Species-specific transcription in mice carrying human chromosome 21 (original) (raw)
. Author manuscript; available in PMC: 2013 Jul 22.
Published in final edited form as: Science. 2008 Sep 11;322(5900):434–438. doi: 10.1126/science.1160930
Abstract
Homologous sets of transcription factors direct conserved tissue-specific gene expression, yet transcription factor binding events diverge rapidly between closely related species. We used hepatocytes from an aneuploid mouse strain carrying human chromosome 21 to determine on a chromosomal scale whether interspecies differences in transcriptional regulation are primarily directed by human genetic sequence or mouse nuclear environment. Virtually all transcription factor binding locations, landmarks of transcription initiation, and the resulting gene expression observed in human hepatocytes were recapitulated across the entire human chromosome 21 in the mouse hepatocyte nucleus. Thus, in homologous tissues, genetic sequence is largely responsible for directing transcriptional programs; interspecies differences in epigenetic machinery, cellular environment, and transcription factors themselves play secondary roles.
BACKGROUND
Higher eukaryotes are organized collections of different cell types, each of which is created from differential transcription of a common genome (1). Evolutionarily conserved sets of tissue-specific transcription factors establish each cell’s transcription during development and maintain it during adulthood by binding to DNA in a sequence-specific manner (1-3). These proteins typically recognize short consensus motifs, often between six and sixteen nucleotides, found at high frequency throughout a genome. How transcription factors discriminate among nearly identical motifs is poorly understood, though chromatin state, cellular environment, and surrounding regulatory sequences have all been suggested to direct transcription factors to specific cognate sites (4, 5). Sequence comparisons alone can identify only a fraction of regulatory regions (6) because the protein-DNA binding events linking transcription factors with genetic control sequences, and thus gene expression, change on a rapid evolutionary timescale (7-10). For instance, the targeted genes and precise binding locations of conserved, tissue-specific transcription factors differ significantly between mouse and human (7). Even when transcription factors bind near orthologous genes in two species, the precise location of the large majority of the binding events do not align (7, 9). In numerous cases, transcription factors frequently bind one highly conserved motif near a gene in one species, and a different conserved motif near the orthologous gene in a second species (7, 9). This divergence of transcription factor binding locations among related species is a widely occurring phenomenon, and similar observations have been made in yeast, Drosophila, and mammals (7-10). Thus, the mechanisms that determine tissue-specific transcriptional regulation must be more complex than simple gain and loss of the immediately-bound local sequence motifs.
The role that DNA sequence plays in directing histone modifications is also not well understood. It has been previously shown on human chromosomes 21 and 22, that, at the sequence level, sites of H3K4 methylation are no more conserved relative to mouse genome than background sequence (11). Genomic locations where H3K4 methylation occurred in both species did not show high levels of overall sequence conservation (11). One interpretation of this observation is that sequence comparisons alone have a limited ability to identify epigenetic landmarks.
Ultimately, transcription factor binding and epigenetic state contribute to tissue-specific gene expression (4, 5). A complete understanding of the mechanisms underlying divergence of transcriptional regulation and transcription itself is central to the debate surrounding the relative roles that cis-regulatory mutations and protein coding mutations play during evolution (12, 13).
Here, we isolate the role that genetic sequence plays in transcription by using a mouse model of Down Syndrome that stably transmits human chromosome 21 (14). In this mouse, we compare transcriptional regulation of orthologous human and mouse sequences in the same nuclei, and thereby eliminate most environmental and experimental variables otherwise inherent to interspecies comparisons.
Tc1 mice are partially mosaic, and approximately 60% of their hepatic cells contain human chromosome 21, which we confirmed by quantitative genotyping (Fig S1). Historically, human chromosome 21 has been extensively studied to explore transcription and transcriptional regulation on a chromosome-wide basis (11, 15, 16), and the corresponding orthologous mouse regions are located primarily in chromosome 16 with additional regions in chromosomes 10 and 17 (14).
