Advances in the Regulation of Osteoclasts and Osteoclast Functions (original) (raw)
Abstract
Osteoclasts are derived from mononuclear hematopoietic myeloid lineage cells, which are formed in the bone marrow and are attracted to the bloodstream by factors, including sphingsine-1 phosphate. These circulating precursors are attracted to bone surfaces undergoing resorption by chemokines and other factors expressed at these sites, where they fuse to form multinucleated bone-resorbing cells. All aspects of osteoclast formation and functions are regulated by macrophage-colony-stimulating factor (M-CSF) and receptor activator of NF-κB ligand (RANKL), cytokines essential for osteoclast formation and expressed by a variety of cell types, including osteoblast lineage cells. Since the discovery of RANKL in the mid-1990s, mouse genetic and molecular studies have revealed numerous signaling pathways activated by RANKL and M-CSF. More recent studies indicate that osteoclasts and their precursors regulate immune responses and osteoblast formation and functions by means of direct cell-cell contact through ligands and receptors, such as ephrins and Ephs, and semaphorins and plexins, and through expression of clastokines. There is also growing recognition that osteoclasts are immune cells with roles in immune responses beyond mediating the bone destruction that can accompany them. This article reviews recent advances in the understanding of the molecular mechanisms regulating osteoclast formation and functions and their interactions with other cells in normal and pathologic states.
Keywords: apoptosis, bone biology, osteoblast(s), bone remodeling/regeneration, cytokine(s), chemokine(s)
Introduction
Osteoclasts (OCs) are multinucleated cells that arise by fusion of myeloid hematopoietic precursors formed in the bone marrow. Osteoclast precursors (OCPs) are attracted from the bone marrow to the bloodstream by chemokines and circulate there until they are attracted back into bones by a variety of factors released at sites undergoing resorption, called bone remodeling units (BRUs), and there they differentiate into OCs (Boyce et al., 2012). These factors include chemokines and cytokines, such as macrophage-colony-stimulating factor (M-CSF) and receptor activator of NF-κB ligand (RANKL), which are expressed by cells in and around BRUs and are required for OCP formation and differentiation in normal and pathologic bone remodeling in which bone resorption is increased generally or locally (Boyce et al., 2012).
Until recently, OCs were considered to be bone-resorbing cells with few other functions. However, studies showing that they secrete cytokines (clastokines) and growth factors, and genetic studies in mice have revealed a number of unanticipated roles for OCs and OCPs (Boyce et al., 2009). These include positive and negative regulation of osteoblast (OB) formation, and immune responses. OCs are also found in a number of pathologic conditions outside of bone – for example, within some cancers. Several major signaling pathways regulate the functions of normal and neoplastic cells, including NF-κB, Wnt-β-catenin, and Notch, but they also play important roles in OC and OB formation and functions. This paper reviews current knowledge of the molecular mechanisms regulating OC formation and functions.
Recruitment of Osteoclast Precursors to Bone Surfaces
OCPs are held in the bone marrow by chemokines, such as stroma-derived factor-1 (SDF-1). This effect can be modulated by tumor necrosis factor (TNF), which inhibits SDF-1 production by marrow cells, allowing OCPs to mobilize to the blood in inflammatory conditions (Boyce et al., 2012). OCPs are also attracted to the blood by the bioactive sphingolipid, sphingosine-1 phosphate (S1P), which is secreted from red blood cells and platelets. OCPs express S1P receptors (S1PRs) 1 and 2, which have chemo-attractant and chemo-repellent functions, respectively, the latter sending them back to the marrow at BRUs (Kikuta et al., 2011). OCs also express S1P, which could attract OCPs to them for fusion. Interestingly, S1P expression is negatively regulated by cathepsin K, the major matrix-degrading collagenase expressed by OCs (Lotinun et al., 2013), suggesting that cathepsin K inhibitors could stimulate bone formation because S1P expressed by OCs enhances OB precursor differentiation.
S1P levels are increased in the synovial fluid of patients with rheumatoid arthritis (RA), suggesting that it could attract OCPs to affected joints. Mice with inflammatory arthritis treated with FTY720, an S1PR1 agonist, had significantly reduced joint destruction and inflammation, consistent with the agonist working to retain OCPs and presumably other immune cells in the bloodstream (Kikuta et al., 2011). S1P has positive and negative regulatory effects on inflammation and cytokine expression and functions; thus, agonists may have unanticipated effects in some conditions. S1P levels have not been reported in crevicular fluids around teeth in periodontal disease, but S1P has been shown to increase the production of inflammatory cytokines in periodontitis (Aarthi et al., 2011). Circulating OCPs are also attracted to actively resorbing primary, but not permanent, teeth, presumably by RANKL and M-CSF, which are expressed by pulp and periodontal ligament cells (Wang and McCauley, 2011).
Regulation of Osteoclast Formation
Myeloid progenitors differentiate into OCPs in response to signaling induced by several transcription factors, including PU.1, and a heterodimeric complex of microphthalmia-associated transcription factor (MITF) and Tfe3 (Fig. 1). PU.1 and MITF activate expression of the M-CSF receptor (M-CSFR) (Mellis et al., 2011), and mice deficient in the genes encoding any of these proteins have osteopetrosis, which develops during endochondral ossification from failed resorption of bone trabeculae in the medullary cavities. Osteopetrosis occurs because of defective OC formation or activity and is also characterized by failure of incisor eruption in rodents and in abnormal tooth eruption and formation in humans because osteoclasts do not resorb normal channels for them in jaw bones (Del Fattore et al., 2008).
Figure 1.

