mouse macrophages, RAW 264.7 cell lysates were prepared after 8 h of IFN-γ treatment and then at 4-h intervals for up to 24 h, and they were immunoblotted with anti-phospho-Ser antibody after immunoprecipitation with anti-L13a antibody (Fig. 2B). L13a phosphorylation was induced between 12 and 16 h post-IFN-γ treatment and remained phosphorylated until 24 h, as shown for human monocytic cells (27). To determine if murine L13a is phosphorylated at Ser77, phosphorylation-defective Ser77-to-Ala (S77A) mutant pcDNA3-Flag-mouse L13a was transiently transfected in U937 and RAW 264.7 cells (Fig. 2C). L13a phosphorylation was determined by immunoprecipitation with anti-Flag antibody and immunoblotting with anti-phospho-Ser antibody. IFN-γ-induced phosphorylation of human and mouse L13a bearing the S77A mutation was completely abrogated in both human and mouse cells (Fig. 2C). To determine if mouse L13a is phosphorylated by the kinase cascade active in human cells, RAW 264.7 cells were transfected with siRNAs targeting DAPK and ZIPK. IFN-γ-induced L13a phosphorylation was assessed with anti-phospho-Ser antibody following immunoprecipitation with anti-L13a antibody. L13a phosphorylation was completely inhibited in DAPK (Fig. 2D, left) and ZIPK (Fig. 2D, right) knockdown cells. To show that murine L13a is in the GAIT complex, RAW 264.7 and U937 cells were treated with IFN-γ for 24 h, metabolically labeled with 32P, and subjected to immunoprecipitation with anti-EPRS antibody. Immunoblot analysis with anti-L13a antibody revealed stimulus-dependent interaction of murine L13a with EPRS (Fig. 2E, top two blots). Autoradiography revealed stimulus-dependent radiolabeled bands consistent in size with phospho-L13a and -EPRS in both murine and human cells (Fig. 2E, bottom). These experiments indicate that the mechanism of IFN-γ activation of L13a in murine macrophages is essentially identical to that observed in human myeloid cells.
Fig 2.

IFN-γ induces DAPK-ZIPK-mediated L13a phosphorylation at Ser77 in mouse macrophages. (A) Multiple-sequence alignment of mammalian L13a protein. The conserved Ser77 phosphorylation site is shaded and the kinase recognition motif underlined. (B) Delayed induction of L13a phosphorylation in mouse macrophages. Lysates from IFN-γ-treated RAW 264.7 cells were immunoprecipitated with anti-L13a antibody and phosphorylation determined by immunoblotting with anti-phospho-Ser (P-Ser) antibody. (C) Mouse L13a is phosphorylated on Ser77. Human and murine Flag-tagged, wild-type (WT) and Ser77-to-Ala (S77A) mutant L13a proteins were transfected in U937 and RAW 264.7 cells and treated with IFN-γ for 16 h. Lysates were immunoprecipitated (IP) with anti-Flag antibody, followed by immunoblotting with anti-P-Ser antibody. (D) DAPK and ZIPK knockdown prevents L13a phosphorylation. RAW 264.7 cells were transfected with siRNAs against DAPK (left) and ZIPK (right) or scrambled control (Cont.) siRNAs. Lysates were immunoprecipitated with anti-L13a antibody, and phosphorylation was determined using anti-P-Ser antibody. (E) Mouse L13a translocates to the GAIT complex. RAW 264.7 and U937 cells were treated with IFN-γ for 24 h in the presence of [32P]orthophosphate. Lysates were subjected to immunoprecipitation with anti-EPRS antibody and analyzed by immunoblotting with antibodies against EPRS and L13a (top two blots) and by autoradiography (bottom).
EPRS is singly phosphorylated at Ser999 in EPRS in murine macrophages.
IFN-γ-induces dual serine phosphorylation of Ser886 and Ser999 in human EPRS, coordinating key events in assembly and activity of the GAIT complex (3). Surprisingly, the Ser999 phosphosite, but not the Ser886 site, is conserved in mouse EPRS, suggesting a modified mechanism of GAIT activation (Fig. 3A). To investigate sites of murine EPRS phosphorylation experimentally, RAW 264.7 cells were treated with IFN-γ and analyzed by phospho-specific antibodies. Stimulus-dependent phosphorylation was detected at both sites in human cells; phosphorylation of only Ser999 was observed in mouse cells (Fig. 3B). To verify this result, pcDNA3-Flag-EPRS linkers (mouse and human) bearing phosphosite Ser-to-Ala mutations were transiently transfected in U937 and RAW 264.7 cells. After induction with IFN-γ, EPRS linker phosphorylation was determined by immunoprecipitation with anti-Flag antibody followed by detection with anti-phospho-Ser antibody. Single-site mutation of Ser999 to Ala (S999A) completely abrogated mouse EPRS linker phosphorylation but approximately halved phosphorylation of the human linker, in which phosphorylation was completely blocked only when both phosphosites were mutated (Fig. 3C). Similar results were seen following transfection of either human or mouse cells. These results show that in contrast to the human protein, only a single site in mouse EPRS linker (Ser999) is phosphorylated following IFN-γ stimulus.