We chose liver as a representative tissue for these experiments because most liver cells are hepatocytes that are easy to isolate and highly conserved in structure and function. A set of conserved, well-characterized transcription factors (including HNF1α, HNF4α, and HNF6) are responsible for hepatocyte development and function (2, 17), and orthologous liver-specific mouse and human transcription factors recognize the same consensus sequences (7). Despite almost perfect conservation in their DNA binding domains, the mouse orthologs of HNF1α, HNF4α, and HNF6 can vary in amino acid composition by up to 5% from their human orthologs in regions that could mediate protein-protein interactions (Table S1) (18, 19). No liver-specific transcription factor genes we profiled reside on HsChr21; therefore, binding events identified are due to mouse transcription factors.
Because approximately three-quarters of the conserved synteny between human chromosome 21 and the mouse genome resides on mouse chromosome 16, we used genomic tiling microarrays to obtain genomic information in four chromosome-nuclear combinations: human chromosome 21 located in human hepatocytes (indicated as WtHsChr21), human chromosome 21 located in Tc1 mouse hepatocytes (TcHsChr21), mouse chromosome 16 located in Tc1 mouse hepatocytes (TcMmChr16), and mouse chromosome 16 located in wild-type mouse hepatocytes (WtMmChr16).
For every experiment, we subtracted all potentially mouse-human degenerate probes computationally as well as experimentally by cross-hybridizing each platform with nucleic acids from the heterologous species (details in Supporting Online Materials). Taken together, our genomic microarrays could in principle interrogate over 28 Mb of human and mouse DNA sequence shared between HsChr21 and MmChr16, which would capture information on approximately 145 genes embedded in their native chromosomal context. After subtraction of regions deleted from TcHsChr21, approximately 20 Mb and 105 genes are interrogated herein.
Three aspects of this system are of particular note: (i) the primary Tc1 hepatocytes used in these experiments are indistinguishable in liver function, tissue architecture, and mouse-genome-based gene expression and transcription factor binding from that profiled from wild-type littermates (see below); (ii) TcHsChr21 and TcMmChr16 are in an identical dietary, developmental, nuclear, organismal, and metabolic environment in Tc1 hepatocytes; (iii) as all profiled transcription factors arise from the mouse genome, species-specific effects are eliminated for antisera used in chromatin immunoprecipitation (ChIP) experiments.
We first confirmed that substantial divergence exists in transcription factor binding between wild-type mouse and human hepatocytes by performing chromatin immunoprecipitations against HNF1α, HNF4α, and HNF6, which are members of three different protein families (Fig 1). As expected, most transcription factor binding events were species-specific (7), and located distal to transcriptional start sites (10, 20). We define human-specific (or human-unique) as ChIP enrichment on the human genome that does not have detectable signal in the orthologous region of the mouse genome (and vice versa) (Fig 1A, Fig S2).
Figure 1.
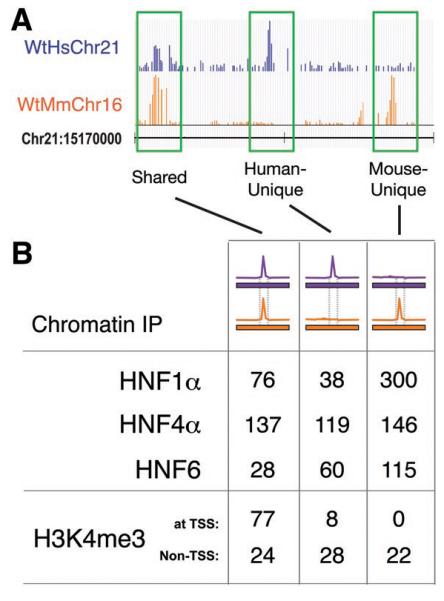
Transcriptional regulation varies between human and mouse hepatocytes across a complete chromosome. (A) Genome track showing chromatin immunoprecipitation enrichment of HNF1α binding in wild-type mouse and human hepatocytes across 30 kb of genomic sequence. The species of bound DNA sequences and ChIP signal is indicated by color: purple represents human, orange represents mouse. Highlighted in green are HNF1α-bound regions that are shared by both species, human-unique, and mouse-unique. (B) The total number of genomic regions occupied by three transcription factors (HNF1α, HNF4α, and HNF6) and the trimethylated form of histone H3K4 (H3K4me3) that are shared between the species, human-unique, or mouse-unique. ChIP data was obtained in wild-type mouse and human hepatocytes across the homologous regions of human chromosome 21 and mouse chromosome 16.