Regulation of osteoclast formation and differentiation. An early requirement for myeloid progenitor differentiation into osteoclast precursors is expression of PU.1 and Mitf, which induce expression of M-CSFR, the receptor for M-CSF. M-CSF induces expression of RANK on these cells, priming them for further differentiation in response to RANKL. TNF also induces expression of RANK, which can enhance osteoclast formation in inflammatory bone disease. In response to RANKL and TNF, expression of a number of transcription factors that regulate further differentiation of RANK-expressing cells is increased. These include NF-κB, c-Fos, and NFATc1. They induce expression of several gene-encoding proteins involved in osteoclast activation, including tartrate-resistant acid (TRAP), cathepsin-K, and the calcitonin receptor, and mediate production of H+ and Cl-, which form HCl under the ruffled borders of osteoclasts. RANKL and TNF also induce activation of c-myc, which promotes further proliferation of these cells, as well as map kinase/kinase 6 (MKK6), p38, and Mitf, along with Src and Erk, which have multiple effects to activate osteoclasts and promote their survival.
One of the earliest effects of M-CSF is to promote expression of RANK by myeloid progenitors (Fig. 1), a property shared by TNF, which primes the cells to respond to RANKL (Boyce et al., 2012). Binding of M-CSF to M-CSFR attracts a signaling complex comprised of phosphorylated DNAX-activating protein 12 (DAP12) and the non-receptor tyrosine kinase, Syk, and this activates ERK/growth-factor-receptor-bound protein 2 (Grb-2) and Akt/PI3K signaling to regulate several other aspects of OCP and OC activities, including proliferation, differentiation, and survival (Ross and Teitelbaum, 2005). M-CSF also promotes OC survival (Tanaka et al., 2006, 2010) and, like RANKL, mediates all aspects of OC functions.
RANKL is expressed by numerous cell types, including osteoblastic, chondroblastic, and T and B cells (Pacifici, 2012). Hypertrophic chondrocytes appear to be the major source of RANKL for removal of trabeculae during endochondral ossification, while osteocytes within trabeculae appear to be the major source during bone remodeling in adult mice and in response to mechanical stress (Xiong and O’Brien, 2012). However, the major cellular sources of RANKL in common bone diseases remain to be identified.
Like other TNF receptor family members, RANK lacks intrinsic kinase activity to phosphorylate and activate downstream signaling molecules and recruits TNF receptor-activating factors (TRAFs), particularly TRAFs 1, 2, 3, 5, and 6, which are adapter proteins that recruit protein kinases (Boyce et al., 2012). In unstimulated OCPs, TRAFs2 and 3 and cIAP1/2 form a complex that degrades NF-κB-inducing kinase (NIK), and this limits OC formation. RANKL induces degradation of TRAF3, thus allowing for the accumulation of NIK and the induction of OC formation (Fig. 2). TRAF2-/- mice die in utero, and TRAF3-/- mice die around 1 wk after birth, making definitive study of their roles in OC formation challenging. TRAF 1-/- and 5-/- mice have no bone phenotype, while TRAF6-/- mice generated separately by 2 groups have either no OCs or dysfunctional OCs (Wada et al., 2006; Boyce, 2013). This apparent dual role for TRAF6 may explain why RANK/TRAF6 signaling activates NF-κB, c-Jun N-terminal kinase (JNK)/activator protein-1 (AP-1, including c-Fos), c-myc, and calcineurin/nuclear factor of activated T cell c1 (NFATc1) signaling to induce OC formation, as well as Src and MKK6/p38/MITF to mediate osteoclast resorption, and Src and ERK to mediate OC survival (Fig. 1) (Boyce, 2013).
Figure 2.

Functions of TRAFs in osteoclast formation and differentiation. Under basal conditions, TRAF2, c-IAP1/2, and TRAF3 form a complex on the intracellular portion of RANK. This complex polyubiquitinates NIK, which is transported to the proteasome for degradation, resulting in very low levels of NIK in unstimulated OCPs. RANKL binding to RANK on OCPs leads to recruitment of TRAFs 1, 5, and 6 and polyubiquitination of TRAF3 by TRAF2/c-IAP1/2 with subsequent lysosomal degradation of TRAF3 (our unpublished observations). This leads to the release of NIK, and signaling through it and TRAF6 results in the activation of NF-κB and subsequent increased expression of NFATc1 and c-Fos to induce further OCP differentiation. TRAF6 also mediates the activation of signaling through JNK, c-Myc, and Src to promote OCP differentiation and activation.
NF-κB signaling is required for osteoclastogenesis, based on NF-κB p50/p52 double-knockout (dKO) mice having marked osteopetrosis due to the failure of OC formation (Boyce, 2013). These dKO, RANKL-/-, and RANK-/- mice also have failure of B-cell and lymph node development and defective T-cell maturation, suggesting that the inhibition of RANK signaling – for example, with Denosumab (a monoclonal antibody to RANKL) – could have adverse effects on immune responses. However, thus far, studies with Denosumab have not reported such adverse effects (Miller, 2011). Interestingly, despite Denosumab and bisphosphonates having different mechanisms of action, osteonecrosis of jawbones has now been reported in a small percentage of patients treated with Denosumab for metastatic bone disease, suggesting that both drugs may be interfering with the functions of similar cells, which could be osteoclasts and/or other immune cells (Kalyan et al., 2013).
There are 5 main stimulatory NF-κB transcription factor proteins: RelA, p50, Rel B, p52, and c-Rel. In many publications, NF-κB refers to RelA/p50 heterodimeric signaling in the canonical NF-κB pathway. RelB/p52 heterodimers activate non-canonical signaling. RANKL activates both canonical and non-canonical signaling, while TNF activates predominantly canonical signaling (DiDonato et al., 2012). One of the earliest responses of OCPs to RANKL is recruitment of RelA/p50 and NFATc2 to the promoter of NFATc1, which has been called the master regulator of osteoclastogenesis. This results in NFATc1 transiently auto-amplifying its own expression (Takayanagi et al., 2002), accompanied by down-regulation of the expression of constitutively active repressors of RANK signaling (Zhao and Ivashkiv, 2011), which allows osteoclastogenesis to proceed. NFATc1 is subsequently activated by c-Fos through TRAF6/NF-κB and CCAAT/enhancer binding protein-α (Chen et al., 2013) signaling to complete OCP differentiation and subsequent OC activation. These early events in osteoclastogenesis are associated with a number of epigenetic phenomena in response to RANKL. These include: histone acetylation and methylation (Takayanagi et al., 2002) as well as histone demethylation on the NFATc1 promoter (Yasui et al., 2011); inhibition of histone acetylation of ATF3 by Jun dimerization protein 2 (Maruyama et al., 2012); and up-regulation of miR-146a, miR-21, and miR-155 to induce OCP differentiation (Delgado-Calle et al., 2012). In this respect, OCs and their precursors are similar to many other types of cells, and no doubt other forms of epigenetic regulation of OC functions will be identified.