Fig 3.

IFN-γ induces single-site EPRS phosphorylation at Ser999. (A) Multiple-sequence alignment of mammalian EPRS protein. Conserved Ser886 and Ser999 phosphorylation sites are shaded, and the kinase recognition motif around human Ser886 is underlined. (B) EPRS is phosphorylated at Ser999 in RAW 264.7 cells. Lysates from IFN-γ-treated cells were probed with phospho-specific antibodies and with anti-EPRS antibody as a loading control. (C) Ser999 is the only EPRS linker site phosphorylated in murine cells. Human and mouse pcDNA3-Flag-tagged, wild-type as well as single and double Ser-to-Ala mutant EPRS linkers were transfected into U937 and RAW 264.7 cells. Cells were treated with IFN-γ for 4 h, and lysates were immunoprecipitated with anti-Flag antibody and probed with anti-Flag and anti-P-Ser antibodies. (D) IFN-γ-activated mouse macrophages phosphorylate human EPRS at both Ser886 and Ser999. Lysates from IFN-γ-treated cells were used to in vitro phosphorylate recombinant EPRS linkers bearing Ser999-to-Ala and Ser886-to-Ala mutations in the presence of [γ-32P]ATP. (E) IFN-γ induces Cdk5 activity in mouse macrophages. U937 and RAW 264.7 cells were treated with IFN-γ for 4 h, and the lysates were immunoprecipitated with anti-Cdk5 antibody and used for in vitro phosphorylation of a Ser886-containing EPRS peptide (QRRDR886SPTRNREPA) substrate (mean ± SEM; n = 4 experiments). (F) Cdk5 knockdown in mouse macrophages blocks EPRS phosphorylation. RAW 264.7 cells were transfected with siRNA against Cdk5 or scrambled control siRNA. Lysates were subjected to immunoblot analysis with antibody against P-EPRS-Ser999. (G) A Cdk5-dependent AGC group kinase phosphorylates Ser999. RAW 264.7 cells were incubated with IFN-γ for 0.5 h and then with specific kinase inhibitors for an additional 3.5 h. Pro-directed kinase group inhibitors were roscovitine (10 μM) and olomoucine (50 μM); AGC kinase group inhibitors were H-7 (10 μM), H-8 (10 μM), staurosporine (20 nM), rottlerin (100 μM), LY294002 (2.5 μM), Akt inhibitor VIII (2.5 μM), and rapamycin (10 nM). Lysates were analyzed by immunoblotting as indicated. (H) Schematic of IFN-γ-induced EPRS phosphorylation in human and mouse macrophages.
The upstream kinase(s) required for phosphorylation of mouse EPRS Ser999 was investigated. In human cells, Cdk5 (with its activation partner, p35) has dual activity in EPRS activation; it is the proximal kinase that phosphorylates Ser886 and is an upstream component of a cascade responsible for Ser999 phosphorylation. In the absence of Ser886 phosphorylation by Cdk5/p35 in the mouse, it is conceivable that this kinase likewise is not a component of the cascade required for phosphorylation of mouse Ser999. To begin to investigate species differences (or similarities), we tested whether mouse cell lysates could in vitro phosphorylate human EPRS linkers bearing mutations permitting single-site phosphorylation. Lysates from IFN-γ-treated RAW 264.7 cells efficiently phosphorylated both Ser886 and Ser999 sites (Fig. 3D). To test Cdk5 activation in mouse macrophages directly, Cdk5 was immunoprecipitated from lysates from IFN-γ-treated cells and used to in vitro phosphorylate the human EPRS peptide substrate containing the Ser886 site. Cdk5 from RAW 264.7 and U937 cells phosphorylated the peptide substrate, confirming Cdk5 activation in mouse macrophages (Fig. 3E). siRNA-mediated knockdown of Cdk5 completely inhibited Ser999 phosphorylation (Fig. 3F). The effect of group-specific pharmacological inhibitors was investigated. Agents targeting Pro-directed kinases, including Cdk5, significantly inhibited IFN-γ-stimulated phosphorylation of EPRS Ser999 (Fig. 3G), as observed in human cells (3). Inhibition of Ser999 phosphorylation by H7, H8, and rapamycin suggests a possible role for a ribosomal S6 kinase family member or a cyclic-nucleotide-dependent protein kinase (protein kinase A [PKA], PKG, etc.). Akt inhibitor VIII, the phosphatidylinositol 3-kinase (PI3K) inhibitor LY294002, and the PKC inhibitor rottlerin were ineffective. Together these results suggest that IFN-γ-inducible Cdk5/p35 activation is required for EPRS phosphorylation in both human and mouse macrophages (Fig. 3H). In humans, IFN-γ-activated Cdk5/p35 directly phosphorylates Ser886 and indirectly phosphorylates Ser999 via activation of an AGC kinase member. In contrast, IFN-γ-activated Cdk5/p35 is required only as an upstream activator of the AGC kinase that phosphorylates Ser999 in mouse macrophages.