To determine the role that human DNA sequence can play in directing mouse transcription factor binding, we performed ChIP experiments against HNF1α, HNF4α, and HNF6 in hepatocytes from the Tc1 mouse (Fig 2). For each transcription factor, we simultaneously hybridized DNA from replicate ChIP-enrichment experiments to microarrays representing human chromosome 21 and mouse chromosome 16 (Supporting Online Materials). We found that transcription factor binding on TcMmChr16 and WtMmChr16 is largely identical; thus, the presence of an extra, human chromosome does not perturb transcription factor binding to the mouse genome (Fig S3).
Figure 2.
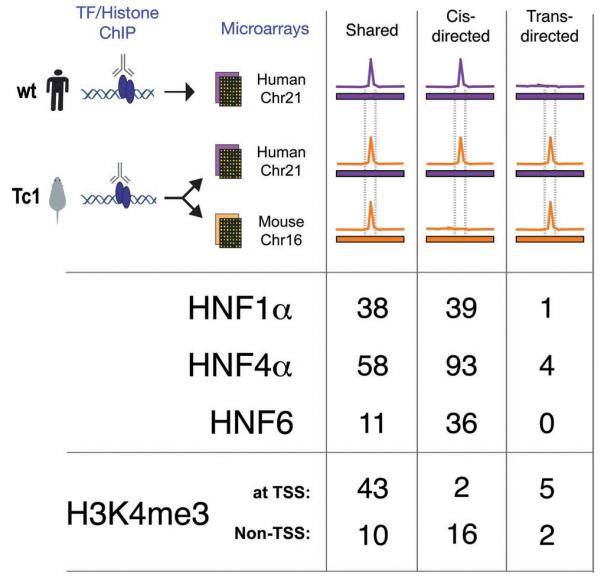
Comparison of the binding of the liver-specific transcription factors HNF1α, HNF4α, and HNF6, and enrichment of H3K4me3 on TcHsChr21 with the corresponding data obtained in mouse TcMmChr16 and human WtHsChr21 regions. The color scheme is the same as in Fig 1; notably, the primary difference from Fig 1 is the addition of the human chromosome in a mouse environment, which is indicated as a purple bar (representing the human chromosomal sequences) with an orange peak (from mouse transcription factor binding). The binding events on TcHsChr21 are sorted into categories based on whether they align with similar peaks in mouse and human (shared), align only with peaks in human (_cis_-directed), or align only with peaks in mice (_trans_-directed).
We then asked whether transcription factor binding to transchromic TcHsChr21 aligned with the positions found on (human) WtHsChr21 or (mouse) TcMmChr16. Though binding events could also be present uniquely on TcHsChr21 that do not align to either WtHsChr21 or TcMmChr16, this was rarely observed. If the transcription factor binding positions on TcHsChr21 align with positions found on WtHsChr21, then this would indicate that this binding is largely determined by cis-acting DNA sequences, as the transcription factors are present in both mouse and human hepatocytes and regulate key liver functions. If more than a small number of binding events on TcHsChr21 were found at locations that align elsewhere in the genome (for instance, with binding events on TcMmChr16), then other mechanistic influences besides genome sequence, such as chromatin structure, interspecies differences in developmental remodeling, diet, and/or environment must contribute substantially towards directing the location of transcription factor binding.
Remarkably, almost all of the transcription factor binding events on HsChr21 are found in both human and Tc1 mouse hepatocytes (85%-92%) (Fig 2A, Fig S4). The few peaks that appear to be unique to WtHsChr21 or TcHsChr21 are generally of lower intensity and difficult to reliably evaluate using standard peak calling algorithms (Fig S5). Indeed, as can be seen in Fig 3, the pattern of conservation and divergence in transcription factor binding found between WtHsChr21 (located in human liver) and WtMmChr16 (located in mouse liver) is recapitulated between TcHsChr21 and TcMmCh16 (both located in mouse liver) (see also Fig S6 and Fig S7). Because transcription factors often bind to regions that do not contain their canonical binding sequences (7, 9, 20), this result is further notable.