Non-canonical NF-κB signaling by RANKL entails degradation of TRAF3, proteasomal processing of NF-κB p100 to p52, and translocation of RelB/p52 heterodimers to nuclei (Yao et al., 2009). It is not required for basal OC formation because NIK-/-, RelB-/-, and p100-/- mice have normal OC numbers and no osteopetrosis. However, it does appear to be required for localized osteolysis in pathologic states (Boyce, 2013), and TNF-transgenic mice deficient in p100 develop more severe joint inflammation and erosion than do controls, indicating that p100 limits TNF-induced OC formation and inflammation (Yao et al., 2009). A peptide inhibitor of the NF-κB essential modulator (NEMO) reduced inflammatory arthritis in mice (Jimi et al., 2004), but it has not been studied in humans to date. Some factors have been shown to stimulate OC formation independent of RANKL, particularly in pathologic conditions, including 1,25(OH)2 Vitamin D3, TGFβ, IL-6, TNF, APRIL, LIGHT, NGF, and IGFs I and II (Hemingway et al., 2011).
Regulation of Osteoclast Activation
Bone resorption begins with OCs attaching to bone surfaces by αVβ3, the integrin vitronectin receptor, resulting in recruitment of Src tyrosine kinase by standard outside-in signaling (Teitelbaum, 2011). This leads to activation of several Src-dependent signaling pathways. For example, Src phosphorylates Syk, which recruits DAP12, a co-stimulatory ITAM protein, and Slp76, and these form an adapter protein complex that activates small Rho family GTPases, including Rac. αVβ3 and the M-CFSR interact physically, and inside-out signaling through αVβ3 leads to integrin activation (Teitelbaum, 2011). RANK also forms a complex with αVβ3 through Src, thus activating Syk, Slp-76, Vav3, and Rac, similar to αVβ3/Src interaction. These interactions lead to the formation of the ruffled border membrane by fusion of lysosomal secretory vesicles with the cytoplasmic membrane (Teitelbaum, 2011). Vesicle fusion is a complex process mediated by several proteins, including Rab7, and synaptotagmin VII, and others involved in autophagy and extracellular protein secretion (Hocking et al., 2012).
H+ ions are pumped through the ruffled border and, along with Cl-, form HCl, which demineralizes bone, and cathepsin K is secreted to degrade the matrix. Cathepsin K inhibitors are being studied in clinical trials as novel anti-resorptive therapy, and a recent study suggests that cathepsin K expressed by OCs negatively regulates osteoblastic RANKL expression (Lotinun et al., 2013). Src expression is required in osteoclasts for resorption, and its over-expression in many cancers enhances proliferation, invasion, and metastasis. Thus, Src-targeted drugs could inhibit both OCs and tumor cells in metastatic bone disease. Saracatamib is a small molecular Src inhibitor under investigation as an adjuvant to standard chemotherapy in metastatic prostate cancer (Boyce and Xing, 2011). Proton pump inhibitors have been developed, but none of these is in clinical trials.
During bone formation, numerous proteins, including TGFβ, BMPs, FGFs, and IGFs, are laid down along with collagen and are released subsequently by OCs during resorption when they can become activated and available locally to promote the resorptive process in normal and pathologic states, such as metastatic bone disease (Kakonen and Mundy, 2003). For example, TGFβ, BMPs, FGFs, and IGFs can induce OC formation by direct and/or indirect (e.g., by modulating expression of RANKL or OPG) mechanisms (Hemingway et al., 2011). TGFβ can also attract osteoblastic cells to resorption sites to act as a coupling factor (Tang et al., 2009).
OCs function most effectively as large multinucleated cells (Fig. 3), such as those seen in osteolytic conditions, including Paget’s disease, giant cell tumors, and reparative granuloma of bone. Their formation does not require them to be attached to bone matrix. For example, only a fraction of the OCs in reparative granuloma or giant cell tumors (Figs. 4, 5) is on the bone surface, and OCs are seen occasionally in primary breast, pancreatic, and other cancers (Fig. 6), where their roles and the mechanisms driving their formation have yet to be determined. In these conditions, tumor cells presumably express RANKL or other osteoclastogenic cytokines (Hemingway et al., 2011). Several proteins, including dendritic-cell-specific transmembrane protein (DC-STAMP), Atp6v0d2, OC-STAMP, and CD9, are involved in the fusion process, and their expression is regulated by PU.1, MITF, c-Fos, and NFATc1 (Fig. 1) (Yagi et al., 2007; Mellis et al., 2011). Vitamin E (α-tocopherol) activates mitogen-activated protein kinase 14 and MITF to induce DC-STAMP expression and OCP fusion (Fujita et al., 2012). These investigators found that doses of α-tocopherol taken by humans as dietary supplements increased OC formation and reduced bone volume in rats, suggesting that excessive Vitamin E consumption could cause osteoporosis.
Figure 3.
