Advances in Autism (original) (raw)
. Author manuscript; available in PMC: 2013 May 6.
Abstract
Autism is a common childhood neurodevelopmental disorder with strong genetic liability. It is not a unitary entity but a clinical syndrome, with variable deficits in social behavior and language, restrictive interests, and repetitive behaviors. Recent advances in the genetics of autism emphasize its etiological heterogeneity, with each genetic susceptibility locus accounting for only a small fraction of cases or having a small effect. Therefore, it is not surprising that no unifying structural or neuropathological features have been conclusively identified. Given the heterogeneity of autism spectrum disorder (ASD), approaches based on studying heritable components of the disorder, or endophenotypes, such as language or social cognition, provide promising avenues for genetic and neurobiological investigations. Early intensive behavioral and cognitive interventions are efficacious in many cases, but autism does not remit in the majority of children. Therefore, development of targeted therapies based on pathophysiologically and etiologically defined subtypes of ASD remains an important and achievable goal of current research.
Keywords: autism spectrum disorders, complex genetics, copy number variation, disconnection syndrome, neuroimaging, neuropathology
INTRODUCTION
Autism was first recognized as a coherent disorder by Kanner in 1943 in a seminal clinical description of 11 boys with an autistic disorder of affective contact (1). In the past 65 years, concepts of this disorder within psychiatry have changed significantly in parallel with the development of the concept that disorders of cognition and behavior have an organic, brain-based etiology. In the 1960s and 1970s, autism was essentially considered a form of psychosis akin to childhood schizophrenia, and the prevailing conventional wisdom was that it could largely be related to parenting styles. The early recognition that a number of rare genetic or biomedical conditions, for example, phenylketonuria (2), could lead to autism should have suggested that these disorders were not caused by bad parenting, as was eventually demonstrated (3). However, these syndromic forms of autism were thought to be the exception. It was not until the early 1980s that autism was classified as a developmental disorder and widely accepted as having a biomedical origin. That so much time had to pass is ironic, in light of Kanner’s (1) closing statements describing autism as “an innate inability to form the usual, biologically provided affective contact … analogous to other innate physical or intellectual handicaps.” The modern conceptualization of autism as a biomedical disorder was largely fueled by the demonstration that it was heritable (4, 5) and was associated with a variety of genetic syndromes (6, 7).
Although diagnostic metrics and categories have changed over the past three decades, the diagnosis of autism has deficits in reciprocal social interaction at its core. In current practice, narrowly defined autism or autistic disorder has been placed under the diagnostic category of pervasive developmental disorder (PPD), along with Asperger syndrome, childhood disintegrative disorder, Rett syndrome, and pervasive developmental disorder not otherwise specified (PDD-NOS) in the Diagnostic and Statistical Manual of Mental Disorders IV (8). The diagnosis of autistic disorder requires three core domains of severe dysfunction with onset prior to the age of three: deficits in language and communication, deficits in social interaction, and the presence of repetitive or restrictive behaviors and interests (8). The current diagnostic criteria reflect a focus on the behavioral and cognitive components of autism; until recently, the neurologic or medical features in children were relatively neglected. In addition to the three core domains necessary for diagnosis, several other areas of clinical dysfunction are observed in a significant proportion of children diagnosed with autism (Table 1). For example, sensory abnormalities are observed in >90% of those with autism and are thought by some to be a core feature. Motor abnormalities such as hypotonia and apraxia are also common, whereas other complaints, such as sleep disturbance and gastrointestinal symptoms, although common, have not been as well characterized. Epilepsy, though variable across the ASD spectrum, is clinically very important to identify and has been consistently identified in a significant subset of cases. The term “autism spectrum disorders (ASDs)” (9) includes Asperger syndrome (normal language; see Table 1) and the broader and more variable category of PDD-NOS, in which patients meet only some of the criteria for autistic disorder.
Table 1.
Domains of impairment in autistic spectrum disorder (ASD)a
| Domain | Autism | Asperger | PDD-NOS | ASD |
|---|---|---|---|---|
| social communication | required | required | required | |
| language | required | – | variable | |
| repetitive, restrictive behaviors | required | required | variable | |
| sensory abnormalities | >90% | 80% | variable | 94% |
| developmental regressionb | 15%–40% | ? | ? | 15%–40% |
| motor signsc | 60%–80% | 60% | 60% | 60%–80% |
| gross motor delay | 10% | ? | ? | 5%–10% |
| sleep disturbance | 55% | 5%–10% | 40% | 50% |
| gastrointestinal disturbanced | 45% | 4% | 50% | 4%–50% |
| epilepsye | 10%–60% | 0%–5% | 5–40% | 6%–60% |
| comorbid psychiatric diagnosisf | 70% | 60% | >25% | 25–70% |
Like many other psychiatric disorders, ASDs are challenging because their clinical definition relies on observation of behavior and cognitive phenotypes without the adjunctive help of laboratory tests, imaging results, or biochemical markers, which cannot currently be tested against a gold-standard postmortem neuropathology. Another major difficulty of classifying patients into diagnostic categories based on the observation of behavior and cognition is that dysfunction in these domains is not absolute, but continuous and quantitative, so that the placement of patients into diagnostic bins is somewhat arbitrary by necessity. In addition, because ASD diagnosis is not currently based on etiology or pathophysiology, autism and ASDs are not unitary conditions, but rather, clinical syndromes. Two children may share the diagnosis of autistic disorder, PDD-NOS, or Asperger syndrome and yet differ quite widely in terms of their clinical manifestations. For example, among those with autistic disorder—the most severe form, involving deficits in all three core domains—IQ varies widely; it may even be normal or above average, although 30%–50% of children with autism also have severe intellectual disability (21). Similar to childhood brain disorders such as epilepsy and intellectual disability (formerly called mental retardation), autism represents many pathophysiological entities (10–12), with both genetic and environmental contributions and many distinct trajectories (13–16). Its conceptualization as “the autisms” or “the ASDs” captures this increasingly recognized broad range of etiology and natural history (17, 18).
Development of diagnostic instruments that reproducibly and reliably classify patients has played an important role in advancing an international research agenda. Coupled with the increased awareness of ASD as a significant public health issue and the availability of new, more powerful techniques, this has led to a significant increase in autism research and subsequent publications. Furthermore, recent findings in genetics permit the identification of specific genetic subsets of ASD based on etiology, which is a significant advance over subjective, DSM-based diagnosis. Despite many challenges, there has been significant progress in the understanding of the genetics and neurobiology of ASD within the last few years.