Figure 3.

Patterns of transcription factor binding and transcription initiation are determined by genetic sequence. ChIP enrichment for (A) HNF1α, (B) HNF4α, (C) HNF6, and (D) H3K4me3 are shown across a 50 kb surrounding the liver-expressed gene CLDN14. The human chromosome 21 coordinates, and the vertebrate sequence conservation track (Seq Cons; genome.ucsc.edu) are shown flanking CLDN14. Each panel shows the species of genetic sequence as a bar colored by species (human: purple, mouse: orange) below a track showing ChIP enrichment, similarly colored by species.
Despite the evolutionary divergence of primate and rodent lineages, mouse-genome encoded transcription factors can bind to human sequences in an identical manner to the human-genome coded transcription factors in a homologous tissue. These data eliminate the possibility that protein concentration differences or small coding variations in the mouse versions of transcription factors (or within larger transcriptional complexes) could redirect transcription factor binding to locations different than those found in human. Taken together, underlying genetic sequences appear to be the dominant influence on where transcription factors bind in homologous mammalian tissues.
We then explored how the mouse chromatin remodeling machinery interacts with TcHsChr21 (Fig 1) (21). Using chromatin immunoprecipitations, we isolated nucleosomes containing the trimethylated lysine 4 of histone H3 (H3K4me3) to identify the genomic anchor points for basal transcriptional machinery (11, 21-24). While most H3K4me3 enrichment occurs at transcription initiation sites and correlates with gene expression, it has been recently shown that most transcription start sites are H3K4me3 enriched, regardless of whether they are being actively elongated (11, 21-24). Depending on the cell type, approximately a quarter of genes can show differential H3K4 methylation and many of these genes have been shown to be cell-type specific (21).
We first identified how well trimethylation of the H3K4 position is shared between the wild-type mouse and human hepatocytes. We found that 77% of the regions of H3K4me3 enrichment were shared between WtHsChr21 and WtMmChr16. These regions are similar in a number of features, including proximity to transcriptional start sites (TSS; 77/101) and presence of CpG islands (80/101). Consistent with H3K4me3 serving as an anchor for the basal transcriptional machinery, for almost every shared region enriched for H3K4me3 in human hepatocytes (97/101), RNA transcripts were found in the liver-derived cell line HepG2 (15).
Regions enriched in trimethylation of H3K4 located distal to known TSS are thought to represent un-annotated promoter regions (11, 24). The vast majority of the species-specific regions enriched in H3K4me3 in human hepatocytes (28/36) and mouse hepatocytes (22/22) were distal to TSS (Fig 1, Fig S8). These species-specific sites of H3K4me3 enrichment were less likely to have CpG islands (3/36 and 2/22 respectively) and showed somewhat lower enrichment than the conserved regions (Fig S8). Consistent with their association with unannotated TSS, human-specific regions enriched for trimethylation of H3K4 also showed evidence of transcription in HepG2 (26/36 and 12/22 respectively). In sum, H3K4me3 enrichment was found to be shared between wild-type mouse and human hepatocytes at the majority of TSS, yet largely divergent elsewhere.
Based on presence of the trimethylated form of H3K4 in both mouse and human we observed at TSS, we expected that a human chromosome subject to mouse developmental remodeling would have enrichment of H3K4me3 at similar positions near transcription start sites. It was unclear, however, whether the mouse transcriptional machinery would successfully recreate the human-specific histone modifications at uncharacterized promoters distal to known TSS. Observing H3K4me3 enrichment on TcHsChr21 at either (i) the human-unique sites on WtHsChr21 or (ii) mouse-unique sites on WtMmChr16 could suggest what mechanisms direct the location of transcriptional initiation.