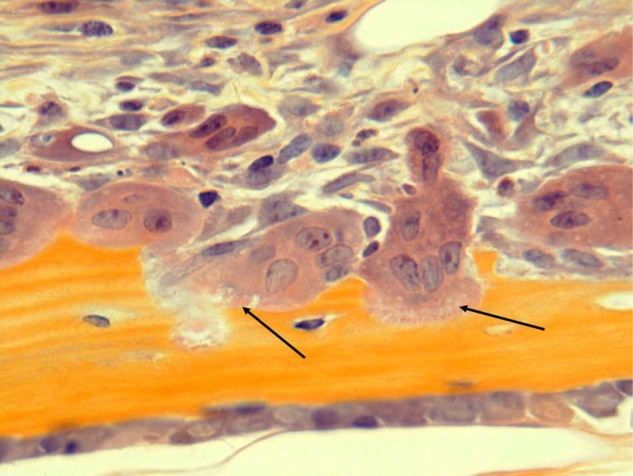
Osteoclasts actively resorbing bone. Osteoclasts with ruffled borders (black arrows) are actively resorbing bone. Note also numerous other mononuclear cells in the marrow adjacent to the osteoclasts. These include immune and stromal cells and osteoclast precursors. H&E, orange G, and phloxine.
Figure 4.
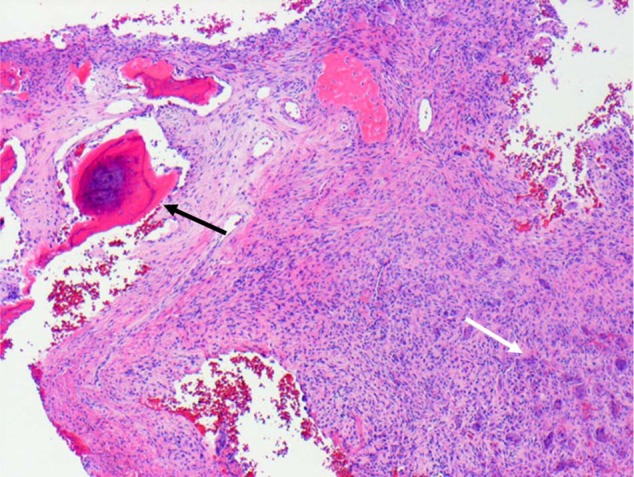
Giant cell reparative granuloma of bone. Biopsy tissue from a lytic lesion in the mandible of a 14-year-old girl, showing reactive new bone (black arrow), adjacent fibrous tissue, and numerous osteoclasts (white arrow) far removed from the bone surface. H&E.
Figure 5.
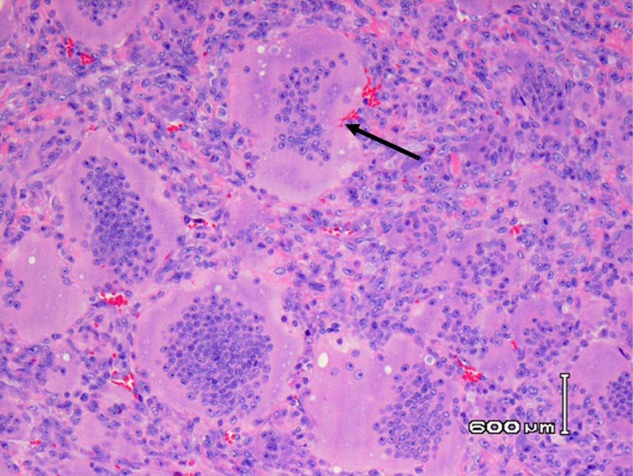
Giant cell tumor of bone. Numerous enormous multinucleated osteoclasts from a giant cell tumor of bone from the distal radius of a 25-year-old man, with associated mononuclear precursors and stromal cells with no bone matrix associated with them. H&E.
Figure 6.
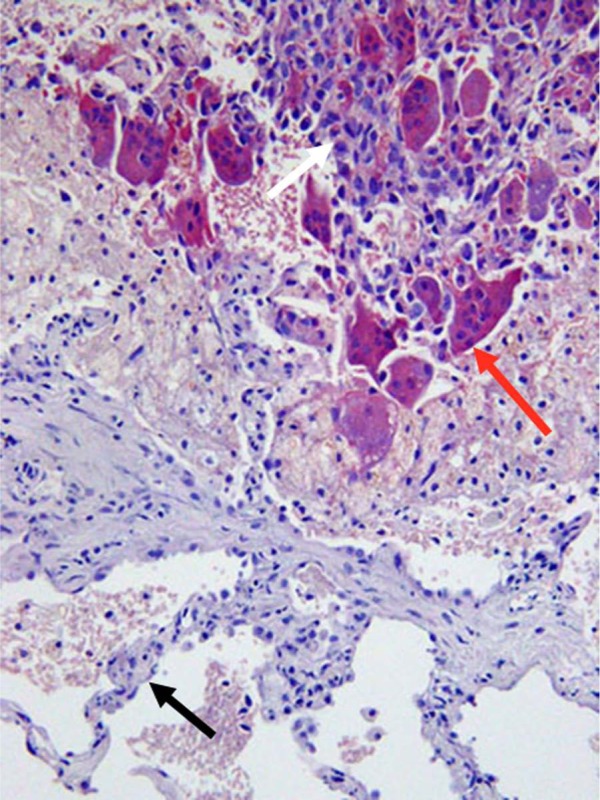
Osteoclasts associated with a sarcoma in the lung. Multinucleated osteoclasts (red arrow) are present in close association with a spindle-cell primary sarcoma in the lung of a 67-year-old woman. Lung alveolar wall (black arrow). Tartrate-resistant acid phosphatase, counterstained with hematoxylin.