Phosphorylation at both Ser886 and Ser999 is required for EPRS release from its residence in the aminoacyl-tRNA multisynthetase complex (MSC) in human monocytes (3). We investigated whether phosphorylation of the sole Ser999 site was sufficient to induce release of mouse EPRS from the MSC. Lysates from IFN-γ-treated RAW 264.7 cells were immunoprecipitated with anti-EPRS antibody and immunoblotted with antibodies against the MSC constituent, aminoacyl-tRNA synthetase-interacting multifunctional protein 3 (AIMP3/p18), and the GAIT complex constituent, L13a. Substantially reduced interaction of EPRS with AIMP3/p18 and increased association with L13a were observed in IFN-γ-treated cells, suggesting that single-site phosphorylation at Ser999 is sufficient for translocation from the MSC to GAIT complex (Fig. 4).
Fig 4.
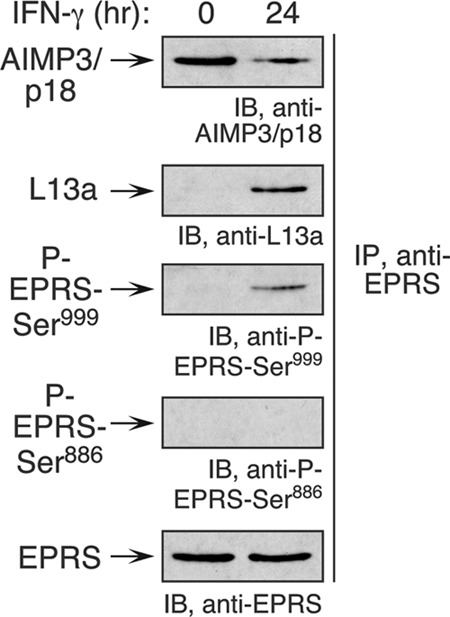
Ser999 phosphorylation induces EPRS translocation from MSC to GAIT complex. Lysates from IFN-γ-treated RAW 264.7 cells were immunoprecipitated with anti-EPRS antibody and subjected to immunoblot analysis with anti-AIMP3/p18 and anti-L13a antibodies to determine association with MSC and GAIT complex, respectively. Phospho-specific antibodies were used to determine EPRS phosphorylation.
Mouse GAIT complex lacks NSAP1.
The differential binding characteristics of the mouse and human GAIT complexes (Fig. 1C) might be attributable to dissimilar protein components. Constituents of the mouse GAIT complex were determined by RNA-EMSA supershift analysis using 32P-labeled Cp GAIT RNA element as a probe. After 24 h of IFN-γ treatment, EPRS, L13a, and GAPDH could bind the GAIT element, but unlike in human cells, NSAP1 did not bind (Fig. 5A). A second species difference was observed, namely, that mouse EPRS binds the GAIT element at 8 h, whereas we previously showed that no human GAIT complex components bind GAIT element RNA until at least 16 h of IFN-γ treatment (21, 27). These results suggest that NSAP1 is not a constituent of the GAIT complex in mouse myeloid cells, a finding consistent with the absence of the EPRS Ser886 phosphosite required for NSAP1 binding in human cells.
Fig 5.

Mouse GAIT complex lacks NSAP1. (A) RNA EMSA supershift investigation of binding of mouse GAIT complex proteins to GAIT element. Lysates from IFN-γ-treated RAW 264.7 cells were incubated with antibodies against GAIT complex constituents and then with a 32P-labeled Cp GAIT element RNA probe. (B) Lysates from IFN-γ-treated U937 and RAW 264.7 cells were immunoblotted with antibodies against human GAIT complex constituents (left). Lysates were immunoprecipitated with anti-EPRS (middle) and anti-NSAP1 (right) antibodies and subjected to immunoblot analysis as indicated.
Immunoblot analysis of cytosolic lysates from RAW 264.7 cells showed that the four human GAIT complex constituents are present in mouse cells and unaffected by IFN-γ treatment (Fig. 5B, left blots). Immunoprecipitation with anti-EPRS antibody verified that unlike in human cells, mouse NSAP1 is not present in a pre-GAIT complex formed at 8 h of IFN-γ treatment, nor is it in a mature GAIT complex formed after 24 h (Fig. 5B, center blots). Immunoprecipitation with anti-NSAP1 antibody confirmed that NSAP1 does not bind any of the GAIT complex proteins in mouse myeloid cells (Fig. 5B, right blots).
Reconstitution of murine GAIT system requires single-site EPRS phosphorylation and not NSAP1.
