Structural Basis of the Cross-Reactivity of Genetically Related Human Anti-HIV-1 Monoclonal Antibodies: Potential Implications for Design of V3-based Immunogens (original) (raw)
. Author manuscript; available in PMC: 2013 Jun 15.
Published in final edited form as: Structure. 2009 Nov 11;17(11):1538–1546. doi: 10.1016/j.str.2009.09.012
SUMMARY
Human monoclonal antibodies (mAbs) 447-52D and 537-10D, both coded by the VH3 gene and specific for the third variable region (V3) of the HIV-1 gp120, were found to share antigen binding structural elements including an elongated CDR H3 forming main-chain interactions with the N-terminus of the V3 crown. However, water-mediated hydrogen bonds and a unique cation-π sandwich stacking allow 447-52D to be broadly reactive with V3 containing both the GPGR and GPGQ crown motifs, while the deeper binding pocket, and a buried Glu in the binding site of 537-10D limit its reactivity to only V3 containing the GPGR motif. Our results suggest that the design of immunogens for anti-V3 antibodies should avoid the Arg at the V3 crown, as GPGR-containing epitopes appear to select for B cells making antibodies of narrower specificity than V3 that carry Gln at this position.
Keywords: HIV-1, gp120, V3, monoclonal antibody, antibody-antigen interactions
INTRODUCTION
One way for human immunodeficiency virus type 1 (HIV-1) to escape antibody-mediated control and eradication is by genetic diversification of its epitopes. Designing immunogens that can elicit broadly neutralizing antibodies able to overcome this diversity remains a major challenge in developing an effective HIV vaccine. The virus’ surface glycoprotein gp120 has five variable regions, named V1-V5, that exhibit high sequence diversities. Among them, V3 is relatively conserved due to its functional importance in the life cycle of the virus – it determines the viral co-receptor usage and viral tropism of the virus by binding to the virus co-receptors on the cell surface, CCR5 or CXCR4, a critical step in viral entry (Ivanoff et al., 1992; McKeating et al., 1989; Resch et al., 2001; Wyatt and Sodroski, 1998). The V3 conservation is reflected both in its sequence and structure. V3 has limited variation in length; it is almost always 35 amino acids long, and it harbors at its tip a highly conserved motif: Gly-Pro-Gly-Arg/Gln (GPGR/Q, residues 312-315 in the HXB2 numbering scheme, Ratner et al., 1987). While the GPGR containing V3 (V3GPGR) is predominant in clade B viruses, V3GPGQ is more often found in clade C and other non-clade B viruses (Kuiken et al., 1999). V3 is structurally characterized in the context of the gp120 core (Huang et al., 2007; Huang et al., 2005). It protrudes ~30 Å from the CD4-bound gp120 core and this extended structure can be divided into 3 regions: the base, the stem and the crown. The conserved base is seated in the gp120 core with a disulfide bridge (Leonard et al., 1990), while the stem extends outwards and is flexible (Huang et al., 2007; Huang et al., 2005). The crown has a unique beta-conformation with the GPGR/Q turn at the distal apex of the crown. Interestingly, all the known human anti-V3 monoclonal antibodies (mAbs) are against the crown (Gorny et al., 1993; Zolla-Pazner et al., 2004; Zwart et al., 1991), and studies have shown that the crown is accessible on the surface of most HIV-1 primary isolates and serves as a neutralization epitope (Gorny et al., 2004).
V3 is highly immunogenic and was named the “principal neutralizing determinant” of HIV-1 (Javaherian et al., 1989). Anti-V3 antibodies are found to exist in the sera of essentially all patients infected with HIV-1 (Carrow et al., 1991; Krachmarov et al., 2005; Krachmarov et al., 2001; Vogel et al., 1994), and immunization of animals with V3-containing immunogens can elicit anti-V3 immune responses (Javaherian et al., 1990; Law et al., 2007; Zolla-Pazner et al., 2008). Importantly, a novel immunization regimen combining DNA primes and protein boosts has recently been shown to focus the rabbit immune response on the V3 epitope and elicit antibodies with cross-clade neutralization capacities (Zolla-Pazner et al., 2009; Zolla-Pazner et al., 2008). Moreover, there are monoclonal and polyclonal anti-V3 antibodies display both intraand inter-subtype neutralization of diverse HIV strains (Binley et al., 2004; Conley et al., 1994; Gorny et al., 1992; Krachmarov et al., 2005). Thus, the V3 epitope is well-suited as a target for HIV-1 vaccine development (Zolla-Pazner, 2004).
Structural characterization of anti-HIV-1 human mAbs targeting immunogenic epitopes on the surface glycoprotein gp120 of HIV-1 can potentially facilitate the design and development of an effective HIV-1 vaccine (Burton, 1997; Douek et al., 2006; Zolla-Pazner, 2004). A structure-aided reverse-engineering strategy of immunogen design requires (i) identification of broadly reactive human mAbs, (ii) determination of their structures in complex with epitopes, and (iii) engineering of the structures of these epitopes onto protein scaffolds. These steps are designed to enable scaffolded epitopes to elicit antibodies with a breadth of neutralizing activity similar to that displayed by the broadly reactive mAbs. Much progress has been made in identifying human anti-V3 mAbs, and a large number of anti-V3 mAbs have been produced (Gorny and Zolla-Pazner, 2003; Gorny et al., 2009).
Many anti-V3 mAbs, especially those selected from animals immunized with V3 peptides (Goudsmit et al., 1988; Palker et al., 1988), are considered “type-specific”. However, a considerable number of anti-V3 mAbs isolated from chronically infected human individuals and selected for specificity with V3 fusion proteins have been found to be broadly reactive (Binley et al., 2004; Gorny et al., 2004; Gorny et al., 2002; Gorny et al., 2006), displaying cross-clade neutralizing activity against both pseudoviruses and primary isolates (Davis et al., 2009; Gorny et al., 2004; Gorny et al., 2002; Gorny et al., 2006; Krachmarov et al., 2006). Efforts have been made to understand the origins of this broad reactivity from a genetic as well as structural basis (Gorny et al., 2009; Stanfield et al., 2004; Stanfield et al., 2006; Stanfield and Wilson, 2005), but many questions remain unanswered.
Monoclonal Ab 447-52D (IgG3, λ), originally derived from an individual infected with clade B HIV-1 (Gorny et al., 1992), is the best-characterized human anti-V3 mAb, and has been used as the prototype of anti-V3 mAbs (Binley et al., 2008; Binley et al., 2004; Burton et al., 2004; Karlsson Hedestam et al., 2008; Kwong and Wilson, 2009). Experiments have shown that 447-52D can neutralize between 30-50% of HIV-1 in panels of tested primary isolates from clades A, B, F, and H (Binley et al., 2004; Gorny et al., 2002; Zolla-Pazner et al., 2004). However, it preferentially neutralizes viruses that harbor the GPGR motif in the V3 crown, with limited capacity against those with the GPGQ motif (Cardozo et al., 2009; Davis et al., 2009; Zolla-Pazner et al., 2004). Its neutralization potency against pseudoviruses with R to Q mutation in the crown motif is decreased ~700-fold (Krachmarov et al., 2006). These data suggest that 447-52D is highly specific for viruses harboring the GPGR V3 motif. The structural basis of its antigen recognition has been investigated using both NMR and protein crystallography, showing that 447-52D has a highly unusual antigen-binding site (Dhillon et al., 2008; Sharon et al., 2003; Stanfield et al., 2004). It possesses an extended complementarity determining region (CDR) H3, which protruds from the body of the molecule and forms main-chain interactions with the N-terminal region of V3 flanking the GPGR turn. Furthermore, the antigen-binding site has a unique structure that forms a 3-residue cation-π stacking which sandwiches the Arg side-chain of the GPGR motif. Although 447-52D has poor or no neutralizing activity against viruses bearing the GPGQ V3 motif, it is able to bind V3 peptides with the GPGQ motif (Gorny et al., 2002; Zolla-Pazner et al., 2004). The structural details of how the active site of 447-52D interacts with V3 containing the GPGQ motif remain unclear.