Osteoimmunology and Co-stimulatory Signaling
NF-κB and NFATc1 signaling in OC formation, coupled with their required roles in lymph node formation, immune responses, and inflammatory arthritis, spawned the new field of osteoimmunology, in which interactions between bone and immune cells are studied (Takayanagi, 2012). Co-stimulatory signaling is a component of normal and aberrant immune responses. It enhances OC formation and activation by activating NFATc1, which, like NF-κB, regulates immune responses, but it also controls neuronal, cardiovascular, muscle, and other cell functions (Hogan et al., 2003). Co-stimulatory signaling activates NFATc1 through ligand binding to immunoglobulin-like receptors, such as TREM-2 (triggering receptor expressed in myeloid cells-2) and osteoclast-associated receptor (OSCAR) (Takayanagi et al., 2002). The ligands for most of these receptors on OCPs have not been identified, but OSCAR is activated by specific parts of collagen, which become exposed during resorption (Barrow et al., 2011). These receptors recruit adapter molecules, including Fc receptor common γ subunit (FcRγ) and DAP12, resulting in phosphorylation of immunoreceptor tyrosine-based activation motifs (ITAMs) within these adapters and the activation of downstream signaling. RANK and co-stimulatory signaling activate phospholipase Cγ (PLCγ) and calcium-calmodulin signaling, resulting in the release of calcium from stores within the cytoplasm and dephosphorylation of NFATc1 by the calcium-dependent phosphatase, calcineurin, and NFATc1 translocates to nuclei (Takayanagi et al., 2002). Thus, co-stimulatory and RANK signaling likely synergize through NFATc1 activation to enhance OC formation and activation, making NFATc1 a strong candidate for therapeutic intervention in inflammatory bone diseases.
NFATc1 activation can be prevented by cyclosporine-A, a calcineurin/NFATc1 inhibitor used clinically as an immunosuppressive drug and which prevented bone loss in a mouse model of RA (Koga et al., 2005). Interestingly, NFATc1 activation through either RANK or OSCAR increases OSCAR expression on OCPs in a positive feedback loop (Asagiri and Takayanagi, 2007). OSCAR and RANKL expression is increased in the synovium from joints of patients with RA (Crotti et al., 2012), but to date there have been no reports of OSCAR expression in inflamed gingival tissues in patients.
NFATc1 was also discovered to positively regulate the expression of osterix, a transcription factor with essential functions downstream of Runx2 in OB precursor differentiation. Treatment of normal mice with cyclosporine A resulted in osteoporosis because of its inhibitory effects on NFATc1-induced OB formation (Koga et al., 2005), suggesting that it has a more important role in OB than OC differentiation during normal bone remodeling. In inflammatory bone diseases, bone formation typically is inhibited by a variety of mechanisms, including TNF- and dickkopf-1-mediated inhibition of osteoblast functions (Boyce et al., 2012), and in these diseases the major observed effects of NFATc1 inhibitors may be reduced bone resorption.
T and B cells are present in inflamed joints of RA patients and in gingival tissues of patients with periodontitis. They also express RANKL to increase osteoclastogenesis in these conditions, but their effects are complex. For example, CD3+ T-helper (Th) cells express RANKL, and Th17 cells induce RANKL expression through IL-17, but T regulatory cells (Tregs) inhibit OC formation in part through expression of IL-4 and IL-10, and Th1 cells express INFγ, which has both inhibitory and stimulatory effects on OC formation, depending upon the stage of OCP differentiation (Zhao and Ivashkiv, 2011). Tregs are present in crevicular fluid from RA patients, but levels of IL-4 and IL-10 were low, and patients with severe periodontitis had the lowest levels (Berthelot and Le Goff, 2010). Overall, the effects of T cells in inflammatory bone disease may be neutral, with synoviocytes being the main source of RANKL (Schett and David, 2010; Takayanagi, 2012). More B cells than T cells are detected in the pericrevicular soft tissues of patients with periodontitis, but there are also conflicting data about the B-cell expression of RANKL (Okamoto and Takayanagi, 2011), and thus further study will be required to determine their roles in inflammatory bone loss.
Negative Regulation of Osteoclast Formation and Activity
Osteoblastic-cell-mediated
OC formation and/or activity is limited by numerous mechanisms, including calcitonin secretion by thyroid C cells, but few of these were identified before the discovery of RANKL, which facilitated the generation of sufficiently high numbers of highly purified OCPs for gene expression profiling. Osteoprotegerin (OPG), which binds to RANKL to inhibit interaction with RANK, is the major negative regulator of all aspects of bone resorption (Appendix Table 1) (Asagiri and Takayanagi, 2007; Boyce et al., 2012). Loss-of-function mutations of TNFRSF11B, the gene encoding OPG, account for most cases of juvenile Paget’s disease, in which unopposed RANKL induces osteoporosis (Whyte et al., 2002). This is similar to the phenotype of OPG-/- mice.
Osteoblastic cells regulate OC formation through additional mechanisms, some of which regulate OPG expression. For example, canonical Wnt/β-catenin signaling, which is required for OB formation, also promotes OPG expression by OBs (Hill et al., 2005). In contrast, Wnt 5a-induced non-canonical signaling promotes OC formation through receptor tyrosine kinase-like orphan receptor proteins expressed in OCPs (Maeda et al., 2012). In addition, Jagged1/Notch1, which also promotes osteoblast differentiation, increases the OPG/RANKL ratio in stromal cells to inhibit OC formation (Appendix Table 1) (Boyce et al., 2012), and Zfp521 inhibits OC formation by reducing RANKL expression and suppressing Ebf-1-induced osteoclastogenesis (Kiviranta et al., 2013).
Semaphorins (Semas), which are expressed widely as secreted and membrane-associated proteins, also regulate OC/OB interactions. Sema3A is secreted by OBs and OCs and inhibits RANKL-induced OC formation by inhibiting ITAM and RhoA signaling. Sema4D is expressed by OCs and inhibits OB differentiation and function (Appendix Table 2) by activating RhoA-ROCK, which inhibits IGF-1 signaling. In contrast, Sema6d induces OC formation through TREM-2/DAP12/PLCγ-induced NFATc1 activation and promotes OC activation by inducing podosome formation through Rac-GTP generation in OCs (Kang and Kumanogoh, 2013). Semaphorins signal through plexins and neuropilins, and knockout mice lacking some of these proteins have either osteopetrosis or osteoporosis, supporting important roles for them in bone cell functions.