Ser886- and Ser999-phosphorylated EPRS and Ser77-phosphorylated L13a (P-L13a) are the key stimulus-dependent constituents of the functional GAIT ribonucleoprotein (RNP) complex in humans (Fig. 6A, top). Previously, using phospho-mimetic human EPRS linker, we reconstituted GAIT system activity with single-site phosphorylation at Ser999 (3). Because NSAP1 is not present in the mouse GAIT complex, we investigated its requirement for activity in the human GAIT complex. The GAIT complex was reconstituted by incubating wild-type, phosphorylated wild-type, and Ser-to-Asp mutant human EPRS linkers with P-L13a in the presence and absence of GAPDH and NSAP1. Activity of the reconstituted GAIT complex was determined by in vitro translation of a Luc-Cp-GAIT element reporter, and control T7 gene 10 RNA, in a wheat germ extract in the presence of [35S]Met. Translation repression of the GAIT element-bearing reporter was observed when NSAP1, P-L13a, and GAPDH were reconstituted with a phosphorylated, wild-type EPRS linker or single S999D or double S886D/S999D phospho-mimetic linkers, confirming the requirement for Ser999 phosphorylation for assembly and activity of human GAIT complex (Fig. 6B). Reconstitution of the S999D EPRS linker with P-L13a and GAPDH, but not NSAP1, also inhibited reporter expression, indicating that NSAP1 is not an essential constituent of a repression-competent human GAIT complex. A similar reconstitution was done using the mouse EPRS linker. Purified, recombinant wild-type mouse EPRS linker and S999A (unmodified and kinase treated) and S999D mutant linkers were reconstituted with P-L13a and GAPDH. Comparable translational silencing of a GAIT element-bearing reporter was observed in the absence or presence of NSAP1 (Fig. 6C). These studies verify the function of the NSAP1-deficient, heterotrimeric GAIT complex in mice (Fig. 6A, bottom).
Fig 6.
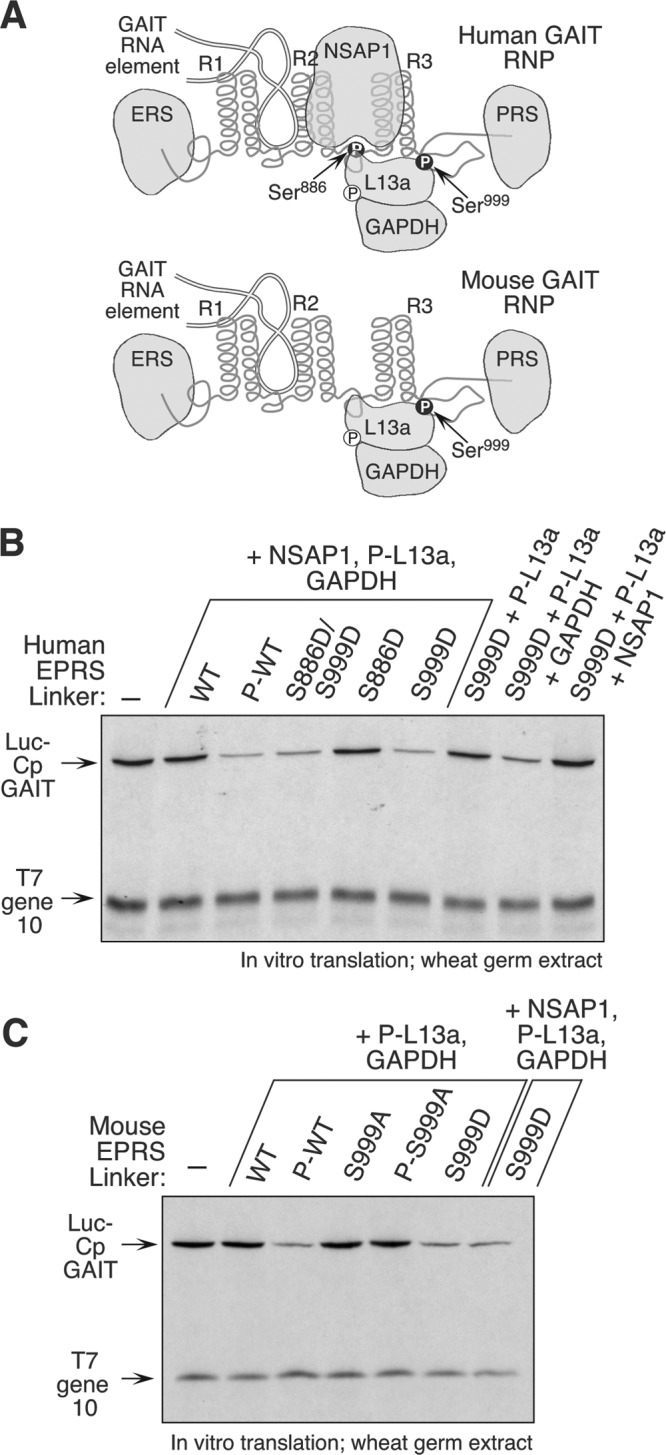
NSAP1 is not essential for reconstitution of GAIT complex function. (A) Schematic of human heterotetrameric (top) and mouse heterotrimeric (bottom) GAIT RNP; (B) reconstitution of GAIT function with human EPRS linker. His-tagged, wild-type (WT), and phospho-mimetic (Ser-to-Asp, S-to-D) human EPRS linker proteins were generated. Phosphorylated WT (P-WT) linker was generated by incubation with IFN-γ-activated cell lysate and repurified by Ni affinity chromatography. Linker proteins were incubated with other GAIT constituents as indicated and added to wheat germ extract for in vitro translation of luciferase (Luc)-Cp-GAIT element and T7 gene 10 reporters. (C) Reconstitution of GAIT function with mouse EPRS linker. His-tagged WT, P-WT, phospho-defective (S999A), phosphorylated S999A (P-S999A; generated by incubation with IFN-γ-activated cell lysate and repurified) and phospho-mimetic (S999D) mouse EPRS linker proteins were incubated other GAIT constituents and used in the in vitro experiment.