Human mAb 537-10D (IgG1, λ) is genetically related to 447-52D; it is also coded by the VH3 Ig gene family: 447-52D and 537-10D use the VH3-15 and VH3-7 Ig gene segments, respectively (Gorny et al., 2009; Gorny et al., 1993). Thus, the two antibodies are highly homologous in their amino acid sequence. Like 447-52D, 537-10D also possesses a long CDR H3 loop that is 20 amino acids in length (Kabat numbering; (Kabat et al., 1991)). However, 537-10D has a much narrower breadth of reactivity and a lower potency of neutralization; it is known to only neutralize HIV-1 strains MN and BaL (both harboring the GPGR motif), with a potency 50 times less than that of 447-52D (Gorny et al., 1992; Gorny et al., 2004; Gorny et al., 1993). The structural basis for this narrower reactivity and reduced potency is not yet understood.
A structural comparison of homologous human mAbs derived from similar genetic origins but with very different antigenic properties would help to further our understanding of the structural basis of antigen recognition, and potentially advance HIV-1 vaccine development by facilitating the design of immunogens inducing broadly neutralizing antibodies against the V3 region of gp120 (Zolla-Pazner, 2005). We have therefore examined the structural differences between these two homologous antibodies by crystallizing Fab fragments of 447-52D and 537-10D in complex with V3 peptides carrying the GPGQ and GPGR motifs, respectively.
RESULTS
Breadth of reactivity of mAbs 447-52D and 537-10D measured by ELISA
Despite the sequence similarities between mAbs 447-52D and 537-10D (Figure 1A, Supplement Figure S1), they were previously shown to display different binding affinities to V3 peptides, and different patterns of peptide cross-reactivity and virus neutralizing activity (Gorny et al., 1992; Gorny et al., 2004; Gorny et al., 1993). To systematically test their reactivity against different V3s, we measured their binding reactivity by Enzyme-Linked Immunosorbent Assay (ELISA) with a panel of 24 V3 peptides representing a wide range of virus isolates from clades A, B, and C (Figure 1B). The 447-52D antibody was able to bind to peptides containing both the GPGR and GPGQ motifs, while the 537-10D showed a binding pattern restricted to the former. Thus, mAb 447-52D exhibited broad reactivity, binding to 22/24 V3 peptides, including those from clades A, B and C (the two non-reactive peptides, CMI.CA7 and UR29, have amino acid sequences unusual in HIV-1 strains at the β-turn of the V3 loop). In contrast, mAb 537-10D had a much narrower pattern of reactivity, only reacting with peptides containing the GPGR apex motif (Figure 1B). Therefore, although anti-V3 mAbs 447-52D and 537-10D have considerable sequence similarities, they display very different reactivity profiles.
Figure 1. 447-52D and 537-10D: sequences of the variable fragments and cross-reactivity with V3 peptides.

(A) Protein sequence alignments of the variable regions for both heavy and light chains of 447-52D and 537-10D. Boxes indicate the CDR regions (Kabat definition), while asterisks and arrowheads mark the residues making direct or water-mediated interactions in the reported structures with the V3 epitopes, respectively. TrpH47 (arrow) of 537-10D has direct contacts with the epitope, although it is not in any of the CDR loops (see Figures 2B and 4B). Note multiple Tyr residues in CDR H3 of both mAbs. (B) The ELISA reactivities of 447-52D and 537-10D against a panel of V3 peptides (sorted according to their ELISA measurements). Only sequences corresponding to the regions of V3 in the structures that have direct contacts with the antibodies are shown (9 amino acids for 447-52D and 11 for 537-10D), and they are numbered at the bottom in the HXB2 numbering scheme. The colors depict the sequence similarities. Note that 447-52D reacts with both V3GPGR and V3GPGQ peptides, while 537-10D only reacts with V3GPGR.
Structure of the Fab fragment of mAb 447-52D in complex with a V3GPGQ peptide
To understand how mAb 447-52D binds V3GPGQ, we determined the crystal structure of the Fab fragment of 447-52D with a 23-mer peptide derived from a clade A virus strain W2RW020 (V3W2R) containing the GPGQ motif. The structure was refined to 2.25Å resolution (Figure 2A, Table 1 and Supplement Figure S2A). There are two Fab/V3W2R complexes in the crystallographic asymmetric unit, but they were highly similar, with a root mean square deviation of less than 0.6Å when superimposed. For clarity, we will therefore refer to only one of the Fab/V3 complexes. We will follow the Kabat and Wu convention (Kabat et al., 1991) for numbering of the heavy and light chains preceded by “H” and “L”, respectively, and designate the residues of the V3 peptide with a “P” and the residue number. Residues P305 to P316 of the V3 crown region, with the sequence KGVRIGPGQA, were observed to have strong electron densities and have thus been built into the final model. There was no visible electron density after AlaP316, reflecting additional flexibility of the C-terminus of V3 in this complex in comparison with the previous mAb 447-52D Fab/V3 peptides structures (Dhillon et al., 2008; Stanfield et al., 2004), of which up to 3 additional residues were visible at an electron density contour level of 0.7σ.
Figure 2. Structures of Fab 447-52D/V3W2R and Fab 537-10D/V3MN complexes.

(A) Stereoview of the 447-52D/V3W2R complex. (B) Stereoview of the 537-10D/V3MN complex. The Fab light chain, heavy chain and the V3 peptide are colored cyan, green and magenta, respectively. Side chains of the residues of the antibodies that have direct contacts with the V3 epitopes are shown. (C) The mAbs 447-52D and (D) 537-10D are shown as surface representations to illustrate the ladle shape of the binding sites. Note that the bowl part of the 447-52D binding site, with an additional spout (arrow), is much shallower than that of 537-10D.
Table 1.