We found that virtually all of the TSS and approximately three-quarters of non-TSS H3K4me3 enriched regions on WtHsChr21 were found at the same location on TcHsChr21 (Fig 2, Fig S4). We found a minority of cases (7/78) where H3K4me3 enrichment occurred at sites on the TcHsChr21 that aligned with H3K4me3 enriched sites on TcMmChr16, without significant signal in WtHsChr21 (Fig 2). While these could be examples where human sequence in a mouse environment is handled in a mouse-specific manner, most are marginally enriched for H3K4me3 (see Supporting Text 1). Taken as a whole, close inspection of the patterns of enrichment of H3K4me3 on TcHsChr21 reveals that 85% of H3K4me3-enriched regions found on WtHsChr21 were reproduced on TcHsChr21 (Fig S4); the remarkable extent of this similarity is shown for the liver-expressed gene CLDN14 as a typical example (Fig 3). Independent ChIP-seq experiments confirmed 93% (77/82) of the sites of H3K4me3 enrichment on TcHsChr21, and 73% of sites on TcMmChr16 (70/95); the majority of non-confirmed sites on TcMmChr16 (20/25) were mouse-unique, half of which (13/25) were found in the Tiam1 gene (see Supporting Text 1 and Fig S9).
In addition to expanding the number of examples of functionally conserved H3K4me3 sites, our results demonstrate that the regions of differential H3K4 methylation between divergent species are primarily dictated by cis-acting genetic sequence. Neither the cellular environment nor differences among the mouse and human chromatin remodeling complexes substantially influence the placement of key chromatin landmarks associated with transcriptionally active regions.
Having shown that transcription factor binding and transcription initiation occurred in positions largely determined by underlying genetic sequences, we finally examined how the Tc1 mouse environment affects gene expression originating from the human chromosome. Using human gene expression microarrays that had been computationally and experimentally confirmed to be unaffected by the presence of mouse transcripts, we identified a distinct set of human genes that was expressed reproducibly in Tc1 mouse hepatocytes (Fig 4A). Genes located in regions known to be deleted from TcHsChr21 were not detected as expressed (Fig S10) (14). Unsupervised clustering and principal component analysis of transcriptional data from the human gene expression microarrays clearly separated Tc1 and wild-type littermates by the presence of TcHsChr21 (Fig S10). Conversely, we asked whether the presence of the human chromosome perturbs mouse-genome based gene expression. No differential expression of mouse hepatocyte mRNA between Tc1 mice and wild-type littermates was detected by mouse-specific Illumina BeadArrays (Fig 4B; note vertical scale). Unsupervised clustering of the normalized mouse array data accurately grouped mice by litter and strain, independently of the absence or presence of the human chromosome (Fig S10).
Figure 4.
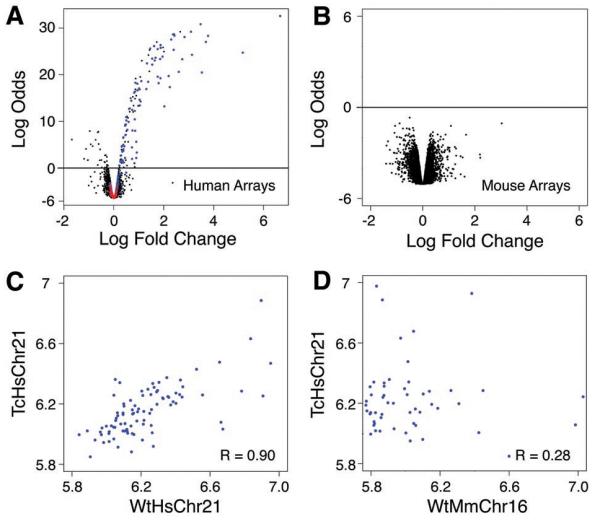
Gene expression in the Tc1 mouse originating from the mouse and human chromosomes is largely indistinguishable from comparable wild-type nuclear environments. Volcano plots (empirical Bayes log odds of differential expression versus average log fold change) demonstrating that (A) Tc1 hepatocytes have high transcription occurring from the transplanted human chromosome 21 using human genomic arrays and wild-type littermate mRNA as a reference (black probes map to human genes; blue probes map to genes located on HsChr21; red probes map to regions absent from TcHsChr21); whereas (B) wild-type and Tc1 mouse gene expression on mouse genomic arrays have indistinguishable patterns of transcription (black probes map to mouse genes). (C) Plot of the log expression of TcHsChr21 (y-axis) transcripts versus WtHsChr21 (x-axis) transcripts (Rcorr ≈ 0.90). (D) Plot of the log expression of TcHsChr21 (y-axis) transcripts versus WtMmChr16 (x-axis) orthologous transcripts (Rcorr ≈ 0.28).