DISCUSSION
Our results reveal important similarities and unexpected differences between the IFN-γ-induced GAIT systems in human and mouse myeloid cells. The human GAIT system is induced by two-site Ser phosphorylation in EPRS at Ser886 by Cdk5 and Ser999 by a cascade of Cdk5 and an unidentified AGC kinase (1–3). EPRS phosphorylation is followed by release from the MSC and interacts with NSAP1 to form the dimeric pre-GAIT complex (Fig. 7, top). Subsequent phosphorylation of L13a at Ser77 by the DAPK-ZIPK cascade releases it from the 60S ribosomal subunit. Phosphorylated L13a binds GAPDH, and together they bind the pre-GAIT complex to form the heterotetrameric GAIT complex. The GAIT complex binds the 3′ UTR GAIT element in the target mRNA, and by a mechanism dependent on end-to-end interactions of the mRNA, P-L13a in GAIT complex interacts with eIF4G of the translation initiation complex, thereby blocking recruitment of the small ribosomal subunit and translation initiation. In mice, IFN-γ similarly activates the GAIT system via induced phosphorylation of EPRS and L13a (Fig. 7, bottom). However, mouse EPRS is subject to single-site phosphorylation at Ser999 only. Despite the specific loss of the Ser886 phosphosite in mouse EPRS, the Ser886 kinase Cdk5 is required for activation of the AGC kinase that phosphorylates Ser999. The lack of Ser886 phosphorylation prevents interaction with NSAP1 but permits release of EPRS from the MSC and joining with L13a and GAPDH to form a functional, heterotrimeric GAIT complex that binds GAIT element in target mRNAs and represses translation.
Fig 7.
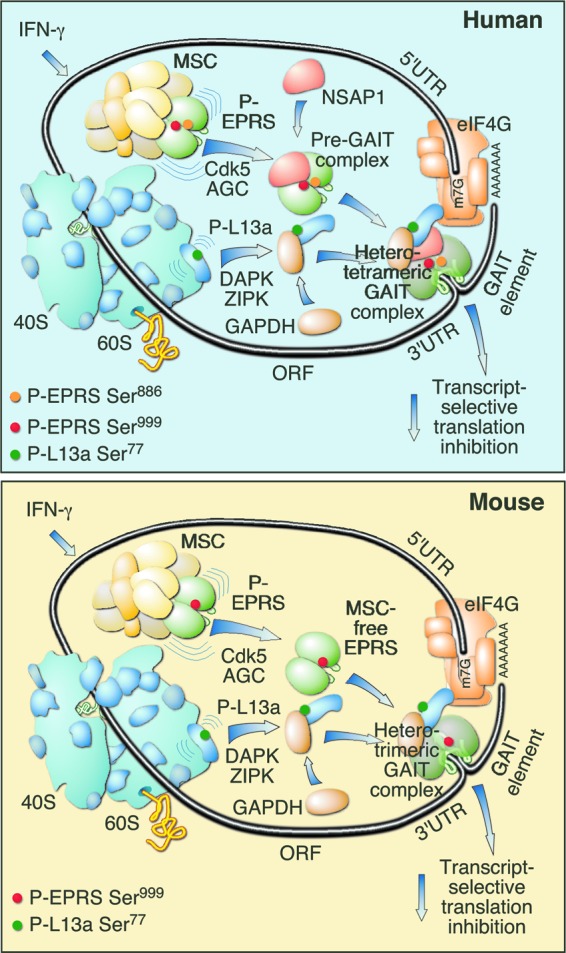
Comparison of human and mouse GAIT systems. Human heterotetrameric (top) and mouse heterotrimeric (bottom) GAIT complexes are shown.