X-ray Diffraction Data and Refinement Statistics
| 447-52D/V3W2R | 537-10D/V3MN | |
|---|---|---|
| Crystal Features | ||
| Space Group | P21 | P21 |
| Unit Cell Dimensions | a = 70.27 Å | a = 46.36 Å |
| b = 76.21 Å | b = 71.32 Å | |
| c = 113.92 Å | c = 77.12 Å | |
| β = 101.25° | β = 104.25° | |
| Fab/V3 Complex per Asymmetric Unit | 2 | 1 |
| Data Quality | ||
| Wavelength (Å) | 0.9791 | 0.9790 |
| Resolution (Å) | 2.25 | 2.4 |
| Total Observations | 428365 | 115181 |
| Unique Reflections | 56738 | 18997 |
| Completeness (%) | 99.9 (99.4) | 97.9 (86.3) |
| I/σ (I) | 18.6 (4.6) | 15.3 (2.0) |
| Rsym (%) | 8.8 (48.5) | 8.0 (33.1) |
| Model Quality | ||
| Rcryst(%) | 19.6 | 18.7 |
| Rfree (%) | 23.2 | 24.1 |
| Total Residues | 921 | 455 |
| Water Molecules | 551 | 322 |
| Mean B value (Å2) | 32.2 | 30.9 |
| Rms deviation for bond lengths (Å) | 0.006 | 0.006 |
| Rms deviation for bond angles (°) | 1.38 | 1.36 |
| Ramachandran plot (Procheck) | ||
| Most favored regions (%) | 88.6 | 88.3 |
| Allowed regions (%) | 10.9 | 10.4 |
| Generously allowed (%) | 0.3 | 0.3 |
| Outlier regions (%) | 0.3 | 1.0 |
The position and orientation of the V3W2R peptide in the complex is very similar to that observed for the structures of 447-52D/V3GPGR complexes reported previously (Dhillon et al., 2008; Stanfield et al., 2004). The antigen-binding site adopts a shape similar to a soup ladle (Figures 2A and 2C). The “handle” of the ladle is formed by the elongated β hairpin structure of the CDR H3 loop extending out from the body of the mAb, while the “bowl” is formed by the base of CDR H3 together with the other CDR loops (Supplemental Figure S2A). The five N-terminal residues flanking the apex of V3 form an anti-parallel 3-stranded β-sheet together with the hairpin of the CDR H3 (Figures 2A, 3A and S2A). There are 4 hydrogen bonds are between the main chain backbones of CDR H3 and the N-terminal side of V3 flanking the apex turn; this is similar to previously described 447-52D/V3GPGR structures (Dhillon et al., 2008; Stanfield et al., 2004) (Figure 3A; Supplemental Table S1A). There is also a side-chain-specific interaction between the hydroxyl group of CDR H3 residue TyrH100J and the side chain of ArgP308, analogous to the TyrH100J-HisP308 hydrogen bond observed for the 447-52D/V3GPGR structures (Dhillon et al., 2008; Stanfield et al., 2004). Four consecutive tyrosine residues in the heavy chain CDR H3 on the β-strand facing V3 provide an extended surface area for binding (Fellouse et al., 2004). Two of these, TyrH100G and TyrH100I, together with TyrL32 from the light chain, interact with ValP307 and IleP309, two V3 positions usually occupied by hydrophobic residues Ile/Val (Kuiken et al., 1999). The anti-parallel configuration of the CDR H3 and V3 loop β-strand orients the V3 β-turn into the 447-52D ladle, where three residues from the light chain CDR L2 loop, TrpL91, AlaL95B and TrpL96, form a ~90° corner for the docking of the GlyP312-ProP313-GlyP314 turn of the V3 apex (Figures 2A and 4A). Here, the pyrrolidine ring of ProP313 is placed parallel to the indole ring of the antibody residue TrpL91 with a distance of ~3.6Å between the aromatic planes (Figure 4A).
Figure 3. Main-chain interactions.

(A) and (B) Main-chain interactions between V3 peptide and CDR H3 in the 447-52D/V3W2R (A), and 537-10D/V3MN (B) complexes. The CDR H3 (green) and V3 (magenta) interactions of the 447-52D and 537-10D Fab/V3 structures are shown as ribbons (top panels) as well as with detailed hydrogen bonds (lower panels).
Figure 4. Structural details of the epitope-binding around the apex of the V3 crown in Fab 447-52D/V3W2R and Fab 537-10D/V3MN complexes.

(A) Stereoview of the 447-52D/V3W2R complex. (B) Stereoview of the 537-10D/V3MN complex. Note the two water molecules (water 1 and 2) at the antigen binding site of 447-52D. Also note that TrpH47, a non-CDR residue, has direct contact with the epitope in 537-10D.
The principal difference between the 447-52D Fab/V3W2R structure and the previously determined Fab/V3GPGR complexes (Dhillon et al., 2008; Stanfield et al., 2004) lies in the interactions between the antibody and the side chains of the ArgP315/GlnP315 of the GPGR/Q motif (Figures 4A, 5A, 5B and Supplemental Figure S3). In the 447-52D/V3GPGR structures, mAb 447-52D uses the residues TrpH33, AspH95 and TyrH100J to make highly unusual specific interactions with ArgP315. The guanidinium moiety of ArgP315 is sandwiched between the indole ring of TrpH33 and the phenyl ring of TyrH100J to form a rare 3-residue cation-π stacking. In addition, ArgP315 is positioned so that its side chain can form a salt bridge with AspH95 (Supplemental Figure S3) (Dhillon et al., 2008; Stanfield et al., 2004). In our 447-52D Fab/V3W2R structure, the shorter GlnP315 is unable to form a cation-π interaction with the TrpH33 and TyrH100J, and is too far away to form a direct hydrogen bond with AspH95 (Figures 4A, 5A and 5B). Instead, GlnP315 forms a water-mediated hydrogen bond with the side chain of AspH95, resulting in a 30% decrease in the buried area at the Fab/V3 interface relative to the mAb interactions with the GPGR sequence (Supplement Table S2A).
Figure 5. Water mediated interactions at the antigen-binding site.
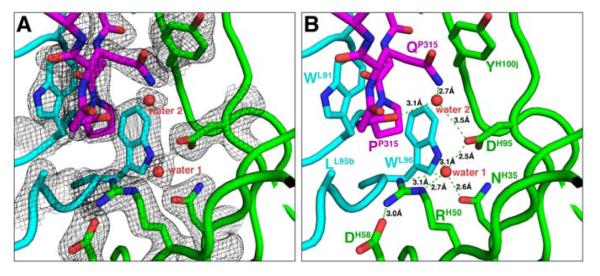
(A) Electron densities (the 2Fo-Fc map contoured at 1.2 σ level) around a well-coordinated water (water 1) molecule at the base of the antigen binding site in the 447-52D/V3W2R complex. (B) Water-mediated hydrogenbinding networks in the 447-52D/V3W2R complex. Note that water 1 is coordinated tetrahedrally by the side chains of four surrounding residues. This water molecule is also present in the 447-52D/V3GPGR complexes (see Figure S3). Note also that GlnP315 does not interact directly with AspH95 but mediated by another water molecule (water 2).
Interestingly, in addition to the AspH95 that is buried in the deep center of the binding site, there are 2 additional charged residues at the 447-52D binding site, ArgH50 and AspH58, with the latter placed at the very outer edge of the binding site (Figures 4A, 5A and 5B). ArgH50 in the middle of this chain of negatively-positively-negatively charged residues forms, on one side, an intra-molecular salt bridge with AspH58 and, on the other side, a water-mediated hydrogen bond with AspH95 (which forms a salt bridge with ArgP315 of V3 in the 447-52D/V3GPGR structures). This water molecule is tightly bound at the bottom of the antigen-binding site, and is coordinated tetrahedrally by the side chains of AsnH35, ArgH50, AspH95 and TrpL96 (Figure 5B).