RANKL also limits OC formation through several mechanisms. For example, although c-Fos mediates NFATc1-induced OC formation, it also promotes secretion of interferon-β, which in turn binds to its receptor on OCPs, leading to reduced c-Fos protein levels (Asagiri and Takayanagi, 2007). c-Fos/NFATc1 signaling also increases the expression of ephrinB2 on the surfaces of OCPs. Ephrins control axon, endothelial, and immune cell functions during embryonic development through direct interaction with Eph receptors on adjacent cells and bi-directional signaling (Davy and Soriano, 2005). Ephrin B2 binding to Eph4 on osteoblastic cells down-regulates c-Fos and NFATc1 expression through reverse signaling to reduce OC formation, and forward signaling through Eph4 promotes OB precursor differentiation by inhibiting the small GTPase, RhoA (Appendix Tables 1, 2) (Zhao et al., 2006).
Many questions remain about exactly how and where in BRUs these various interactions take place and which ones are between OCs and OBs or their precursors. Intuitively, it would make sense to have OB precursors positively regulating OC formation and fusion at the resorbing edges of BRUs and also promoting OC apoptosis in reversal sites where OCPs could enhance OB precursor differentiation. In contrast, mature OB secretion of OPG and expression of other negative regulators of OC formation would keep OCs away from bone-building sites.
Constitutive Transcriptional Repression of RANK Signaling
There are also constitutive mechanisms to inhibit basal OC formation. For example, in the absence of RANKL, Bcl6 is recruited to the NFATc1, cathepsin K, and DC-STAMP promoters to inhibit osteoclastogenesis. RANKL induces the removal of Bcl6 from these promoters and its replacement by NFATc1 to mediate osteoclastogenesis (Miyauchi et al., 2010). Interferon regulatory factor-8 (IRF-8), Eos, and v-maf musculoaponeurotic fibrosarcoma oncogene family protein B are other constitutively expressed transcriptional repressors (Zhao and Ivashkiv, 2011), and their expression is down-regulated by RANK signaling. They are also direct targets of B-lymphocyte-induced maturation protein-1, deletion of which in OCs results in osteopetrosis due to up-regulation of Bcl6 and impaired osteoclastogenesis (Miyauchi et al., 2010). In contrast, Bcl6-/- mice have increased OC formation and severe osteoporosis. Thus, RANKL/RANK activation of NFATc1 in OCPs not only promotes osteoclastogenesis directly, but also facilitates it indirectly by repressing expression of these negative regulators.
Pro-inflammatory Cytokines
Cytokines, such as TNF, are important inducers of bone resorption and inflammation in numerous disorders, including RA and periodontal disease (Boyce et al., 2012), but they also activate mechanisms to restrict the destruction. For example, most of the factors that induce expression of RANKL also induce expression of OPG, albeit to a lesser degree, the net effect being increased bone resorption (Kearns et al., 2008). Similarly, although TNF induces resorption through and independently of RANKL (Yao et al., 2009), it also limits OC formation by several mechanisms. These include preventing the degradation of non-canonical NF-κB inhibitory proteins (Yao et al., 2009) and inducing expression of IRF-8 and the Notch-induced DNA-binding molecule, RBP-Jκ, in OCPs (Zhao and Ivashkiv, 2011). TNF also promotes secretion by OCPs and OCs of TNF-stimulated gene 6, which synergizes with OPG to limit OC activity through an autocrine mechanism (Mahoney et al., 2011). IL-10, an anti-inflammatory cytokine, which functions to resolve inflammation, limits OC formation by inhibiting expression of c-Fos, c-Jun, TREM-2, and NFATc1 in OCPs. IL-4 limits bone resorption by promoting OPG expression and suppressing expression of RANKL, RANK, NF-κB, c-Fos, NFATc1, MAPK, and calcium signaling during OC formation (Zhao and Ivashkiv, 2011). During co-stimulatory immune reactions, ITAM-bearing proteins partner with proteins containing immunoreceptor tyrosine-based inhibitory motifs (ITIMs), and these negatively regulate osteoclastogenesis (Mori et al., 2008).
Osteoclast Apoptosis and Survival
OCs undergo apoptosis at reversal sites in BRUs where new bone is laid down by OBs. Estrogen maintains bone mass in part by promoting OC apoptosis mediated by TGFβ (Boyce, 2013) and increasing Fas-ligand expression in OCs, but also by inhibiting expression of genes regulating OC activity, without affecting OCP proliferation or fusion (Nakamura et al., 2007). Bisphosphonates induce OC apoptosis, in part by inhibiting the activity of enzymes in the mevalonate pathway and promoting caspase cleavage of Mammalian Sterile 20-like (Mst) kinase 1 (Rogers et al., 2011), although some amino-bisphosphonates can inhibit bone resorption without inducing OC apoptosis (Matsumoto et al., 2011). Denosumab (Hanley et al., 2012) and raloxifene (Krum et al., 2008) also induce OC apoptosis, but calcitonin and cathepsin K inhibitors do not (Boonen et al., 2012).