The absence of NSAP1 from the murine GAIT complex indicates that its function is secondary rather than essential in the human system. This concept was verified experimentally by the in vitro reconstitution of GAIT activity by the human (and mouse) EPRS linker with GAPDH and P-L13a (Fig. 6B and C). The function of NSAP1 in the human GAIT system has been investigated before (3, 13). NSAP1 binding to human EPRS requires phosphorylation of Ser886 in the spacer between the second (R2) and third (R3) of the three helix-turn-helix WHEP repeats (named after the aminoacyl-tRNA synthetases that contain them, i.e., Trp(W)RS, His(H)RS, and EPRS [12, 41]) that populate the human and mouse EPRS linker domains (Fig. 6A). NSAP1 binding to human EPRS blocks the binding of GAIT element-bearing mRNA to the first (R1) and second (R2) WHEP repeats. The NSAP1-mediated inhibition is reversed by the subsequent interaction of P-L13a and GAPDH to EPRS that induces a conformational shift in the linker and exposes the RNA binding sites of EPRS. In view of the nearly 16-h residence in the cell before maturation as the GAIT complex, it is possible, if not likely, that the human pre-GAIT complex exhibits a GAIT-independent function. It is also possible that the human NSAP1-containing, pre-GAIT complex binds non-GAIT mRNA targets and affects a different function, for example, mRNA stability. This possibility is supported by reports of multiple RNA-related regulatory functions of NSAP1, also known as heterogeneous nuclear ribonucleoprotein (hnRNP) Q or synaptotagmin-binding cytoplasmic RNA interacting protein (SYNCRIP), including editing, splicing, stabilization, transport, surveillance by nonsense-mediated mRNA decay, and internal ribosome entry site (IRES)-dependent translation (4–6, 11, 17, 25, 29). Like the human pre-GAIT complex, mouse phospho-EPRS might exhibit GAIT-independent functions until joined by other constituents of the functional GAIT complex. Two important structural and functional differences distinguish the released murine phospho-EPRS from its human counterpart. First, the absence of NSAP1 binding to murine P-EPRS eliminates any role of the former in regulating the RNA-binding property of the latter. Second, unlike the human pre-GAIT complex, free murine P-EPRS can bind GAIT element-bearing target mRNAs, but it is unable to silence translation until P-L13a and GAPDH join to form the active trimeric complex. Free murine phospho-EPRS therefore could influence GAIT mRNA targets by GAIT-independent mechanisms, for example, by influencing mRNA stability or by binding to other, non-GAIT translational control complexes.
The absence of the Ser886 phosphorylation site in mice is not particularly exceptional. Global alignment of orthologs of functional phosphorylation sites indicates that about 80% of phospho-Ser and -Thr sites are conserved in mammals, suggesting that phosphorylation sites are frequently gained or lost during evolution, most likely to direct species-specific activities (10). Conservation of the Ser886 phosphosite in primates, rat, horse, and opossum (an early-appearing placental mammal) suggests that its absence in mice is due to a species-specific loss (Fig. 3A). Several differences between mouse and human phosphosites have been investigated. In one example, the Thr157 phosphosite in human p27kip1 is not conserved in mice (28). Phosphorylation at this site has been implicated in protein-protein interactions and nuclear localization and is critical for regulation of cell cycle progression (24, 40, 42). Likewise, phosphorylation of Ser216 in the dual-specificity protein phosphatase Cdc25C is essential for G2 arrest after DNA damage but is absent in mice (19, 30).
Expression patterns and functions of the vast majority of genes are common to humans and mice (16). However, 65 million to 75 million years of evolutionary divergence have introduced significant functional differences in both orthologous and paralogous genes in the two species (23). In fact, mitochondrial DNAs from museum skins of white-footed mice living in the same location from 1855 to the present exhibit surprisingly rapid microevolution (31). Multiple species-specific differences related to immune- or inflammation-related responses have been reported, driven in part by challenges to widely different pathogens in distinct ecological niches (23, 39). Several examples involve differential responses to specific agonists and inflammatory challenges. For example, inducible nitric oxide synthase (iNOS) is induced by IFN-γ and lipopolysaccharide in mouse but not in human macrophages (38). Also, in mice a higher concentration of endotoxin is required to induce an acute inflammatory response, e.g., induction of plasma interleukin-6 (7). More specifically, Toll-like receptors in murine macrophages trigger direct antimicrobial activity against intracellular bacteria compared to the indirect, vitamin D-dependent activation in human monocytes/macrophages (18). Important constitutive, stimulus-independent differences between the species have been reported. For example, human blood is neutrophil rich, whereas mouse blood is lymphocyte rich (23); moreover, only human neutrophils produce defensins (23, 35). These species-specific differences in inflammatory responses are not just of evolutionary interest; they also profoundly affect the clinical applicability of therapeutic agents successfully tested in mouse models, e.g., in septic shock (20, 32, 39). Thus, it is neither exceptional nor surprising that the protein constituents and activation mechanisms of the GAIT system differ in mouse and human myeloid cells. It is also possible, if not likely, that the mRNA targets differ between species. Although VEGF-A is targeted in both species, secondary-structure analysis by Mfold suggests that the GAIT element is not conserved in mouse Cp (33). Our validation and characterization of the murine GAIT system lay a solid foundation for future in vivo studies. However, our results certainly recommend a cautious approach in extrapolating to humans the findings from genetic mouse models of defective or absent GAIT system components.