Structure of the Fab fragment of mAb 537-10D in complex with a V3GPGR peptide
In order to compare the structures of the broadly reactive 447-52D with that of a “type-specific” human mAb, we determined the structure of Fab 537-10D in complex with a 23-mer peptide corresponding to the clade B viral isolate MN (V3MN), a GPGR containing V3, refined it to 2.4Å resolution (Figures 2B and S2B; Table 1). There was only one Fab/V3MN complex in the crystallographic asymmetric unit, and we again labeled the V3 peptide, and the antibody heavy and light chains with residue numbers preceded by P, H and L, respectively. Strong electron densities were found for 15 amino acids in the V3 peptide (P304 to P320) with sequence RKRIHIGPGRAFYAT, and were built into the final model. The structure of Fab 537-10D/V3MN is strikingly similar to that of the Fab 447-52D/V3W2R complex (Figure 2, Supplement Figure S2), permitting again the use of the soup ladle analogy for its antigen binding site. The comparable shape of the two mAb’s binding sites is determined by the related Ig genes coding for the variable fragments (see Supplemental Figure S1). However, the bowl of the 537-10D binding site is considerably deeper, measuring about 6Å in depth (Figure 2D), in contrast to the 2Å depth of the 447-52D antigen binding site (Figure 2C). This deeper bowl allows for the complete burial of the apex GPGR of the V3MN peptide, except the carbonyl oxygen of GlyP314 that is pointing outward.
Mab 537-10D CDR H3 forms 6 main-chain hydrogen bonds with the N-terminal strand of the V3MN peptide (Figure 3B; Supplement Table S1B), which is one and two more than observed for the 447-52D Fab/V3MN and the Fab/V3W2R crystal structures, respectively (Figure 3A, and (Dhillon et al., 2008; Stanfield et al., 2004)). Together with the C-terminal region flanking the V3 apex, V3 in 537-10D forms a well-ordered 4-stranded anti-parallel CDR H3/V3 β-sheet, rather than the 3-stranded sheet observed for 447-52D (Figure 3). Like 447-52D, interactions are also observed in the 537-10D/V3MN with residues IleP307 and IleP309 MN (the highly conserved hydrophobic positions), and at the GlyP312-ProP313-GlyP314 turn of the crown. There are 5 tyrosine residues in the 537-10D CDR H3, including the conserved TyrH100G and TyrH100I residues that, together with TyrL32, provide a hydrophobic surface for interaction with the hydrophobic IleP307 and IleP309 residues. The GlyP312-ProP313-GlyP314 turn of the V3 crown is docked into a hydrophobic, non-polar corner formed by ValL96, TrpH47 and MetH100L (Figures 2B and 4B). Interestingly, TrpH47 interacts directly with the V3 epitope, although it does not belong to any of the CDR loops (Figure 1A), contrary to the common perception that only residues in the CDR loops interact directly with epitopes.
MAb 537-10D’s interactions with the key residue ArgP315 of the V3 crown are similar to those of mAb 447-52D (Figures 4 and S3). Residues TrpH33 and TyrH100J that sandwich ArgP315 in 447-52D are conserved in 537-10D. The guanidinium moiety of ArgP315 stacks closely (~3.2Å) and almost parallel to the indole ring of the TrpH33 of 537-10D to allow a 2-residue cation-π stacking. However, there are some crucial differences. First, in contrast to 447-52D, the phenyl ring of TyrH100J is not able to sandwich ArgP315 on the other side. Instead, it is tilted ~60° and displaced to the side (Figure 4B). Second, although ArgP315 forms a salt bridge (2.72Å) with the negatively charged GluH95, a residue that is homologous to, but has a longer side chain than, the AspH95 of 447-52D, the salt bridge is completely buried at the 537-10D/V3 interface due to the deeper binding site. Furthermore, different from 447-52D, there are no alternately charged residues and mediating water molecules at the bottom of the 537-10D binding site.
DISCUSSION
The genetic origin of a human anti-V3 mAb may correlate with its capacity to neutralize HIV-1 viruses (Gorny et al., 2009), and we present here a structural comparison of two genetically related human anti-V3 mAbs: the broadly reactive 447-52D and the narrowly reactive (or “type specific”) 537-10D, both encoded by VH3 gene but with very different spectra of cross-reactivity (Figure 1). Our structures show that they do have highly similar antigen binding sites, shaped like a soup ladle, with the three key structural determinants (Figure 2). (i) The long CDR H3 – the handle of the ladle – binds to the V3 epitope through main-chain interactions (Figures 2-3). The long tyrosine-lined CDR H3 loop extends ~25Å away from the body of the antibody and interacts with the N-terminal end of the V3 crown. (ii) The hydrophobic corner – the left half of the ladle’s bowl – interacts with the GlyP312-ProP313-GlyP314 turn of the V3 crown (Figure 4). (iii) The polar/charged corner – the right half of the bowl – interacts specifically with the side chain of residue ArgP315/GlnP315 of V3 (Figures 4-5 and S3). The fact that a negatively charged residue, AspH95 in 447-52D and GluH95 in 537-10D, resides at the base of this binding corner and it can form a salt bridge with the side chain of ArgP315 of the V3 epitiopes indicates that both mAbs are specific for V3 containing the GPGR motif, or V3GPGR. This is consistent with the fact that both mAbs were derived from patients infected with clade B viruses, within which the V3GPGR is predominant (Gaschen et al., 1999).
The structural details of the three epitope-binding determinants of 447-52D and 537-10D can explain the breadth of their cross-reactivities against V3 peptides as measured by ELISA (Figure 1B). Both mAbs have a long protruding CDR H3 that interacts, through main-chain and hydrophobic interactions, with the N-terminal side of the V3 loop flanking the β-turn; hence they can tolerate sequence variations in this region of the V3 crown. Mab 447-52D binds V3GPGQ peptides in addition to V3GPGR peptides (Figures 1B, left panel) because the apex residue GlnP315 can interact with the antibody through water-mediated hydrogen bonds. However, the relative affinity measured by 50% maximal binding in ELISA of 447-52D with V3GPGQ peptides (data not shown) is lower than with the V3GPGR peptides, as V3GPGQ can not form the salt bridge nor the unique cation-π sandwich stacking. In contrast, mAb 537-10D can only bind V3GPGR peptides (Figure 1B, right panel) because the buried negatively charged GluH95 at its binding site must be paired with the positively charged ArgP315.
Our structural data also provide clues for understanding the substantial differences in epitope binding, in terms of _K_D values, between 447-52D and 537-10D (Supplemental Table S3A). The V3-binding affinities of these two antibodies are known to be quite different: the binding affinity of 537-10D is many times lower than that of 447-52D measured for six V3 peptides (Gorny et al., 1993). The higher affinity of 447-52D against V3GPGR peptides may be attributed to two of its three epitope-binding determinants: the hydrophobic corner and the cation-π stacking that sandwiches the side chain of ArgP315. In the former determinant, 447-52D appears to have an ideal shape for the GPG turn: a square corner formed by mAb residues TrpL91, AlaL95B and TrpL96 which fits perfectly the peptide plane formed by GlyP312 and the pyrrolidine ring of ProP313 of the V3 GPG turn (Figure 4A). In the latter determinant, 447-52D can sandwich the guanidinium group of ArgP315 by a unique 3-residue cation-π interaction stacking, while 537-10D can only provide half of it (a 2-residue stacking).