OC survival is enhanced by cytokines, including M-CSF, RANKL, TNF, IL-1, and VEGF-A, through up-regulation of Rho family small G-protein Ras/Rac1/Erk and PI3 kinase/mTOR/S6K signaling (Tanaka et al., 2006), while cytokine withdrawal leads to reduced expression of the anti-apoptotic protein, Bcl-2, and rapid OC apoptosis (Tanaka et al., 2010). An early effect of RANKL signaling is NF-κB p65-mediated prevention of Bid- and caspase 3-induced OCP apoptosis (Vaira et al., 2008). M-CSF prevents OC apoptosis by several mechanisms, including: activation of MITF, which increases Bcl-2 expression (Tanaka et al., 2006, 2010); increased degradation of Bim by c-Cbl, an ubiquitin ligase; up-regulation of Bcl-XL expression, which inhibits cleavage of procaspase-9; and inhibition of caspases 3 and 9, which initiate apoptosis. Bim is a pro-apoptotic Bcl-2 family member whose expression is down-regulated by IL-3 signaling through the Raf/Erk and/or PI3K/mTOR pathways. Bim-/- mice have decreased OC activity, despite increased OC survival (Tanaka et al., 2010). However, enhanced OC survival overall is associated with increased bone resorption.
Supplementary Material
Supplementary material
Acknowledgments
The author thanks Nora Plumeri for secretarial assistance.
Footnotes
The author acknowledges grant support from the NIH (National Institute of Arthritis and Musculoskeletal and Skin Diseases, grant number AR43510-17).
The author declares no potential conflicts of interest with respect to the authorship and/or publication of this article.
References
- Aarthi JJ, Darendeliler MA, Pushparaj PN. (2011). Dissecting the role of the S1P/S1PR axis in health and disease. J Dent Res 90:841-854. [DOI] [PubMed] [Google Scholar]
- Asagiri M, Takayanagi H. (2007). The molecular understanding of osteoclast differentiation. Bone 40:251-264. [DOI] [PubMed] [Google Scholar]
- Barrow AD, Raynal N, Andersen TL, Slatter DA, Bihan D, Pugh N, et al. (2011). OSCAR is a collagen receptor that costimulates osteoclastogenesis in DAP12-deficient humans and mice. J Clin Invest 121:3505-3516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berthelot JM, Le Goff B. (2010). Rheumatoid arthritis and periodontal disease. Joint Bone Spine 77:537-541. [DOI] [PubMed] [Google Scholar]
- Boonen S, Rosenberg E, Claessens F, Vanderschueren D, Papapoulos S. (2012). Inhibition of cathepsin K for treatment of osteoporosis. Curr Osteoporos Rep 10:73-79. [DOI] [PubMed] [Google Scholar]
- Boyce BF. (2013). Advances in osteoclast biology reveal potential new drug targets and new roles for osteoclasts. J Bone Miner Res 28:711-722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boyce BF, Xing L. (2011). Src inhibitors in the treatment of metastatic bone disease: rationale and clinical data. Clin Investig (Lond) 1:1695-1706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boyce BF, Yao Z, Xing L. (2009). Osteoclasts have multiple roles in bone in addition to bone resorption. Crit Rev Eukaryot Gene Expr 19:171-180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boyce BF, Zuscik MJ, Xing L. (2012). Biology of bone and cartilage. In: Genetics of bone biology and skeletal disease. Thakker RV, Eisman J, Igarashi T, Whyte MP, editors. London, UK: Elsevier, pp. 3-24. [Google Scholar]
- Chen W, Zhu G, Hao L, Wu M, Ci H, Li YP. (2013). C/EBPalpha regulates osteoclast lineage commitment. Proc Natl Acad Sci USA 110:7294-7299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crotti TN, Dharmapatni AA, Alias E, Zannettino AC, Smith MD, Haynes DR. (2012). The immunoreceptor tyrosine-based activation motif (ITAM) -related factors are increased in synovial tissue and vasculature of rheumatoid arthritic joints. Arthritis Res Ther 14:R245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davy A, Soriano P. (2005). Ephrin signaling in vivo: look both ways. Dev Dyn 232:1-10. [DOI] [PubMed] [Google Scholar]
- Del Fattore A, Cappariello A, Teti A. (2008). Genetics, pathogenesis and complications of osteopetrosis. Bone 42:19-29. [DOI] [PubMed] [Google Scholar]
- Delgado-Calle J, Garmilla P, Riancho JA. (2012). Do epigenetic marks govern bone mass and homeostasis? Curr Genomics 13:252-263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DiDonato JA, Mercurio F, Karin M. (2012). NF-kappaB and the link between inflammation and cancer. Immunol Rev 246:379-400. [DOI] [PubMed] [Google Scholar]
- Fujita K, Iwasaki M, Ochi H, Fukuda T, Ma C, Miyamoto T, et al. (2012). Vitamin E decreases bone mass by stimulating osteoclast fusion. Nat Med 18:589-594. [DOI] [PubMed] [Google Scholar]
- Hanley DA, Adachi JD, Bell A, Brown V. (2012). Denosumab: mechanism of action and clinical outcomes. Int J Clin Pract 66:1139-1146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hemingway F, Taylor R, Knowles HJ, Athanasou NA. (2011). RANKL-independent human osteoclast formation with APRIL, BAFF, NGF, IGF I and IGF II. Bone 48:938-944. [DOI] [PubMed] [Google Scholar]
- Hill TP, Spater D, Taketo MM, Birchmeier W, Hartmann C. (2005). Canonical Wnt/beta-catenin signaling prevents osteoblasts from differentiating into chondrocytes. Dev Cell 8:727-738. [DOI] [PubMed] [Google Scholar]
- Hocking LJ, Whitehouse C, Helfrich MH. (2012). Autophagy: a new player in skeletal maintenance? J Bone Miner Res 27:1439-1447. [DOI] [PubMed] [Google Scholar]