PREVALENCE
Initial assessments of the rate of autism indicated that it was a rare disorder, with a prevalence of about 4 in 10,000 children (19). Over time and as diagnostic criteria have embraced a more sophisticated and broader notion of ASD, this estimate has increased from ~1/1000 in 1988 (20) to the currently accepted prevalence of 1/150–1/200 in 2002 for ASD (21) and ~1/500 for the narrow diagnosis of strict autism (22). This figure of 1/150–1/200 is concordant with analyses of data from multiple sources and population bases using different methods (25–27) and is considered a reliable figure. This means that ASD is more prevalent than many other childhood disorders that are considered common (10).
Although diagnostic factors clearly are contributory, the extent to which the increase in ASD prevalence is due to changes in diagnosis and ascertainment is not known. The increase in ASD prevalence estimates has led to concerns about environmental factors—most prominently, various forms of vaccines, including the MMR (measles, mumps, and rubella vaccine) and those containing the preservative thimerosal. Multiple, high-quality epidemiologic studies provide strong evidence against a role for vaccines or thimerosal in autism etiology or its increase in prevalence (23–31), and there is no connection between regressive forms of autism and the MMR vaccine, which was an initial concern (24). However, given the association of congenital infections (25, 26) and perinatal factors (27–28) in some cases and the less-than-100% heritability of ASD (see below), it is likely that as yet undetermined environmental factors do contribute to some forms of the disorder. In this regard, the fascination with increasing paternal age with ASD clearly warrants further pursuit (see Sidebar, “Paternal Age and ASD”).
A more thorough understanding of the genetic factors, which compose a significantly larger proportion of ASD risk than environmental factors, will facilitate identification of environmental contributions by suggesting mechanisms and providing more homogeneous etiological subtypes in which to examine gene-environment interactions.
PATHOLOGY AND ANATOMY
Most studies on brain growth and structure in ASD have been conducted using in vivo neuroimaging methods. Many diverse findings have been reported, but few have been replicated until recently. This is likely due in part to small sample sizes and methodological differences, but may also reflect true heterogeneity of the syndrome, as most studies involve <20 subjects with ASD. The majority of research has been performed in high-functioning autistic individuals (IQ > 70), boys, or subjects older than age seven, and it is not yet known whether findings in a particular clinically defined subgroup can be generalized. The trajectory in brain growth appears to be far more indicative of intellectual level both in typical development and neuropsychiatric disease (29) than measurements taken at one time during development (30).
Brain Overgrowth and Macrocephaly
The findings of early postnatal brain “overgrowth” in ASD followed by normalization or relative growth arrest in childhood are intriguing (31, 32) and have been independently replicated (33). This potentially important observation clearly implicates an early neurodevelopmental process. The time course of brain development argues against vaccine or postnatal toxin involvement, as the process underlying brain overgrowth likely has its onset prior to these purported exposures. Whether the observation of early overgrowth is universal and how it relates to other phenotypic or etiologic factors in specific subsets of children with an ASD are not known. In addition, macrocephaly or head circumference above the ninety-eighth percentile is observed in ~20% of autistic children in mid-childhood (34), and macrocephaly itself is a risk factor for ASD (35). But macrocephaly is not observed at birth, consistent with imaging data showing postnatal growth acceleration. Furthermore, macrocephaly is familial and is observed at ~20% in ASD probands; it is seen at a similar rate in the unaffected siblings and parents of macrocephalic children with ASD (36; S. Spence and D.H. Geschwind, unpublished observations). These data suggest that large head size observed in childhood is more akin to a risk factor than to a common pathophysiological anomaly, and is not related to the early-overgrowth phenotype, which may be more pervasive.
The brain overgrowth pattern in ASD is not observed throughout all brain regions. So far, it appears to be most prominent in the frontal lobes and anterior temporal regions (32) and may involve interconnected parietal areas—key components of the circuits underlying the development of higher cognitive specializations (37), such as language and social cognition, that are disrupted in ASD (38, 39). These same frontal and temporal regions and their subcortical partners have been implicated by recent genetic findings in ASD (40), providing an important convergence around brain circuitry involved in social cognition and language development that is likely to be involved in autism. The abnormalities in brain structures include both the gray and white matter, the latter of which is involved in both short- and long-range connections between brain regions. An emerging theory is that short-range connections may be overgrown, whereas longer-range connections between different brain lobules are reduced (41, 42). Studies using various electrophysiological methods have also noted reduced size of the corpus callosum, which connects the two cerebral hemispheres via long-range connections; reduced cerebellar connections with other widely spread brain regions (43); and evidence of reduced functional and structural connectivity between frontal and temporal regions in subjects with ASD. The perseverance of normal or supranormal short-range connections could also explain some of the preserved or improved local processing functions, such as certain aspects of visual perception or attention to detail, in ASD subjects (41). How global changes in brain connectivity relate to focal findings, for example, cerebellar hypoplasia or morphological changes in frontal cortical language-related regions (44, 45), remains to be elucidated. Viewing ASD as a developmental disconnection syndrome encompassing brain regions involved in language, emotional reciprocity, and social cognition could provide a unifying theme for autism pathophysiology (18).
Pathological Investigations
Early investigations highlighted cerebellar and brainstem pathology. Several independent studies observed decreased cerebellar Purkinje cells in ASD cases (46). More recent studies also suggest widespread abnormalities in cerebral cortex, most prominently, higher-order association areas such as frontal cortex and anterior temporal cortex. Bailey and colleagues performed one of the first modern anatomical studies of six brains, describing subtle abnormalities of neuronal morphology and position that, although widespread, were most pronounced in the frontal and temporal lobes (47). Careful investigation of cortical organization has also suggested another subtle deficit, a disruption in frontal cortical mini-columnar organization (48, 49). Mini-columns are thought to provide a fundamental building block of cortical information processing, and changes in this cortical microarchitecture could account for deficits in complex information processing. Though promising, these studies require specialized expertise and have only been performed in a small number of cases. A recent stereological study of the amygdala in nine ASD brains showed consistently fewer neurons in the ASD amygdala, which is involved in the regulation of emotion (50). How this or other neuropathological findings relates to the early-overgrowth phenotype observed in neuroimaging studies, as well as clinical phenotypes in patients, is not yet known.
Most studies of postmortem autistic brain have found no evidence of inflammation, but rather evidence of an early developmental process. However, a recent careful postmortem study of brain and cerebrospinal fluid (CSF) identified evidence of glial activation, increased expression of the complement cascade, and several other potential immunomodulatory molecules, including macrophage chemo-attractant protein-1 in both tissue and CSF (51). The vasculature was intact, again suggesting a noninflammatory process. The classical complement cascade has been shown to regulate synaptic remodeling (52), and microglia, TNF-α, and other immune-related molecules have been implicated in normal synaptic development and developmental apoptosis (53). So an alternative explanation for these findings is that these putative immune markers do not reflect immune system activation but rather ongoing alterations in synaptic plasticity.