ACKNOWLEDGMENTS
This work was supported in part by National Institutes of Health grants P01 HL029582, P01 HL076491, and R01 GM086430 (to P.L.F.) and by a National Center Scientist Development Grant (10SDG3930003) from the American Heart Association (to A.A.).
Footnotes
Published ahead of print 15 October 2012
REFERENCES
- 1.Arif A. 2012. Extraneuronal activities and regulatory mechanisms of the atypical cyclin-dependent kinase Cdk5. Biochem. Pharmacol. 84:985–993 [DOI] [PubMed] [Google Scholar]
- 2.Arif A, Jia J, Moodt RA, DiCorleto PE, Fox PL. 2011. Phosphorylation of glutamyl-prolyl tRNA synthetase by cyclin-dependent kinase 5 dictates transcript-selective translational control. Proc. Natl. Acad. Sci. U. S. A. 108:1415–1420 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Arif A, et al. 2009. Two-site phosphorylation of EPRS coordinates multimodal regulation of noncanonical translational control activity. Mol. Cell 35:164–180 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bannai H, et al. 2004. An RNA-interacting protein, SYNCRIP (heterogeneous nuclear ribonuclear protein Q1/NSAP1) is a component of mRNA granule transported with inositol 1,4,5-trisphosphate receptor type 1 mRNA in neuronal dendrites. J. Biol. Chem. 279:53427–53434 [DOI] [PubMed] [Google Scholar]
- 5.Blanc V, et al. 2001. Identification of GRY-RBP as an apolipoprotein B RNA-binding protein that interacts with both apobec-1 and apobec-1 complementation factor to modulate C to U editing. J. Biol. Chem. 276:10272–10283 [DOI] [PubMed] [Google Scholar]
- 6.Chen CY, Shyu AB. 2003. Rapid deadenylation triggered by a nonsense codon precedes decay of the RNA body in a mammalian cytoplasmic nonsense-mediated decay pathway. Mol. Cell. Biol. 23:4805–4813 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Copeland S, Warren HS, Lowry SF, Calvano SE, Remick D. 2005. Acute inflammatory response to endotoxin in mice and humans. Clin. Diagn. Lab. Immunol. 12:60–67 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gebauer F, Hentze MW. 2004. Molecular mechanisms of translational control. Nat. Rev. Mol. Cell Biol. 5:827–835 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gibbons DL, Spencer J. 2011. Mouse and human intestinal immunity: same ballpark, different players; different rules, same score. Mucosal Immunol. 4:148–157 [DOI] [PubMed] [Google Scholar]
- 10.Gnad F, et al. 2007. PHOSIDA (phosphorylation site database): management, structural and evolutionary investigation, and prediction of phosphosites. Genome Biol. 8:R250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Grosset C, et al. 2000. A mechanism for translationally coupled mRNA turnover: interaction between the poly(A) tail and a c-fos RNA coding determinant via a protein complex. Cell 103:29–40 [DOI] [PubMed] [Google Scholar]
- 12.Guo M, Yang XL, Schimmel P. 2010. New functions of aminoacyl-tRNA synthetases beyond translation. Nat. Rev. Mol. Cell Biol. 11:668–674 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Jia J, Arif A, Ray PS, Fox PL. 2008. WHEP domains direct noncanonical function of glutamyl-prolyl tRNA synthetase in translational control of gene expression. Mol. Cell 29:679–690 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Jia J, et al. 2012. Protection of extraribosomal RPL13a by GAPDH and dysregulation by S-nitrosylation. Mol. Cell 47:656–663 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kapasi P, et al. 2007. L13a blocks 48S assembly: role of a general initiation factor in mRNA-specific translational control. Mol. Cell 25:113–126 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Liao BY, Zhang J. 2006. Evolutionary conservation of expression profiles between human and mouse orthologous genes. Mol. Biol. Evol. 23:530–540 [DOI] [PubMed] [Google Scholar]
- 17.Liu HM, et al. 2009. SYNCRIP (synaptotagmin-binding, cytoplasmic RNA-interacting protein) is a host factor involved in hepatitis C virus RNA replication. Virology 386:249–256 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Liu PT, et al. 2006. Toll-like receptor triggering of a vitamin D-mediated human antimicrobial response. Science 311:1770–1773 [DOI] [PubMed] [Google Scholar]
- 19.Liu Q, et al. 2000. Chk1 is an essential kinase that is regulated by Atr and required for the G(2)/M DNA damage checkpoint. Genes Dev. 14:1448–1459 [PMC free article] [PubMed] [Google Scholar]
- 20.López A, et al. 2004. Multiple-center, randomized, placebo-controlled, double-blind study of the nitric oxide synthase inhibitor 546C88: effect on survival in patients with septic shock. Crit. Care Med. 32:21–30 [DOI] [PubMed] [Google Scholar]