We asked how well the transcripts originating from TcHsChr21 correlated with the transcripts originating from WtHsChr21 in human hepatocytes (Fig 4C, Fig S11). Gene expression in Tc1 mouse hepatocytes originating from the human chromosome was determined using the probes representing the 121 genes present on TcHsChr21, and then compared with matching gene expression data for the same 121 genes obtained from human hepatocytes. We found a strong correlation between the expression levels of the human genes located in Tc1 mouse hepatocytes and their counterparts located in wild-type human hepatocytes (Fig 4C, Fig S11). This correlation (Rcorr ≈ 0.90) was slightly lower than that found between replicate individual human livers (Fig S12), yet appears to be higher than similar correlations previously reported between human and other primates (25, 26). The expression of orthologous genes within Tc1 hepatocytes (i.e. TcHsChr21 vs TcMmChr16) is substantially more divergent, with Rcorr ≈ 0.28 (Fig 4D). It is possible that the correlation between mouse and human orthologs could be influenced by the experimental differences between platforms as well as microarray design peculiarities. To address this concern, we determined the relative rank-order of expression among the genes on WtHsChr21, TcHsChr21 and TcMmChr16, and then compared the ranked results. We found correlation trends similar to the above (Fig S11, Supporting Online Material).
Our results test the hypothesis that variation in gene expression is dictated by regulatory regions, extending recent expression quantitative trait loci mapping studies and comparative expression studies that have been confined to closely related species (25-29). The apparent absence of overt trans influences could be explained by the modest amount of human DNA provided by a single copy of human chromosome 21 when compared to the complete mouse genome as well as the absence of liver-specific transcriptional regulators on chromosome 21. The extent that protein coding and cis-regulatory mutations contribute to changes in morphology, physiology and behavior is actively debated in evolutionary biology (3, 12, 13). Myriad points of control influence gene expression; however, it has also been an unresolved question as to which of these mechanisms has the most influence globally. Here we show that each layer of transcriptional regulation within the adult hepatocyte, from the binding of liver master regulators and chromatin remodeling complexes to the output of the transcriptional machinery, is directed primarily by DNA sequence. Although conservation of motifs alone cannot predict transcription factor binding, we show that within the genetic sequence there must be embedded adequate instructions to direct species-specific transcription.
Supplementary Material
Supplemental_Information
Acknowledgments
We are grateful to E. Jacobsen, R. Stark, I. Spiteri, B. Liu, J. Marioni, A. Lynch, the CRI Genomics Core, CRI Bioinformatics Core, and Camgrid for technical assistance, and B. Gottgens and J. Ferrer for insightful advice. Supported by the European Research Council (DTO), Royal Society Wolfson Research Award (ST), Hutchinson Whampoa (DTO, ST), Medical Research Council (EF, VT), Wellcome Trust (EF, VT), University of Cambridge (DTO, DS, NBM, ST), Cancer Research UK (DTO, MDW, NBM, ST, DS). Data deposited under ArrayExpress accession numbers E-TABM-473 and E-TABM-474.
Footnotes
The authors declare no competing interests.