Although a universal neuropathology and neuroimaging phenotype has not yet emerged in ASD, there are several features that have been repeatedly observed. These features, including early brain overgrowth and subtle neuropathological alterations consistent with early developmental processes, appear to converge most convincingly on frontal and anterior temporal association cortices and the amygdala. More clinical-neuropathological correlation is needed to define a potential role for neuroinflammation, including parsing out cases with epilepsy, which itself leads to significant pathological alterations. However, it should be emphasized that given the clear etiological heterogeneity of ASD (see “Genetics,” below), one does not expect uniform pathological and imaging findings, especially in the relatively small cohorts studied thus far. These disparate findings may not be contradictory; rather, these data and the underlying heterogeneity of the condition underscore the need for much larger cohorts in future studies. In addition, it will be critical to carefully define subtypes that can be connected to imaging and other biomarkers, as well as neuropsychological findings that correspond to common neurobiological or neuropathological processes.
GENETICS
The emerging notion of ASD as “the autisms,” a collection of dozens or perhaps hundreds of etiologic forms that converge on common behavioral and cognitive phenotypes, is largely a result of advances in autism genetics, which have been reviewed recently in detail (10).
ASDs are highly heritable. Estimates based on twin studies suggest a heritability of 0.7 for autism and as high as 0.9 for the broader ASD phenotype. Family studies also show a significant increase in autism in first-degree relatives of those with autism; that is, siblings of autistic probands have an approximately 25-fold higher risk than those in the general population, consistent with familial predisposition (10). There appears to be a range in the forms of genetic susceptibility from rare, apparently Mendelian causes to more complex genetic risk involving the interaction of multiple genes and the environment. But the magnitude of the contribution from the different forms of genetic risk to the overall genetic architecture of ASD within the population is unknown (10).
Several recent studies have provided a remarkable advance in our knowledge of the genetic causes of ASD. The first set of studies, based largely on whole-genome DNA microarrays, demonstrated a significant role for structural chromosomal abnormalities, ranging from submicroscopic to microscopic, in ASD. A seminal experiment by Sebat and colleagues (54) showed that such de novo copy number variation (CNV) might cause 10% of ASD in sporadic cases and a lower frequency, 2%, of familial (multiplex) cases. This experiment built on previous work by Jacquemont and colleagues (55), who identified apparent de novo chromosomal anomalies (mostly deletions) in ~25% of children with ASD in a dysmorphic, low-IQ population. The work of Sebat et al. and subsequent work by Marshall and colleagues (56) showed that such de novo structural chromosomal variation was not limited to cases with intellectual disability. Furthermore, these data suggest a different genetic architecture for sporadic versus familial ASD, as CNVs, predominantly deletions, were far more frequent in sporadic cases of ASD.
In addition to rare de novo CNVs, recurrent CNVs have been identified, including duplications of chromosomes 15q11–13, duplications and deletions of 16p11, and deletions of 22q11–13, each accounting for ~0.5%–1% of cases in large series (54–58). Based on these and previous data on other genetic causes of ASD, it is estimated that at least 10%–20% of children being evaluated for ASD (higher in those with low IQ or dysmorphic features) harbor an identifiable genetic lesion (10). Given the implications for recurrent risk and potentially different outcomes, a consensus is emerging that careful physical and neurological examination (including Woods lamp evaluation for tuberous sclerosis), genetic testing for fragile X, and high-resolution DNA microarray comparative genomic hybridization should be performed as part of the normal clinical evaluation of ASD. Genetic counseling is a critical component of this evaluation, as some of these genetic lesions—for example, 16p deletions/duplications—are not specific for autism and are observed at even higher rates in patients with other forms of global developmental delay (57).
In contrast to studies of structural chromosomal variation, genetic linkage and association studies have been slower to identify common contributory loci for ASD. This is probably because of heterogeneity at the phenotypic and genetic levels, and small effect sizes for individual risk alleles. Sample sizes several times larger than the 1000 families published are likely needed (58). An instructive parallel is the genetics of type II diabetes, in which common risk variants were not reproducibly identified until sample sizes of several thousand cases and controls were studied (59).
AUTISM AS A CONTINUUM
In addition to raw sample size, are there refinements in our concept of the ASD phenotype that could improve power in genetic and neurobiological studies? Should ASD be viewed as the tail of normal population variation in cognition and behavior, or does it instead represent a set of discrete entities? Support for its discrete nature might be inferred from genetic studies showing that numerous single-gene mutations can result in ASD. However, none of these single-gene disorders are specific for ASD, and all cause general intellectual disability, raising the additional question of how ASD is biologically distinct from intellectual disability. In fact, few would argue that general intellectual function does not follow a normal distribution in the general population, despite the existence of many rare forms of severe intellectual disability. Similarly, the existence of rare, syndromic forms of common metabolic disorders such as hyperlipidemia and diabetes is not usually interpreted as signifying that these conditions are not part of the continuum of normal variation related to common genetic polymorphisms.
Familial studies of ASD support the notion that it is composed of separate components that segregate in families and are observed in unaffected family members (60–63), sometimes referred to as the “broader phenotype” (63). These features, which involve elements of the core diagnostic components, such as language (64), social responsiveness (65), and restrictive behaviors (63, 64), are distinct and observed more frequently in the parents and siblings of ASD probands than in the general population. That the same holds true for comorbid psychiatric diagnoses, such as attention deficit/hyperactivity disorder (ADHD) (Table 1), again highlights the continuity with other supposedly distinct disorders defined by the DSM.
Thus, it seems parsimonious to posit that a significant proportion of the genetic liability to ASD is related to the same factors that modulate cognitive function and behavior in the general population. From this viewpoint, there are few genes causing ASD per se; genetic variation modulates language, social cognition, and one’s range of interests or repetitive behaviors, and it is a combination of genetic variation in these features that results in ASD susceptibility (Figure 1).
Figure 1.