- 21.Mazumder B, et al. 2003. Regulated release of L13a from the 60S ribosomal subunit as a mechanism of transcript-specific translational control. Cell 115:187–198 [DOI] [PubMed] [Google Scholar]
- 22.Mazumder B, Seshadri V, Fox PL. 2003. Translational control by the 3′-UTR: the ends specify the means. Trends Biochem. Sci. 28:91–98 [DOI] [PubMed] [Google Scholar]
- 23.Mestas J, Hughes CC. 2004. Of mice and not men: differences between mouse and human immunology. J. Immunol. 172:2731–2738 [DOI] [PubMed] [Google Scholar]
- 24.Morishita D, Katayama R, Sekimizu K, Tsuruo T, Fujita N. 2008. Pim kinases promote cell cycle progression by phosphorylating and down-regulating p27Kip1 at the transcriptional and posttranscriptional levels. Cancer Res. 68:5076–5085 [DOI] [PubMed] [Google Scholar]
- 25.Mourelatos Z, Abel L, Yong J, Kataoka N, Dreyfuss G. 2001. SMN interacts with a novel family of hnRNP and spliceosomal proteins. EMBO J. 20:5443–5452 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Mukhopadhyay R, Jia J, Arif A, Ray PS, Fox PL. 2009. The GAIT system: a gatekeeper of inflammatory gene expression. Trends Biochem. Sci. 34:324–331 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mukhopadhyay R, et al. 2008. DAPK-ZIPK-L13a axis constitutes a negative-feedback module regulating inflammatory gene expression. Mol. Cell 32:371–382 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Nacusi LP, Sheaff RJ. 2006. Akt1 sequentially phosphorylates p27kip1 within a conserved but non-canonical region. Cell Div. 1:11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Park SM, et al. 2011. Translation-competent 48S complex formation on HCV IRES requires the RNA-binding protein NSAP1. Nucleic Acids Res. 39:7791–7802 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Peng CY, et al. 1997. Mitotic and G2 checkpoint control: regulation of 14-3-3 protein binding by phosphorylation of Cdc25C on serine-216. Science 277:1501–1505 [DOI] [PubMed] [Google Scholar]
- 31.Pergams OR, Barnes WM, Nyberg D. 2003. Mammalian microevolution: rapid change in mouse mitochondrial DNA. Nature 423:397. [DOI] [PubMed] [Google Scholar]
- 32.Poli-de-Figueiredo LF, Garrido AG, Nakagawa N, Sannomiya P. 2008. Experimental models of sepsis and their clinical relevance. Shock 30(Suppl 1):53–59 [DOI] [PubMed] [Google Scholar]
- 33.Ray PS, Fox PL. 2007. A post-transcriptional pathway represses monocyte VEGF-A expression and angiogenic activity. EMBO J. 26:3360–3372 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ray PS, et al. 2009. A stress-responsive RNA switch regulates VEGFA expression. Nature 457:915–919 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Risso A. 2000. Leukocyte antimicrobial peptides: multifunctional effector molecules of innate immunity. J. Leukoc. Biol. 68:785–792 [PubMed] [Google Scholar]
- 36.Sampath P, Mazumder B, Seshadri V, Fox PL. 2003. Transcript-selective translational silencing by gamma interferon is directed by a novel structural element in the ceruloplasmin mRNA 3′ untranslated region. Mol. Cell. Biol. 23:1509–1519 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sampath P, et al. 2004. Noncanonical function of glutamyl-prolyl-tRNA synthetase: gene-specific silencing of translation. Cell 119:195–208 [DOI] [PubMed] [Google Scholar]
- 38.Schneemann M, Schoedon G. 2002. Species differences in macrophage NO production are important. Nat. Immunol. 3:102. [DOI] [PubMed] [Google Scholar]
- 39.Schroder K, et al. 2012. Conservation and divergence in Toll-like receptor 4-regulated gene expression in primary human versus mouse macrophages. Proc. Natl. Acad. Sci. U. S. A. 109:E944–E953 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Sekimoto T, Fukumoto M, Yoneda Y. 2004. 14-3-3 suppresses the nuclear localization of threonine 157-phosphorylated p27(Kip1). EMBO J. 23:1934–1942 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Shiba K. 2002. Intron positions delineate the evolutionary path of a pervasively appended peptide in five human aminoacyl-tRNA synthetases. J. Mol. Evol. 55:727–733 [DOI] [PubMed] [Google Scholar]
- 42.Shin I, Rotty J, Wu FY, Arteaga CL. 2005. Phosphorylation of p27Kip1 at Thr-157 interferes with its association with importin alpha during G1 and prevents nuclear re-entry. J. Biol. Chem. 280:6055–6063 [DOI] [PubMed] [Google Scholar]
- 43.Vyas K, et al. 2009. Genome-wide polysome profiling reveals an inflammation-responsive posttranscriptional operon in gamma interferon-activated monocytes. Mol. Cell. Biol. 29:458–470 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wilkie GS, Dickson KS, Gray NK. 2003. Regulation of mRNA translation by 5′- and 3′-UTR-binding factors. Trends Biochem. Sci. 28:182–188 [DOI] [PubMed] [Google Scholar]