REFERENCES
- 1.Davidson EH, Erwin DH. Science. 2006 Feb 10;311:796. doi: 10.1126/science.1113832. [DOI] [PubMed] [Google Scholar]
- 2.Zaret KS. Mech Dev. 2000 Mar 15;92:83. doi: 10.1016/s0925-4773(99)00326-3. [DOI] [PubMed] [Google Scholar]
- 3.Wray GA. Nat Rev Genet. 2007 Mar;8:206. doi: 10.1038/nrg2063. [DOI] [PubMed] [Google Scholar]
- 4.Li B, Carey M, Workman JL. Cell. 2007 Feb 23;128:707. doi: 10.1016/j.cell.2007.01.015. [DOI] [PubMed] [Google Scholar]
- 5.Guccione E, et al. Nat Cell Biol. 2006 Jul;8:764. doi: 10.1038/ncb1434. [DOI] [PubMed] [Google Scholar]
- 6.Elnitski L, Jin VX, Farnham PJ, Jones SJ. Genome Res. 2006 Dec;16:1455. doi: 10.1101/gr.4140006. [DOI] [PubMed] [Google Scholar]
- 7.Odom DT, et al. Nat Genet. 2007 Jun;39:730. doi: 10.1038/ng2047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Moses AM, et al. PLoS Comput Biol. 2006 Oct;2:e130. doi: 10.1371/journal.pcbi.0020130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Borneman AR, et al. Science. 2007 Aug 10;317:815. doi: 10.1126/science.1140748. [DOI] [PubMed] [Google Scholar]
- 10.Birney E, et al. Nature. 2007 Jun 14;447:799. [Google Scholar]
- 11.Bernstein BE, et al. Cell. 2005 Jan 28;120:169. doi: 10.1016/j.cell.2005.01.001. [DOI] [PubMed] [Google Scholar]
- 12.Hoekstra HE, Coyne JA. Evolution. 2007 May;61:995. doi: 10.1111/j.1558-5646.2007.00105.x. [DOI] [PubMed] [Google Scholar]
- 13.Carroll SB. Cell. 2008 Jul 11;134:25. doi: 10.1016/j.cell.2008.06.030. [DOI] [PubMed] [Google Scholar]
- 14.O’Doherty A, et al. Science. 2005 Sep 23;309:2033. [Google Scholar]
- 15.Kampa D, et al. Genome Res. 2004 Mar;14:331. doi: 10.1101/gr.2094104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Carroll JS, et al. Cell. 2005 Jul 15;122:33. [Google Scholar]
- 17.Cereghini S. Faseb J. 1996 Feb;10:267. [PubMed] [Google Scholar]
- 18.Eeckhoute J, Oxombre B, Formstecher P, Lefebvre P, Laine B. Nucleic Acids Res. 2003 Nov 15;31:6640. doi: 10.1093/nar/gkg850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sladek FM, Ruse MD, Jr., Nepomuceno L, Huang SM, Stallcup MR. Mol Cell Biol. 1999 Oct;19:6509. doi: 10.1128/mcb.19.10.6509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Rada-Iglesias A, et al. Hum Mol Genet. 2005 Nov 15;14:3435. doi: 10.1093/hmg/ddi378. [DOI] [PubMed] [Google Scholar]
- 21.Guenther MG, Levine SS, Boyer LA, Jaenisch R, Young RA. Cell. 2007 Jul 13;130:77. doi: 10.1016/j.cell.2007.05.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Vermeulen M, et al. Cell. 2007 Oct 5;131:58. [Google Scholar]
- 23.Sims RJ, 3rd, et al. Mol Cell. 2007 Nov 30;28:665. doi: 10.1016/j.molcel.2007.11.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Barski A, et al. Cell. 2007 May 18;129:823. doi: 10.1016/j.cell.2007.05.009. [DOI] [PubMed] [Google Scholar]
- 25.Gilad Y, Oshlack A, Smyth GK, Speed TP, White KP. Nature. 2006 Mar 9;440:242. doi: 10.1038/nature04559. [DOI] [PubMed] [Google Scholar]
- 26.Khaitovich P, et al. Science. 2005 Sep 16;309:1850. doi: 10.1126/science.1108296. [DOI] [PubMed] [Google Scholar]
- 27.Wittkopp PJ, Haerum BK, Clark AG. Nat Genet. 2008 Mar;40:346. doi: 10.1038/ng.77. [DOI] [PubMed] [Google Scholar]
- 28.Park CC, et al. Nat Genet. 2008 Apr;40:421. doi: 10.1038/ng.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gilad Y, Rifkin SA, Pritchard JK. Trends Genet. 2008 Aug;24:408. doi: 10.1016/j.tig.2008.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental_Information