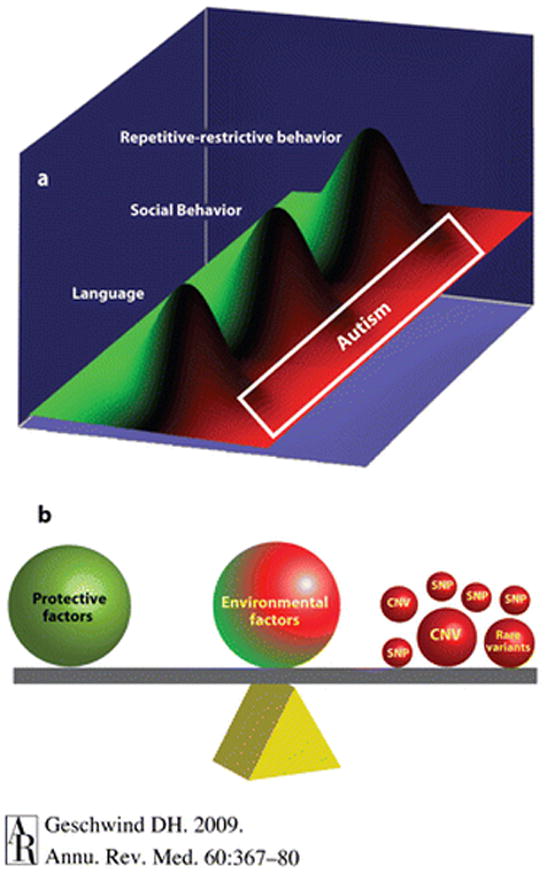
Autism heterogeneity in the context of common and rare genetic variation. Adapted from Reference 65 a with permission. (a) Susceptibility to autism spectrum disorder (ASD) may in some cases reflect the contribution of normal variation in heritable, potentially distinct core components of autism (endophenotypes). Normal variations in language, social, and repetitive restrictive behaviors are illustrated as overlapping but distinct functions, with the green and red demarcating the highest level of functioning and progressive abnormality, respectively. Under such a scheme, moderate abnormality in all three areas is necessary for a diagnosis of autism, but abnormality in one area, e.g., language, leads to a more circumscribed condition, such as specific language impairment. Patients with Asperger syndrome would have normal language but lie in the red zone for the other two components. Each of these heritable cognitive-behavioral components, or endophenotypes, although genetically complex, is likely to be less heterogeneous than the syndrome of autism. These endophenotypes can be studied separately to increase power to detect common genetic risk variants. (b) A simplified model of how a variety of risk variants, large and small, may contribute to ASDs in aggregate. The contribution of genetic or environmental factors is emphasized by the size or position of each representative weight. Mendelian mutations, such as de novo copy number variations, are depicted as a large ball because they are considered causal in many cases and would tip the balance, placing an individual essentially in the red in all categories shown in panel (a) (pleiotropy), resulting in autism. It is also likely that in some cases, common variation would modulate such major gene effects on phenotype.
This paradigm is supported by the observation that the core components of ASD—abnormalities in language, social cognition, and the domain of repetitive restrictive behaviors—are discretely heritable in the general population (66). It is further supported by recent genetic studies relying on ASD endophenotypes, such as language or social cognition, rather than the broad categorical diagnosis of autism itself (67, 68). In some cases, these approaches based on quantitative endophenotypes have led to identification of specific genes such as CNTNAP2, which appears to modulate a language endophenotype in ASD (40). The same genomic region of CNTNAP2 associated with ASD has recently been associated with specific language impairment (68a). This finding is consistent with the assertion that genetic risk variants are likely to affect normal variation in cognitive endophenotypes that cut across current qualitative definitions of psychiatric disorders (10). The comorbidity of ASD with other neuropsychiatric disorders, including ADHD (Table 1), further emphasizes the need to look beyond categorical diagnostic boundaries. Additional definition and study of quantitative ASD-related endophenotypes—from cognitive, electrophysiological, and structural imaging variables to biochemical measures, such as gene expression (69)—will be important for progress in delineating ASD genetic risk and understanding its underlying pathophysiology in relation to other brain disorders.
A FINAL NOTE ON TREATMENT AND EARLY INTERVENTION: HOPE FOR THE FUTURE
A wide variety of potential interventions for ASD have been championed, but few have been subjected to the rigors of controlled clinical trials. Only one pharmacological treatment for ASD, risperidone, has been approved by the U.S. Food and Drug Administration, and it does not target the core symptoms but rather certain maladaptive behaviors.
Despite the relative paucity of controlled treatment trials, an emerging body of evidence supports the efficacy of early, intensive behavioral and cognitive intervention strategies, especially in the improvement of language and social functioning (70, 71). Of the methods studied, applied behavior analysis (ABA) has the most experimental support, probably owing to its wide use. Overall, it appears that earlier and more intensive interventions are more powerful (72). Although significant improvement can be seen, few children treated with cognitive or behavioral therapy attain actual remission of ASD. Longitudinal studies of ASD started in the 1980s and 1990s indicate that the long-term prognosis of children diagnosed with an ASD is poor, with >70% not achieving independent status (73, 74). There is an urgent need for more efficacious pharmacologic and cognitive-behavioral therapies and a better notion of which therapy is most appropriate for which child. The identification of genetic and other biomarkers for specific subtypes and their relation to trajectory and treatment response constitute another critical area of research.
The recent demonstrations in animal models that certain forms of neurodevelopmental disorders associated with autism, such as fragile X, tuberous sclerosis, and Rett syndrome, can be largely reversed in adulthood represent a paradigm shift in our concept of developmental disorders (75–77). Should these findings generalize to humans, genetically identified pathway therapeutics would become the most important area of future treatment research in ASD.
PATERNAL AGE AND ASD.
Two recent studies identify advancing paternal age as a significant risk factor for ASD (78, 79). One explanation is that men with features of the broader phenotype, who carry more genetic liability, may marry and have children later. Another potential explanation could be the increase in mutation burden observed in older sperm (80). These two hypotheses, which are not mutually exclusive, can be rigorously tested with whole-genome CNV data, study of epigenetic factors, or gene resequencing, as well as behavioral testing. It is interesting to note that, in either case, delays in having children would lead to an increase in ASD prevalence. Another recent study shows increasing risk for ASD with increasing age in mothers as well, albeit less than the risk associated with fathers (81). These important observations highlight an area of potential environmental contribution to ASD. Should the increased mutation burden or epigenetic alterations in the male or female gamete be implicated, it would provide a new window for understanding gene-environment interactions.
SUMMARY POINTS.
- Autism is a syndrome, not an etiologically defined disorder. The term autism spectrum disorder (ASD) accommodates less severe forms of autism and the wide variability in its clinical presentation.
- At least half of children with ASDs have an IQ > 70 and are not considered intellectually disabled.
- The risk for autism has a high genetic component, typically estimated at between 70% and 90%.
- Genetic mutations causing autism can be identified in ~10%–20% of cases using current methods, many of which detect copy number variants (CNVs).
- No single genetic mutation accounts for more than ~1% of ASD, suggesting that it is etiologically very heterogeneous.
- No unifying pathology has been identified in autism, although changes in brain growth trajectory and involvement of frontal and anterior temporal lobes have been replicated.
FUTURE ISSUES.