The difference in neutralizing capacity between the two mAbs (Supplemental Table S3B) is a more complex issue. First, the neutralization capacity of 537-10D is narrower than that of 447-52D; it only neutralized 2 of the 7 viruses (all with the GPGR motif) tested. This may be attributed to the more restrictive antigen-binding site of 537-10D, i.e., the structurally shallow antigen-binding site of 447-52D can better tolerate flexibility of the V3 crown, while the deep pocket of 537-10D requires a “closer fit” in order to bind (Figure 2; Supplemental Table S2). Second for the two viruses that both can neutralize, the 50% neutralizing dose of 537-10D for HIV-1MN isolates was 54 times that of 447-52D as reported in one study and 17-fold in another previous study (Gorny et al., 1992; Gorny et al., 1993). The origin of this difference may lie in the binding kinetics of these two mAbs in neutralization. The charge states of the negatively charged residue (AspH95 for 447-52D and GluH95 for 537-10D) in the binding pockets of the two antibodies are likely very different before the epitope approaching the binding site. In the case of 537-10D, GluH95 at its epitope-binding site is probably shielded by solvent molecules before epitope binding. These solvent molecules have to be stripped away when V3 binds, likely slowing down the binding kinetics. In contrast, AspH95 of the 447-52D antigen-binding site is hydrated by a stably bound water molecule1, which is not removed by epitope binding. In addition, the bowl of 447-52D has a “spout” (Figure 2C), which can “leak out” any additional solvent molecules upon epitope binding.
The insights gained through the structural understanding of the antibody-antigen interactions of human anti-V3 mAbs should contribute to the rational design of immunogens that will elicit broadly neutralizing antibodies (Zolla-Pazner, 2005). The structures of 447-52D and 537-10D in complex with V3 peptides show that two of the three structural determinants – the long CDR H3 making main-chain interactions with the N-terminus of V3 crown, and the docking of the relatively conserved GPG region of the V3 crown – can maximally tolerate the sequence variation that occurs in the central portion of the V3 loop. However, the third structural element of these two antibodies – the polar/charged corner of the binding pocket – imposes limitations on their specificities. B cells making antibodies with this charged pocket appear to be preferentially selected by V3 epitopes that contain ArgP315 at the tip of the V3 crown, giving rise to anti-V3 antibodies that are less broadly reactive than those induced by viruses carrying V3 epitopes that contain GlnP315 (Krachmarov, 2005; Gorny, 2006). Structurally, there seem to be two families of anti-V3 human antibodies: one family binds squarely on the GPGR/Q motif (like 447-52D and 537-10D) and the other family avoids this motif (such as 2219). The presence of Arg draws immune response to this motif, and GPGR-containing epitopes tend to elicit antibodies belong to the first family. Although 447-52D is broadly reactive, its antigen binding site possesses rare structural determinants highly optimized for binding the GPGR motif and capable of tolerating sequence variation and flexibility of the epitopes. The chance of eliciting antibodies like 447-52D is hence small. It follows that immunogens mimicking V3 neutralization epitopes that do not contain ArgP315 may be more advantageous than those that bear ArgP315. Undoubtedly, a successful V3-based vaccine will have to mimic several canonical V3 conformations representing V3 structures that are recognized by broadly neutralizing antibodies such as 447-52D and 2219 (Stanfield et al., 2006) and F425-b4e8 (Bell et al., 2008). Comparison of the structure of these anti-V3 mAbs to those of mAbs with narrow specificity, like 537-10D, will contribute to the design of better V3 epitopes useful in inducing broadly neutralizing antibodies.
EXPERIMENTAL PROCEDURES
Antibody production, sequencing and Fab preparation
Human mAbs 447-52D and 537-10D were produced as previously described (Gorny et al., 1989). Briefly, peripheral blood mononuclear cells derived from HIV-1 infected individuals were transformed with Epstein-Barr virus, and the cells reactive with a 23-mer V3MN peptide were fused with heteromyeloma cells SHM-D33 (Teng et al., 1983). The resulting hybridomas were cloned by limiting dilution to monoclonality. MAbs 447-52D and 537-10D were purified from culture supernatants on protein G and protein A columns (GE Healthcare, Piscataway, NJ), respectively.
The nucleotide sequences of the variable fragments of the heavy and light chains of mAb 447-52D and the heavy chain of mAb 537-10D were taken from published data (Gorny et al., 2009; Lewis et al., 1995). The 537-10D light chain variable fragment was sequenced using cDNA prepared from previous studies (Gorny et al., 2009). A combination of two methods was employed: 5′ RACE amplification of cDNA ends (Invitrogen) and TOPO TA cloning method (Invitrogen). Briefly, using the 5′RACE method poly-C tails were added to 3′ ends of the cDNA with terminal deoxynucleotidyl transferase followed by PCR amplification using 5′RACE Abridged Anchor primer and reverse primer specific for the constant domain of the λ light chain gene (5′-CACTGTCTTCTCCACGGTGCTCCCTTC-3′). Then, PCR products were directly cloned into a PCR 2.1-TOPO vector. Clones were verified by enzyme mapping. Six colonies were selected and digested with EcoRI. The plasmids with correct insert size were sequenced at the institutional DNA Sequencing Facility. The sequence data were analyzed using the International ImMunoGene Tics (IMGT) information system (http://imgt.cines.fr).
To produce the antigen-binding fragments (Fabs), papain (Worthington, Lakewood, NJ) in 100 mM sodium acetate (NaOAc) pH 5.5 was activated with 10 mM cysteine, and incubated with the mAbs at a 1:20 molar ratio for 4 h at 37°C. Papain was then deactivated using iodoacetamide and the Fabs were then purified with protein G (for 447-52D) or protein A (for 537-10D) columns. The Fabs were further purified by size exclusion chromatography after dialysis overnight against 50 mM sodium acetate pH 5.5/100 mM NaCl.
V3 peptide panel
Twenty-four 19- to 24-mer peptides that span the crown of the V3 region from group M (clade A, B and C) HIV-1 viruses were used. Five V3 peptides representing the sequences of RF, SF2, NY/5, CDC4 and WM52 were purchased from Intracel, Inc. (Cambridge, MA). Four peptides, MN, BZ167, SF162 and VI191 were synthesized by Sigma-Genosys (Woodland, TX). Three peptides, D687, D808 and D757, were synthesized by Genemed Biotechnologies (South San Francisco, CA), and peptide W2RW020 was synthesized by Macromolecular Resource (Ft. Collins, CO). The remaining peptides were synthesized by Lawrence Loomis-Price at the H. M. Jackson Foundation (Rockville, MD) by standard solid-phase methods using an ABI 433 automated peptide synthesizer, and were >70% pure as assessed by HPLC.
Enzyme-linked immunosorbent assay (ELISA)
The reactivity of 447-52D and 537-10D against a panel of V3 peptides derived from various clades was measured by an enzyme-linked immunosorbent assay (ELISA). Briefly, ELISA plates were coated overnight at 4°C with V3 peptides at 1.0 μg/ml. The plates were washed 3 times with phosphate buffered saline (PBS) with 0.05% Tween-20, pH 7.4 (PBST) and incubated with mAbs at a concentration of 10 μg/ml for 1.5 h at 37°C. The plates were washed to remove any unbound antibodies and incubated with alkaline phosphatase-conjugated goat anti-human IgG (Southern Biotech, Birmingham, AL) for 1.5 h at 37°C. The plates were then washed 3 times with PBST, the substrate (_p_-nitrophenyl phosphate in 10% diethanolamine) was added, and color was read at 405 nm after 30-min incubation at room temperature. Negative controls consisted of V3 peptide-coated wells reacted with an irrelevant human anti-parvovirus B19 mAb 1418 (Gigler et al., 1999); cut-off values were defined as the mean plus 3 × SD obtained from three negative control wells.