- Large-scale studies of clinically well-characterized cohorts need to be completed to better define different clinical subtypes using neuroimaging and biomarker discovery. Such cohorts will also be useful for genetic studies, as thousands of cases and controls will be needed for adequate power to detect common and rare genetic susceptibility factors. Distinct subtypes of ASD are also likely to have different trajectories and respond differently to therapies, so the definition of ASD subtypes will inform treatment studies in critical ways.
- Key aspects of autism neuropathology need to be investigated using detailed stereological anatomic study, as well as biochemical, cellular, and molecular investigation in a large group of postmortem brains. These elements can be related back to clinical, imaging, and genetic features to define a neurobiology of ASD.
- Many investigators are currently working to develop biomarkers that will allow earlier identification of those at risk for ASD and help define distinct etiologic subtypes, so as to facilitate early therapeutic intervention.
- More validated and accepted animal models should be produced to accelerate targeted development of pathway-based pharmacotherapy. Understanding evolutionary differences between humans and animal models will clinically inform such studies.
Acknowledgments
The author is grateful to Portia Iverson and Jonathan Shestack, founders of Cure Autism Now, who challenged him to enter the field of autism, as well as his colleagues and students in the Center for Autism Research and Treatment at UCLA, who have also helped guide his work. Research in the Geschwind laboratory has been supported by funding from NIMH, Cure Autism Now, Autism Speaks, and the Tourette Syndrome Foundation. The author also thanks Li Hong and Lauren Kawaguchi for their editorial assistance.
Glossary
Asperger syndrome
a form of ASD less severe than autistic disorder, in which the domain of language is spared and there is no clinically significant cognitive deficit
Pervasive developmental delay not otherwise specified (PDD-NOS)
Diagnosis for patients with qualitative impairment in social function, who do not meet criteria for autistic disorder owing to late age of onset, atypical or subthreshold symptomatology, or both
Heritability
the extent to which a particular trait is due to inherited, genetic factors, usually scaled between zero and one, where 1.0 indicates 100% heritability
DNA microarray
an ordered microscopic array of DNA fragments, usually oligonucleotides, that allows one to assess gene abundance or genetic polymorphisms throughout the whole genome in a single hybridization step
De novo
a de novo genetic alteration is one that arises either in the gamete or in the fertilized egg, rather than being present in the parental somatic genome
Endophenotypes
component, measurable features associated with an underlying psychiatric disorder, which are heritable and observed in nonaffected family members at a higher rate than in the general population
Footnotes
DISCLOSURE STATEMENT
Dr. Geschwind is a volunteer on the Autism Speaks Scientific Advisory Council.
LITERATURE CITED
- 1.Kanner L. Autistic disturbances of affective contact. Nerv Child. 1943;2:217–50. [PubMed] [Google Scholar]
- 2.Friedman E. The autistic syndrome and phenylketonuria. Schizophrenia. 1969;1:249–61. [Google Scholar]
- 3.Cantwell DP, Baker L, Rutter M. Families of autistic and dysphasic children. I Family life and interaction patterns. Arch Gen Psychiatry. 1979;36:682–87. doi: 10.1001/archpsyc.1979.01780060072008. [DOI] [PubMed] [Google Scholar]
- 4.Folstein S, Rutter M. Infantile autism: a genetic study of 21 twin pairs. J Child Psychol Psychiatry. 1977;18:297–321. doi: 10.1111/j.1469-7610.1977.tb00443.x. [DOI] [PubMed] [Google Scholar]
- 5.Ritvo ER, Freeman BJ, Mason-Brothers A, et al. Concordance for the syndrome of autism in 40 pairs of afflicted twins. Am J Psychiatry. 1985;142:74–77. doi: 10.1176/ajp.142.1.74. [DOI] [PubMed] [Google Scholar]
- 6.Gillberg C, Wahlström J. Chromosome abnormalities in infantile autism and other childhood psychoses: a population study of 66 cases. Dev Med Child Neurol. 1985;27:293–304. doi: 10.1111/j.1469-8749.1985.tb04539.x. [DOI] [PubMed] [Google Scholar]
- 7.Wahlström J, Gillberg C, Gustavson KH, et al. Infantile autism and the fragile X. A Swedish multicenter study. Am J Med Genet. 1986;23:403–8. doi: 10.1002/ajmg.1320230132. [DOI] [PubMed] [Google Scholar]
- 8.American Psychiatric Association. Diagnostic and Statistical Manual of Mental Disorders. Washington, DC: Am. Psychiatr. Assoc; 2000. [Google Scholar]
- 9.Wing L, Gould J. Severe impairments of social interaction and associated abnormalities in children: epidemiology and classification. J Autism Dev Disord. 1979;9:11–29. doi: 10.1007/BF01531288. [DOI] [PubMed] [Google Scholar]
- 10.Abrahams BS, Geschwind DH. Advances in autism genetics: on the threshold of a new neurobiology. Nat Rev Genet. 2008;9:341–55. doi: 10.1038/nrg2346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Happe F, Ronald A, Plomin R. Time to give up on a single explanation for autism. Nat Neurosci. 2006;9:1218–20. doi: 10.1038/nn1770. [DOI] [PubMed] [Google Scholar]
- 12.Rutter M. The development of infantile autism. Psychol Med. 1974;4:147–63. doi: 10.1017/s0033291700041982. [DOI] [PubMed] [Google Scholar]
- 13.Howlin P, Goode S, Hutton J, et al. Adult outcome for children with autism. J Child Psychol Psychiatry. 2004;45:212–29. doi: 10.1111/j.1469-7610.2004.00215.x. [DOI] [PubMed] [Google Scholar]
- 14.Szatmari P, Bryson SE, Streiner DL, et al. Two-year outcome of preschool children with autism or Asperger’s syndrome. Am J Psychiatry. 2000;157:1980–87. doi: 10.1176/appi.ajp.157.12.1980. [DOI] [PubMed] [Google Scholar]
- 15.Venter A, Lord C, Schopler E. A follow-up study of high-functioning autistic children. J Child Psychol Psychiatry. 1992;33:489–507. doi: 10.1111/j.1469-7610.1992.tb00887.x. [DOI] [PubMed] [Google Scholar]
- 16.Charman T, Taylor E, Drew A, et al. Outcome at 7 years of children diagnosed with autism at age 2: predictive validity of assessments conducted at 2 and 3 years of age and pattern of symptom change over time. J Child Psychol Psychiatry. 2005;46:500–13. doi: 10.1111/j.1469-7610.2004.00377.x. [DOI] [PubMed] [Google Scholar]
- 17.Coleman M. Studies of the autistic syndromes. Res Publ Assoc Res Nerv Ment Dis. 1979;57:265–75. [PubMed] [Google Scholar]
- 18.Geschwind DH, Levitt P. Autism spectrum disorders: developmental disconnection syndromes. Curr Opin Neurobiol. 2007;17:103–11. doi: 10.1016/j.conb.2007.01.009. [DOI] [PubMed] [Google Scholar]
- 19.Wing L, Yeates SR, Brierley LM, et al. The prevalence of early childhood autism: comparison of administrative and epidemiological studies. Psychol Med. 1976;6:89–100. doi: 10.1017/s0033291700007522. [DOI] [PubMed] [Google Scholar]
- 20.Bryson SE, Clark BS, Smith IM. First report of a Canadian epidemiological study of autistic syndromes. J Child Psychol Psychiatry. 1988;29:433–45. doi: 10.1111/j.1469-7610.1988.tb00735.x. [DOI] [PubMed] [Google Scholar]
- 21.Autism and Developmental Disabilities Monitoring Network Surveillance Year; Principal Investigators, Centers for Disease Control and Prevention. 2007 Prevalence of autism spectrum disorders—autism and developmental disabilities monitoring network, 14 sites, United States, 2002. MMWR Surveill Summ. 2002;56:12–28. [PubMed] [Google Scholar]
- 22.Chakrabarti S, Fombonne E. Pervasive developmental disorders in preschool children. JAMA. 2001;285:3093–99. doi: 10.1001/jama.285.24.3093. [DOI] [PubMed] [Google Scholar]
- 23.Fombonne E, Zakarian R, Bennett A, et al. Pervasive developmental disorders in Montreal, Quebec, Canada: prevalence and links with immunizations. Pediatrics. 2006;118:139–50. doi: 10.1542/peds.2005-2993. [DOI] [PubMed] [Google Scholar]
- 24.Richler J, Luyster R, Risi S, et al. Is there a ‘regressive phenotype’ of autism spectrum disorder associated with the measles-mumps-rubella vaccine? A CPEA study. J Autism Dev Disord. 2006;36:299–316. doi: 10.1007/s10803-005-0070-1. [DOI] [PubMed] [Google Scholar]
- 25.Sweeten TL, Posey DJ, McDougle CJ. Brief report: autistic disorder in three children with cytomegalovirus infection. J Autism Dev Disord. 2004;34:583–86. doi: 10.1007/s10803-004-2552-y. [DOI] [PubMed] [Google Scholar]
- 26.Fombonne E, Du Mazaubrun C, Cans C, et al. Autism and associated medical disorders in a French epidemiological survey. J Am Acad Child Adolesc Psychiatry. 1997;36:1561–69. doi: 10.1016/S0890-8567(09)66566-7. [DOI] [PubMed] [Google Scholar]
- 27.Hultman CM, Sparen P, Cnattingius S. Perinatal risk factors for infantile autism. Epidemiology. 2002;13:417–23. doi: 10.1097/00001648-200207000-00009. [DOI] [PubMed] [Google Scholar]
- 28.Larsson HJ, Eaton WW, Madsen KM, et al. Risk factors for autism: perinatal factors, parental psychiatric history, and socioeconomic status. Am J Epidemiol. 2005;161:916–25. 26–28. doi: 10.1093/aje/kwi123. [DOI] [PubMed] [Google Scholar]
- 29.Giedd JN, Lenroot RK, Shaw P, et al. Trajectories of anatomic brain development as a phenotype. Novartis Found Symp. 2008;289:101–12. 12–18, 93–95. doi: 10.1002/9780470751251.ch9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Shaw P, Greenstein D, Lerch J, et al. Intellectual ability and cortical development in children and adolescents. Nature. 2006;440:676–79. doi: 10.1038/nature04513. [DOI] [PubMed] [Google Scholar]
- 31.Courchesne E, Carper R, Akshoomoff N. Evidence of brain overgrowth in the first year of life in autism. JAMA. 2003;290:337–44. doi: 10.1001/jama.290.3.337. [DOI] [PubMed] [Google Scholar]
- 32.Courchesne E, Pierce K, Schumann CM, et al. Mapping early brain development in autism. Neuron. 2007;56:399–413. doi: 10.1016/j.neuron.2007.10.016. [DOI] [PubMed] [Google Scholar]
- 33.Hazlett HC, Poe M, Gerig G, et al. Magnetic resonance imaging and head circumference study of brain size in autism: birth through age 2 years. Arch Gen Psychiatry. 2005;62:1366–76. doi: 10.1001/archpsyc.62.12.1366. [DOI] [PubMed] [Google Scholar]
- 34.Fombonne E, Roge B, Claverie J, et al. Microcephaly and macrocephaly in autism. J Autism Dev Disord. 1999;29:113–19. doi: 10.1023/a:1023036509476. [DOI] [PubMed] [Google Scholar]
- 35.Bolton PF, Roobol M, Allsopp L, et al. Association between idiopathic infantile macrocephaly and autism spectrum disorders. Lancet. 2001;358:726–27. doi: 10.1016/S0140-6736(01)05903-7. [DOI] [PubMed] [Google Scholar]
- 36.Miles JH, Hadden LL, Takahashi TN, et al. Head circumference is an independent clinical finding associated with autism. Am J Med Genet. 2000;95:339–50. [PubMed] [Google Scholar]
- 37.Abrahams BS, Tentler D, Perederiy JV, et al. Genome-wide analyses of human perisylvian cerebral cortical patterning. Proc Natl Acad Sci USA. 2007;104:17849–54. doi: 10.1073/pnas.0706128104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Amaral DG, Schumann CM, Nordahl CW. Neuroanatomy of autism. Trends Neurosci. 2008;31:137–45. doi: 10.1016/j.tins.2007.12.005. [DOI] [PubMed] [Google Scholar]
- 39.Mundy P. Annotation: the neural basis of social impairments in autism: the role of the dorsal medial-frontal cortex and anterior cingulate system. J Child Psychol Psychiatry. 2003;44:793–809. doi: 10.1111/1469-7610.00165. [DOI] [PubMed] [Google Scholar]
- 40.Alarcón M, Abrahams BS, Stone JL, et al. Linkage, association, and gene-expression analyses identify CNTNAP2 as an autism-susceptibility gene. Am J Hum Genet. 2008;82:150–59. doi: 10.1016/j.ajhg.2007.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Baron-Cohen S, Belmonte MK. Autism: a window onto the development of the social and the analytic brain. Annu Rev Neurosci. 2005;28:109–26. doi: 10.1146/annurev.neuro.27.070203.144137. [DOI] [PubMed] [Google Scholar]