Crystallization, data collection, structure determination, and refinement
Commercially synthesized 23-mer V3 peptides corresponding to clade A strain W2RW020, with amino acid sequence NNTRKGVRIGPGQAFYATGGIG (V3W2R), and clade B strain MN, with amino acid sequence YNKRKRIHIGPGRAFYTTKNIIG (V3MN), were dissolved in water to 1 mg/ml and added in a 5:1 molar ratio to purified Fab fragments of 447-52D and 537-10D, respectively. The mixtures were then concentrated to ~16 mg/ml for crystallization. Conditions for crystallization were screened with the hanging drop method and initial crystals for both complexes were observed within 48 hours. Well diffracted crystals were obtained in 2-3 weeks. The 447-52D/V3W2R complex was crystallized with a well solution of 1.4 M ammonium sulfate, 0.1M Tris pH 7.5, and the 537-10D/V3MN complex with 20% PEG 4000, 5% 2-propanol, 0.1 M Hepes pH 7.5.
Selected crystals were briefly soaked in their respective mother liquor with additional 25% glycerol (v/v) prior to mounting onto the X-ray beam. Diffraction data were collected at beamlines X4A and X4C, National Synchrotron Light Source, Brookhaven National Laboratory. All datasets were processed using the HKL package (Otwinowski and Minor, 1997), and structures determined in CNS (Brunger et al., 1998) by molecular replacement using the 447-52D Fab/V3MN structure (Protein Data Bank entry 1Q1J) as the starting model. Cycles of refinement for each model were carried out in CNS and O (Jones, 2004). Non-crystallographic symmetry restraints were employed in the early stage of the structural refinement for the 447-52D Fab/V3GPGQ W2R model. Final structural analysis was carried out using ICM (Abagyan et al., 1994), and figures were generated using PyMOL (DeLano, 2002) and ICM.
Supplementary Material
01
ACKNOWLEDGEMENTS
We thank Dr. Xunqing Jiang for useful discussions, Jared Sampson for help in preparing the manuscript, Dr. John Schwanof and Randy Abramowitz at beamlines X4A and X4C, National Synchrotron Light Source, Brookhaven National Laboratory for help with X-ray diffraction data collection, and Drs. Tim Cardozo, Catarina Hioe, and Phillipe Nyambi for critical reading of the manuscript. This study was supported in part by the Bill and Melinda Gates Foundation, NIH grants AI36085, HL59725 and AI77451, by the Immunology Core of the NYU Center for AIDS Research (NIH grant AI27742), and by research funds from the Department of Veterans Affairs.
Footnotes
1
This water molecule was also present in the previous 447-52D/V3GPGR complexes although it was not discussed.
ACCESSION NUMBERS The atomic coordinates of both structures have been deposited in RCSB Protein Data Bank with PDB IDs 3GHB (447-52D/V3W2R complex) and 3GHE (537-10D/V3MN complex).
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- Abagyan RA, Totrov M, Kuznetsov D. ICM - A new method for protein modeling and design: Applications to docking and structure prediction from the distorted native conformation. Journal of Computational Chemistry. 1994;15:488–506. [Google Scholar]
- Bell CH, Pantophlet R, Schiefner A, Cavacini LA, Stanfield RL, Burton DR, Wilson IA. Structure of antibody F425-B4e8 in complex with a V3 peptide reveals a new binding mode for HIV-1 neutralization. J Mol Biol. 2008;375:969–978. doi: 10.1016/j.jmb.2007.11.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Binley JM, Lybarger EA, Crooks ET, Seaman MS, Gray E, Davis KL, Decker JM, Wycuff D, Harris L, Hawkins N, et al. Profiling the specificity of neutralizing antibodies in a large panel of plasmas from patients chronically infected with human immunodeficiency virus type 1 subtypes B and C. J Virol. 2008;82:11651–11668. doi: 10.1128/JVI.01762-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Binley JM, Wrin T, Korber B, Zwick MB, Wang M, Chappey C, Stiegler G, Kunert R, Zolla-Pazner S, Katinger H, et al. Comprehensive Cross-Clade Neutralization Analysis of a Panel of Anti-Human Immunodeficiency Virus Type 1 Monoclonal Antibodies. J. Virol. 2004;78:13232–13252. doi: 10.1128/JVI.78.23.13232-13252.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brunger AT, Adams PD, Clore GM, DeLano WL, Gros P, Grosse-Kunstleve RW, Jiang JS, Kuszewski J, Nilges M, Pannu NS, et al. Crystallography & NMR system: A new software suite for macromolecular structure determination. Acta Crystallogr D Biol Crystallogr. 1998;54:905–921. doi: 10.1107/s0907444998003254. [DOI] [PubMed] [Google Scholar]
- Burton DR. A vaccine for HIV type 1: the antibody perspective. Proc Natl Acad Sci U S A. 1997;94:10018–10023. doi: 10.1073/pnas.94.19.10018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burton DR, Desrosiers RC, Doms RW, Koff WC, Kwong PD, Moore JP, Nabel GJ, Sodroski J, Wilson IA, Wyatt RT. HIV vaccine design and the neutralizing antibody problem. Nat Immunol. 2004;5:233–236. doi: 10.1038/ni0304-233. [DOI] [PubMed] [Google Scholar]
- Cardozo T, Swetnam J, Pinter A, Krachmarov C, Nadas A, Almond D, Zolla-Pazner S. Worldwide distribution of HIV-1 epitopes recognized by human anti-V3 monoclonal antibodies. AIDS Res Hum Retroviruses. 2009 doi: 10.1089/aid.2008.0188. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carrow EW, Vujcic LK, Glass WL, Seamon KB, Rastogi SC, Hendry RM, Boulos R, Nzila N, Quinnan GV., Jr. High prevalence of antibodies to the gp120 V3 region principal neutralizing determinant of HIV-1MN in sera from Africa and the Americas. AIDS Res Hum Retroviruses. 1991;7:831–838. doi: 10.1089/aid.1991.7.831. [DOI] [PubMed] [Google Scholar]
- Conley AJ, Gorny MK, Kessler JA, 2nd, Boots LJ, Ossorio-Castro M, Koenig S, Lineberger DW, Emini EA, Williams C, Zolla-Pazner S. Neutralization of primary human immunodeficiency virus type 1 isolates by the broadly reactive anti-V3 monoclonal antibody, 447-52D. J Virol. 1994;68:6994–7000. doi: 10.1128/jvi.68.11.6994-7000.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis KL, Bibollet-Ruche F, Li H, Decker JM, Kutsch O, Morris L, Salomon A, Pinter A, Hoxie JA, Hahn BH, et al. Human immunodeficiency virus type 2 (HIV-2)/HIV-1 envelope chimeras detect high titers of broadly reactive HIV-1 V3-specific antibodies in human plasma. J Virol. 2009;83:1240–1259. doi: 10.1128/JVI.01743-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeLano WL. The PyMOL User’s Manual. DeLano Scientific; Palo Alto: 2002. [Google Scholar]
- Dhillon AK, Stanfield RL, Gorny MK, Williams C, Zolla-Pazner S, Wilson IA. Structure determination of an anti-HIV-1 Fab 447-52D-peptide complex from an epitaxially twinned data set. Acta Crystallogr D Biol Crystallogr. 2008;64:792–802. doi: 10.1107/S0907444908013978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Douek DC, Kwong PD, Nabel GJ. The rational design of an AIDS vaccine. Cell. 2006;124:677–681. doi: 10.1016/j.cell.2006.02.005. [DOI] [PubMed] [Google Scholar]