- 42.Just MA, Cherkassky VL, Keller TA, et al. Functional and anatomical cortical underconnectivity in autism: evidence from an FMRI study of an executive function task and corpus callosum morphometry. Cereb Cortex. 2007;17:951–61. doi: 10.1093/cercor/bhl006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Catani M, Jones DK, Daly E, et al. Altered cerebellar feedback projections in Asperger syndrome. Neuroimage. 2008;41:1184–91. doi: 10.1016/j.neuroimage.2008.03.041. [DOI] [PubMed] [Google Scholar]
- 44.Nordahl CW, Dierker D, Mostafavi I, et al. Cortical folding abnormalities in autism revealed by surface-based morphometry. J Neurosci. 2007;27:11725–35. doi: 10.1523/JNEUROSCI.0777-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Levitt JG, Blanton RE, Smalley S, et al. Cortical sulcal maps in autism. Cereb Cortex. 2003;13:728–35. doi: 10.1093/cercor/13.7.728. [DOI] [PubMed] [Google Scholar]
- 46.Bauman ML, Kemper TL. The neuropathology of the autism spectrum disorders: What have we learned? Novartis Found Symp. 2003;251:112–22. discussion 22–28, 281–97. [PubMed] [Google Scholar]
- 47.Bailey A, Luthert P, Dean A, et al. A clinicopathological study of autism. Brain. 1998;121(Pt. 5):889–905. doi: 10.1093/brain/121.5.889. [DOI] [PubMed] [Google Scholar]
- 48.Casanova MF. The neuropathology of autism. Brain Pathol. 2007;17:422–33. doi: 10.1111/j.1750-3639.2007.00100.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Buxhoeveden DP, Semendeferi K, Buckwalter J, et al. Reduced minicolumns in the frontal cortex of patients with autism. Neuropathol Appl Neurobiol. 2006;33:720–21. doi: 10.1111/j.1365-2990.2006.00745.x. [DOI] [PubMed] [Google Scholar]
- 50.Schumann CM, Amaral DG. Stereological analysis of amygdala neuron number in autism. J Neurosci. 2006;26:7674–79. doi: 10.1523/JNEUROSCI.1285-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Vargas DL, Nascimbene C, Krishnan C, et al. Neuroglial activation and neuroinflammation in the brain of patients with autism. Ann Neurol. 2005;57:67–81. doi: 10.1002/ana.20315. [DOI] [PubMed] [Google Scholar]
- 52.Stevens B, Allen NJ, Vazquez LE, et al. The classical complement cascade mediates CNS synapse elimination. Cell. 2007;131:1164–78. doi: 10.1016/j.cell.2007.10.036. [DOI] [PubMed] [Google Scholar]
- 53.Bessis A, Bechade C, Bernard D, et al. Microglial control of neuronal death and synaptic properties. Glia. 2007;55:233–38. doi: 10.1002/glia.20459. [DOI] [PubMed] [Google Scholar]
- 54.Sebat J, Lakshmi B, Malhotra D, et al. Strong association of de novo copy number mutations with autism. Science. 2007;316:445–49. doi: 10.1126/science.1138659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Jacquemont ML, Sanlaville D, Redon R, et al. Array-based comparative genomic hybridisation identifies high frequency of cryptic chromosomal rearrangements in patients with syndromic autism spectrum disorders. J Med Genet. 2006;43:843–49. doi: 10.1136/jmg.2006.043166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Marshall CR, Noor A, Vincent JB, et al. Structural variation of chromosomes in autism spectrum disorder. Am J Hum Genet. 2008;82:477–88. doi: 10.1016/j.ajhg.2007.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Weiss LA, Shen Y, Korn JM, et al. Autism Consortium. Association between microdeletion and microduplication at 16p11.2 and autism. N Engl J Med. 2008;358:667–75. doi: 10.1056/NEJMoa075974. [DOI] [PubMed] [Google Scholar]
- 58.Szatmari P, Paterson AD, Zwaigenbaum L, et al. Autism Genome Project Consortium. Mapping autism risk loci using genetic linkage and chromosomal rearrangements. Nat Genet. 2007;39:319–28. doi: 10.1038/ng1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Frayling TM. Genome-wide association studies provide new insights into type 2 diabetes aetiology. Nat Rev Genet. 2007;8:657–62. doi: 10.1038/nrg2178. [DOI] [PubMed] [Google Scholar]
- 60.Bolton P, Macdonald H, Pickles A, et al. A case-control family history study of autism. J Child Psychol Psychiatry. 1994;35:877–900. doi: 10.1111/j.1469-7610.1994.tb02300.x. [DOI] [PubMed] [Google Scholar]
- 61.Fombonne E, Bolton P, Prior J, et al. A family study of autism: cognitive patterns and levels in parents and siblings. J Child Psychol Psychiatry. 1997;38:667–83. doi: 10.1111/j.1469-7610.1997.tb01694.x. [DOI] [PubMed] [Google Scholar]
- 62.Pickles A, Starr E, Kazak S, et al. Variable expression of the autism broader phenotype: findings from extended pedigrees. J Child Psychol Psychiatry. 2000;41:491–502. [PubMed] [Google Scholar]
- 63.Piven J, Palmer P, Jacobi D, et al. Broader autism phenotype: evidence from a family history study of multiple-incidence autism families. Am J Psychiatry. 1997;154:185–90. doi: 10.1176/ajp.154.2.185. [DOI] [PubMed] [Google Scholar]
- 64.Le Couteur A, Bailey A, Goode S, et al. A broader phenotype of autism: the clinical spectrum in twins. J Child Psychol Psychiatry. 1996;37:785–801. doi: 10.1111/j.1469-7610.1996.tb01475.x. [DOI] [PubMed] [Google Scholar]
- 65.Constantino JN, Lajonchere C, Lutz M, et al. Autistic social impairment in the siblings of children with pervasive developmental disorders. Am J Psychiatry. 2006;163:294–96. doi: 10.1176/appi.ajp.163.2.294. [DOI] [PubMed] [Google Scholar]
- 65a.Geschwind DH. Autism: Many genes, common pathways? Cell. 2008 doi: 10.1016/j.cell.2008.10.016. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Ronald A, Happe F, Bolton P, et al. Genetic heterogeneity between the three components of the autism spectrum: a twin study. J Am Acad Child Adolesc Psychiatry. 2006;45:691–99. doi: 10.1097/01.chi.0000215325.13058.9d. [DOI] [PubMed] [Google Scholar]
- 67.Alarcón M, Cantor RM, Liu J, et al. Autism Genetic Research Exchange Consortium. Evidence for a language quantitative trait locus on chromosome 7q in multiplex autism families. Am J Hum Genet. 2002;70:60–71. doi: 10.1086/338241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Duvall JA, Lu A, Cantor RM, et al. A quantitative trait locus analysis of social responsiveness in multiplex autism families. Am J Psychiatry. 2007;164:656–62. doi: 10.1176/ajp.2007.164.4.656. [DOI] [PubMed] [Google Scholar]
- 68a.Vernes SC, Newbury DF, Abrahams BS, et al. A functional genetic link between distinct developmental language disorders. N Engl J Med. 2008 doi: 10.1056/NEJMoa0802828. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Nishimura Y, Lese-Martin C, Vazquez-Lopez A, et al. Genome-wide expression profiling of lymphoblastoid cell lines distinguishes different forms of autism and reveals shared pathways. Hum Mol Genet. 2007;16:1682–98. doi: 10.1093/hmg/ddm116. [DOI] [PubMed] [Google Scholar]
- 70.Landa RJ. Diagnosis of autism spectrum disorders in the first 3 years of life. Nat Clin Pract Neurol. 2008;4:138–47. doi: 10.1038/ncpneuro0731. [DOI] [PubMed] [Google Scholar]
- 71.Rogers SJ, Vismara LA. Evidence-based comprehensive treatments for early autism. J Clin Child Adolesc Psychol. 2008;37:8–38. doi: 10.1080/15374410701817808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Eikeseth S. Outcome of comprehensive psycho-educational interventions for young children with autism. Res Dev Disabil. 2008 doi: 10.1016/j.ridd.2008.02.003. In press. [DOI] [PubMed] [Google Scholar]
- 73.Billstedt E, Gillberg IC, Gillberg C. Autism after adolescence: population-based 13- to 22-year follow-up study of 120 individuals with autism diagnosed in childhood. J Autism Dev Disord. 2005;35:351–60. doi: 10.1007/s10803-005-3302-5. [DOI] [PubMed] [Google Scholar]
- 74.Cederlund M, Hagberg B, Billstedt E, et al. Asperger syndrome and autism: a comparative longitudinal follow-up study more than 5 years after original diagnosis. J Autism Dev Disord. 2008;38:72–85. doi: 10.1007/s10803-007-0364-6. [DOI] [PubMed] [Google Scholar]
- 75.Dolen G, Osterweil E, Rao BS, et al. Correction of fragile X syndrome in mice. Neuron. 2007;56:955–62. doi: 10.1016/j.neuron.2007.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Ehninger D, Han S, Shilyansky C, et al. Reversal of learning deficits in a Tsc2(+/−) mouse model of tuberous sclerosis. Nat Med. 2008;14:843–48. doi: 10.1038/nm1788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Guy J, Gan J, Selfridge J, et al. Reversal of neurological defects in a mouse model of Rett syndrome. Science. 2007;315:1143–47. doi: 10.1126/science.1138389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Cantor RM, Yoon JL, Furr J, et al. Paternal age and autism are associated in a family-based sample. Mol Psychiatry. 2007;12:419–21. doi: 10.1038/sj.mp.4001966. [DOI] [PubMed] [Google Scholar]
- 79.Reichenberg A, Gross R, Weiser M, et al. Advancing paternal age and autism. Arch Gen Psychiatry. 2006;63:1026–32. doi: 10.1001/archpsyc.63.9.1026. [DOI] [PubMed] [Google Scholar]
- 80.Crow JF. The origins, patterns and implications of human spontaneous mutation. Nat Rev Genet. 2000;1:40–47. doi: 10.1038/35049558. [DOI] [PubMed] [Google Scholar]
- 81.Croen LA, Najjar DV, Fireman B, et al. Maternal and paternal age and risk of autism spectrum disorders. Arch Pediatr Adolesc Med. 2007;161:334–40. doi: 10.1001/archpedi.161.4.334. [DOI] [PubMed] [Google Scholar]
- 82.Gillberg C, Billstedt E. Autism and Asperger syndrome: coexistence with other clinical disorders. Acta Psychiatr Scand. 2000;102:321–30. doi: 10.1034/j.1600-0447.2000.102005321.x. [DOI] [PubMed] [Google Scholar]
- 83.Hansen RL, Ozonoff S, Krakowiak P, et al. Regression in autism: prevalence and associated factors in the CHARGE Study. Ambul Pediatr. 2008;8:25–31. doi: 10.1016/j.ambp.2007.08.006. [DOI] [PubMed] [Google Scholar]
- 84.Jansiewicz EM, Goldberg MC, Newschaffer CJ, et al. Motor signs distinguish children with high functioning autism and Asperger’s syndrome from controls. J Autism Dev Disord. 2006;36:613–21. doi: 10.1007/s10803-006-0109-y. [DOI] [PubMed] [Google Scholar]
- 85.Krakowiak P, Goodlin-Jones B, Hertz-Picciotto I, et al. Sleep problems in children with autism spectrum disorders, developmental delays, and typical development: a population-based study. J Sleep Res. 2008;17:197–206. doi: 10.1111/j.1365-2869.2008.00650.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Leekam SR, Nieto C, Libby SJ, et al. Describing the sensory abnormalities of children and adults with autism. J Autism Dev Disord. 2007;37:894–910. doi: 10.1007/s10803-006-0218-7. [DOI] [PubMed] [Google Scholar]
- 87.Malow BA, Marzec ML, McGrew SG, et al. Characterizing sleep in children with autism spectrum disorders: a multidimensional approach. Sleep. 2006;29:1563–71. doi: 10.1093/sleep/29.12.1563. [DOI] [PubMed] [Google Scholar]
- 88.Ming X, Brimacombe M, Wagner GC. Prevalence of motor impairment in autism spectrum disorders. Brain Dev. 2007;29:565–70. doi: 10.1016/j.braindev.2007.03.002. [DOI] [PubMed] [Google Scholar]
- 89.Simonoff E, Pickles A, Charman T, et al. Psychiatric disorders in children with autism spectrum disorders: prevalence, comorbidity, and associated factors in a population-derived sample. J Am Acad Child Adolesc Psychiatry. 2008;47:921–29. doi: 10.1097/CHI.0b013e318179964f. [DOI] [PubMed] [Google Scholar]
- 90.Tuchman R, Rapin I. Epilepsy in autism. Lancet Neurol. 2002;1:352–58. doi: 10.1016/s1474-4422(02)00160-6. [DOI] [PubMed] [Google Scholar]
- 91.Volkmar FR, Nelson DS. Seizure disorders in autism. J Am Acad Child Adolesc Psychiatry. 1990;29:127–29. doi: 10.1097/00004583-199001000-00020. [DOI] [PubMed] [Google Scholar]
- 92.Xue M, Brimacombe M, Chaaban J, et al. Autism spectrum disorders: concurrent clinical disorders. J Child Neurol. 2008;23:6–13. doi: 10.1177/0883073807307102. [DOI] [PubMed] [Google Scholar]