- Fellouse FA, Wiesmann C, Sidhu SS. Synthetic antibodies from a four-aminoacid code: a dominant role for tyrosine in antigen recognition. Proc Natl Acad Sci U S A. 2004;101:12467–12472. doi: 10.1073/pnas.0401786101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaschen B, Korber BT, Foley BT. Global variation in the HIV-1 V3 region. In: Kuiken CL, Foley B, Hahn B, Marx PA, McCutchan F, Mellors JW, Mullins JI, Wolinsky S, Korber B, editors. Human retroviruses and AIDS. Los Alamos National Laboratory; Los Alamos: 1999. pp. 593–789. 1999. [Google Scholar]
- Gigler A, Dorsch S, Hemauer A, Williams C, Kim S, Young NS, Zolla-Pazner S, Wolf H, Gorny MK, Modrow S. Generation of neutralizing human monoclonal antibodies against parvovirus B19 proteins. J Virol. 1999;73:1974–1979. doi: 10.1128/jvi.73.3.1974-1979.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorny M, Zolla-Pazner S. In: Human Monoclonal Antibodies that Neutralize HIV-1 In HIV Immunology and HIV/SIV Vaccine Databases. Korber B, Brander C, Haynes BF, Koup RA, Moore JP, Walker B, Watkins DI, editors. Los Alamos National Laboratory; Los Alamos: 2003. pp. 37–51. 2003. [Google Scholar]
- Gorny MK, Conley AJ, Karwowska S, Buchbinder A, Xu JY, Emini EA, Koenig S, Zolla-Pazner S. Neutralization of diverse human immunodeficiency virus type 1 variants by an anti-V3 human monoclonal antibody. J Virol. 1992;66:7538–7542. doi: 10.1128/jvi.66.12.7538-7542.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorny MK, Gianakakos V, Sharpe S, Zolla-Pazner S. Generation of human monoclonal antibodies to human immunodeficiency virus. Proc Natl Acad Sci U S A. 1989;86:1624–1628. doi: 10.1073/pnas.86.5.1624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorny MK, Revesz K, Williams C, Volsky B, Louder MK, Anyangwe CA, Krachmarov C, Kayman SC, Pinter A, Nadas A, et al. The v3 loop is accessible on the surface of most human immunodeficiency virus type 1 primary isolates and serves as a neutralization epitope. J Virol. 2004;78:2394–2404. doi: 10.1128/JVI.78.5.2394-2404.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorny MK, Wang XH, Williams C, Volsky B, Revesz K, Witover B, Burda S, Urbanski M, Nyambi P, Krachmarov C, et al. Preferential use of the VH5-51 gene segment by the human immune response to code for antibodies against the V3 domain of HIV-1. Mol Immunol. 2009;46:917–926. doi: 10.1016/j.molimm.2008.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorny MK, Williams C, Volsky B, Revesz K, Cohen S, Polonis VR, Honnen WJ, Kayman SC, Krachmarov C, Pinter A, Zolla-Pazner S. Human monoclonal antibodies specific for conformation-sensitive epitopes of V3 neutralize human immunodeficiency virus type 1 primary isolates from various clades. J Virol. 2002;76:9035–9045. doi: 10.1128/JVI.76.18.9035-9045.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorny MK, Williams C, Volsky B, Revesz K, Wang XH, Burda S, Kimura T, Konings FA, Nadas A, Anyangwe CA, et al. Cross-clade neutralizing activity of human anti-V3 monoclonal antibodies derived from the cells of individuals infected with non-B clades of human immunodeficiency virus type 1. J Virol. 2006;80:6865–6872. doi: 10.1128/JVI.02202-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorny MK, Xu JY, Karwowska S, Buchbinder A, Zolla-Pazner S. Repertoire of neutralizing human monoclonal antibodies specific for the V3 domain of HIV-1 gp120. J Immunol. 1993;150:635–643. [PubMed] [Google Scholar]
- Goudsmit J, Debouck C, Meloen RH, Smit L, Bakker M, Asher DM, Wolff AV, Gibbs CJ, Jr., Gajdusek DC. Human immunodeficiency virus type 1 neutralization epitope with conserved architecture elicits early type-specific antibodies in experimentally infected chimpanzees. Proc Natl Acad Sci U S A. 1988;85:4478–4482. doi: 10.1073/pnas.85.12.4478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang CC, Lam SN, Acharya P, Tang M, Xiang SH, Hussan SS, Stanfield RL, Robinson J, Sodroski J, Wilson IA, et al. Structures of the CCR5 N terminus and of a tyrosine-sulfated antibody with HIV-1 gp120 and CD4. Science. 2007;317:1930–1934. doi: 10.1126/science.1145373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang CC, Tang M, Zhang MY, Majeed S, Montabana E, Stanfield RL, Dimitrov DS, Korber B, Sodroski J, Wilson IA, et al. Structure of a V3-containing HIV-1 gp120 core. Science. 2005;310:1025–1028. doi: 10.1126/science.1118398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ivanoff LA, Dubay JW, Morris JF, Roberts SJ, Gutshall L, Sternberg EJ, Hunter E, Matthews TJ, Petteway SR., Jr. V3 loop region of the HIV-1 gp120 envelope protein is essential for virus infectivity. Virology. 1992;187:423–432. doi: 10.1016/0042-6822(92)90444-t. [DOI] [PubMed] [Google Scholar]
- Javaherian K, Langlois AJ, LaRosa GJ, Profy AT, Bolognesi DP, Herlihy WC, Putney SD, Matthews TJ. Broadly neutralizing antibodies elicited by the hypervariable neutralizing determinant of HIV-1. Science. 1990;250:1590–1593. doi: 10.1126/science.1703322. [DOI] [PubMed] [Google Scholar]
- Javaherian K, Langlois AJ, McDanal C, Ross KL, Eckler LI, Jellis CL, Profy AT, Rusche JR, Bolognesi DP, Putney SD, et al. Principal neutralizing domain of the human immunodeficiency virus type 1 envelope protein. Proc Natl Acad Sci U S A. 1989;86:6768–6772. doi: 10.1073/pnas.86.17.6768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones TA. Interactive electron-density map interpretation: from INTER to O. Acta Crystallogr D Biol Crystallogr. 2004;60:2115–2125. doi: 10.1107/S0907444904023509. [DOI] [PubMed] [Google Scholar]
- Kabat EA, Wu TT, Perry HM, Gottesman KS. Sequences of Proteins of Immunological Interest. 5th edn National Institutes of Health; Bethesda, MD: 1991. [Google Scholar]
- Karlsson Hedestam GB, Fouchier RA, Phogat S, Burton DR, Sodroski J, Wyatt RT. The challenges of eliciting neutralizing antibodies to HIV-1 and to influenza virus. Nat Rev Microbiol. 2008;6:143–155. doi: 10.1038/nrmicro1819. [DOI] [PubMed] [Google Scholar]
- Krachmarov C, Pinter A, Honnen WJ, Gorny MK, Nyambi PN, Zolla-Pazner S, Kayman SC. Antibodies that are cross-reactive for human immunodeficiency virus type 1 clade a and clade B v3 domains are common in patient sera from Cameroon, but their neutralization activity is usually restricted by epitope masking. J Virol. 2005;79:780–790. doi: 10.1128/JVI.79.2.780-790.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krachmarov CP, Honnen WJ, Kayman SC, Gorny MK, Zolla-Pazner S, Pinter A. Factors determining the breadth and potency of neutralization by V3-specific human monoclonal antibodies derived from subjects infected with clade A or clade B strains of human immunodeficiency virus type 1. J Virol. 2006;80:7127–7135. doi: 10.1128/JVI.02619-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krachmarov CP, Kayman SC, Honnen WJ, Trochev O, Pinter A. V3-specific polyclonal antibodies affinity purified from sera of infected humans effectively neutralize primary isolates of human immunodeficiency virus type 1. AIDS Res Hum Retroviruses. 2001;17:1737–1748. doi: 10.1089/08892220152741432. [DOI] [PubMed] [Google Scholar]
- Kuiken CL, Foley B, Hahn B, Marx PA, McCutchan F, Mellors JW, Mullins JI, Wolinsky S, Korber B. Human Retroviruses and AIDS 1999: A Compilation and Analysis of Nucleic Acid and Amino Acid Sequences. Theoretical Biology and Biophysics Group, Los Alamos National Laborator; Los Alamos, NM: 1999. [Google Scholar]
- Kwong PD, Wilson IA. HIV-1 and influenza antibodies: seeing antigens in new ways. Nat Immunol. 2009;10:573–578. doi: 10.1038/ni.1746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Law M, Cardoso RM, Wilson IA, Burton DR. Antigenic and immunogenic study of membrane-proximal external region-grafted gp120 antigens by a DNA prime-protein boost immunization strategy. J Virol. 2007;81:4272–4285. doi: 10.1128/JVI.02536-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leonard CK, Spellman MW, Riddle L, Harris RJ, Thomas JN, Gregory TJ. Assignment of intrachain disulfide bonds and characterization of potential glycosylation sites of the type 1 recombinant human immunodeficiency virus envelope glycoprotein (gp120) expressed in Chinese hamster ovary cells. J Biol Chem. 1990;265:10373–10382. [PubMed] [Google Scholar]
- Lewis CM, Hollis GF, Mark GE, 3rd, Tung JS, Ludmerer SW. Use of a novel mutagenesis strategy, optimized residue substitution, to decrease the off-rate of an antigp120 antibody. Mol Immunol. 1995;32:1065–1072. doi: 10.1016/0161-5890(95)00079-8. [DOI] [PubMed] [Google Scholar]
- McKeating JA, Gow J, Goudsmit J, Pearl LH, Mulder C, Weiss RA. Characterization of HIV-1 neutralization escape mutants. Aids. 1989;3:777–784. doi: 10.1097/00002030-198912000-00001. [DOI] [PubMed] [Google Scholar]
- Otwinowski Z, Minor W. Processing of X-ray Diffraction Data Collected in Oscillation Mode. In: Carter CW Jr., Sweet RM, editors. Methods in Enzymology: Macromolecular Crystallography Part A. Academic Press; New York: 1997. pp. 307–326. [DOI] [PubMed] [Google Scholar]
- Palker TJ, Clark ME, Langlois AJ, Matthews TJ, Weinhold KJ, Randall RR, Bolognesi DP, Haynes BF. Type-specific neutralization of the human immunodeficiency virus with antibodies to env-encoded synthetic peptides. Proc Natl Acad Sci U S A. 1988;85:1932–1936. doi: 10.1073/pnas.85.6.1932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ratner L, Fisher A, Jagodzinski LL, Mitsuya H, Liou RS, Gallo RC, Wong-Staal F. Complete nucleotide sequences of functional clones of the AIDS virus. AIDS Res Hum Retroviruses. 1987;3:57–69. doi: 10.1089/aid.1987.3.57. [DOI] [PubMed] [Google Scholar]
- Resch W, Hoffman N, Swanstrom R. Improved success of phenotype prediction of the human immunodeficiency virus type 1 from envelope variable loop 3 sequence using neural networks. Virology. 2001;288:51–62. doi: 10.1006/viro.2001.1087. [DOI] [PubMed] [Google Scholar]
- Sharon M, Kessler N, Levy R, Zolla-Pazner S, Gorlach M, Anglister J. Alternative conformations of HIV-1 V3 loops mimic beta hairpins in chemokines, suggesting a mechanism for coreceptor selectivity. Structure. 2003;11:225–236. doi: 10.1016/s0969-2126(03)00011-x. [DOI] [PubMed] [Google Scholar]
- Stanfield RL, Gorny MK, Williams C, Zolla-Pazner S, Wilson IA. Structural rationale for the broad neutralization of HIV-1 by human monoclonal antibody 447-52D. Structure. 2004;12:193–204. doi: 10.1016/j.str.2004.01.003. [DOI] [PubMed] [Google Scholar]
- Stanfield RL, Gorny MK, Zolla-Pazner S, Wilson IA. Crystal structures of human immunodeficiency virus type 1 (HIV-1) neutralizing antibody 2219 in complex with three different V3 peptides reveal a new binding mode for HIV-1 cross-reactivity. J Virol. 2006;80:6093–6105. doi: 10.1128/JVI.00205-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stanfield RL, Wilson IA. Structural studies of human HIV-1 V3 antibodies. Hum Antibodies. 2005;14:73–80. [PubMed] [Google Scholar]
- Teng NN, Lam KS, Calvo Riera F, Kaplan HS. Construction and testing of mouse--human heteromyelomas for human monoclonal antibody production. Proc Natl Acad Sci U S A. 1983;80:7308–7312. doi: 10.1073/pnas.80.23.7308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vogel T, Kurth R, Norley S. The majority of neutralizing Abs in HIV-1-infected patients recognize linear V3 loop sequences. Studies using HIV-1MN multiple antigenic peptides. J Immunol. 1994;153:1895–1904. [PubMed] [Google Scholar]
- Wyatt R, Sodroski J. The HIV-1 envelope glycoproteins: fusogens, antigens, and immunogens. Science. 1998;280:1884–1888. doi: 10.1126/science.280.5371.1884. [DOI] [PubMed] [Google Scholar]
- Zolla-Pazner S. Identifying epitopes of HIV-1 that induce protective antibodies. Nat Rev Immunol. 2004;4:199–210. doi: 10.1038/nri1307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zolla-Pazner S. Improving on nature: focusing the immune response on the V3 loop. Hum Antibodies. 2005;14:69–72. [PubMed] [Google Scholar]
- Zolla-Pazner S, Cohen S, Pinter A, Krachmarov C, Wrin T, Wang S, Lu S. Cross-clade neutralizing antibodies against HIV-1 induced in rabbits by focusing the immune response on a neutralizing epitope. Virology. 2009 doi: 10.1016/j.virol.2009.05.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zolla-Pazner S, Cohen SS, Krachmarov C, Wang S, Pinter A, Lu S. Focusing the immune response on the V3 loop, a neutralizing epitope of the HIV-1 gp120 envelope. Virology. 2008;372:233–246. doi: 10.1016/j.virol.2007.09.024. [DOI] [PubMed] [Google Scholar]
- Zolla-Pazner S, Zhong P, Revesz K, Volsky B, Williams C, Nyambi P, Gorny MK. The cross-clade neutralizing activity of a human monoclonal antibody is determined by the GPGR V3 motif of HIV type 1. AIDS Res Hum Retroviruses. 2004;20:1254–1258. doi: 10.1089/aid.2004.20.1254. [DOI] [PubMed] [Google Scholar]
- Zwart G, Langedijk H, van der Hoek L, de Jong JJ, Wolfs TF, Ramautarsing C, Bakker M, de Ronde A, Goudsmit J. Immunodominance and antigenic variation of the principal neutralization domain of HIV-1. Virology. 1991;181:481–489. doi: 10.1016/0042-6822(91)90880-k. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
01