- Hogan PG, Chen L, Nardone J, Rao A. (2003). Transcriptional regulation by calcium, calcineurin, and NFAT. Genes Dev 17:2205-2232. [DOI] [PubMed] [Google Scholar]
- Jimi E, Aoki K, Saito H, D’Acquisto F, May MJ, Nakamura I, et al. (2004). Selective inhibition of NF-kappa B blocks osteoclastogenesis and prevents inflammatory bone destruction in vivo. Nat Med 10:617-624. [DOI] [PubMed] [Google Scholar]
- Kakonen SM, Mundy GR. (2003). Mechanisms of osteolytic bone metastases in breast carcinoma. Cancer 97(3 Suppl):834S-839S. [DOI] [PubMed] [Google Scholar]
- Kalyan S, Quabius ES, Wiltfang J, Monig H, Kabelitz D. (2013). Can peripheral blood gammadelta T cells predict osteonecrosis of the jaw? An immunological perspective on the adverse drug effects of aminobisphosphonate therapy. J Bone Miner Res 28:728-735. [DOI] [PubMed] [Google Scholar]
- Kang S, Kumanogoh A. (2013). Semaphorins in bone development, homeostasis, and disease. Semin Cell Dev Biol 24:163-171. [DOI] [PubMed] [Google Scholar]
- Kearns AE, Khosla S, Kostenuik PJ. (2008). Receptor activator of nuclear factor kappaB ligand and osteoprotegerin regulation of bone remodeling in health and disease. Endocr Rev 29:155-192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kikuta J, Iwai K, Saeki Y, Ishii M. (2011). S1P-targeted therapy for elderly rheumatoid arthritis patients with osteoporosis. Rheumatol Int 31:967-969. [DOI] [PubMed] [Google Scholar]
- Kiviranta R, Yamana K, Saito H, Ho DK, Laine J, Tarkkonen K, et al. (2013). Coordinated transcriptional regulation of bone homeostasis by Ebf1 and Zfp521 in both mesenchymal and hematopoietic lineages. J Exp Med 210:969-985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koga T, Matsui Y, Asagiri M, Kodama T, de Crombrugghe B, Nakashima K, et al. (2005). NFAT and Osterix cooperatively regulate bone formation. Nat Med 11:880-885. [DOI] [PubMed] [Google Scholar]
- Krum SA, Miranda-Carboni GA, Hauschka PV, Carroll JS, Lane TF, Freedman LP, et al. (2008). Estrogen protects bone by inducing Fas ligand in osteoblasts to regulate osteoclast survival. EMBO J 27:535-545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lotinun S, Kiviranta R, Matsubara T, Alzate JA, Neff L, Luth A, et al. (2013). Osteoclast-specific cathepsin K deletion stimulates S1P-dependent bone formation. J Clin Invest 123:666-681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maeda K, Kobayashi Y, Udagawa N, Uehara S, Ishihara A, Mizoguchi T, et al. (2012). Wnt5a-Ror2 signaling between osteoblast-lineage cells and osteoclast precursors enhances osteoclastogenesis. Nat Med 18:405-412. [DOI] [PubMed] [Google Scholar]
- Mahoney DJ, Swales C, Athanasou NA, Bombardieri M, Pitzalis C, Kliskey K, et al. (2011). TSG-6 inhibits osteoclast activity via an autocrine mechanism and is functionally synergistic with osteoprotegerin. Arthritis Rheum 63:1034-1043. [DOI] [PubMed] [Google Scholar]
- Maruyama K, Fukasaka M, Vandenbon A, Saitoh T, Kawasaki T, Kondo T, et al. (2012). The transcription factor Jdp2 controls bone homeostasis and antibacterial immunity by regulating osteoclast and neutrophil differentiation. Immunity 37:1024-1036. [DOI] [PubMed] [Google Scholar]
- Matsumoto T, Nagase Y, Iwasawa M, Yasui T, Masuda H, Kadono Y, et al. (2011). Distinguishing the proapoptotic and antiresorptive functions of risedronate in murine osteoclasts: role of the Akt pathway and the ERK/Bim axis. Arthritis Rheum 63:3908-3917. [DOI] [PubMed] [Google Scholar]
- Mellis DJ, Itzstein C, Helfrich MH, Crockett JC. (2011). The skeleton: a multi-functional complex organ: the role of key signalling pathways in osteoclast differentiation and in bone resorption. J Endocrinol 211:131-143. [DOI] [PubMed] [Google Scholar]
- Miller PD. (2011). A review of the efficacy and safety of denosumab in postmenopausal women with osteoporosis. Ther Adv Musculoskelet Dis 3:271-282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyauchi Y, Ninomiya K, Miyamoto H, Sakamoto A, Iwasaki R, Hoshi H, et al. (2010). The Blimp1-Bcl6 axis is critical to regulate osteoclast differentiation and bone homeostasis. J Exp Med 207:751-762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mori Y, Tsuji S, Inui M, Sakamoto Y, Endo S, Ito Y, et al. (2008). Inhibitory immunoglobulin-like receptors LILRB and PIR-B negatively regulate osteoclast development. J Immunol 181:4742-4751. [DOI] [PubMed] [Google Scholar]
- Nakamura T, Imai Y, Matsumoto T, Sato S, Takeuchi K, Igarashi K, et al. (2007). Estrogen prevents bone loss via estrogen receptor alpha and induction of Fas ligand in osteoclasts. Cell 130:811-823. [DOI] [PubMed] [Google Scholar]
- Okamoto K, Takayanagi H. (2011). Regulation of bone by the adaptive immune system in arthritis. Arthritis Res Ther 13:219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pacifici R. (2012). Role of T cells in ovariectomy induced bone loss—revisited. J Bone Miner Res 27:231-239. [DOI] [PubMed] [Google Scholar]
- Rogers MJ, Crockett JC, Coxon FP, Monkkonen J. (2011). Biochemical and molecular mechanisms of action of bisphosphonates. Bone 49:34-41. [DOI] [PubMed] [Google Scholar]
- Ross FP, Teitelbaum SL. (2005). alphavbeta3 and macrophage colony-stimulating factor: partners in osteoclast biology. Immunol Rev 208:88-105. [DOI] [PubMed] [Google Scholar]
- Schett G, David JP. (2010). The multiple faces of autoimmune-mediated bone loss. Nat Rev Endocrinol 6:698-706. [DOI] [PubMed] [Google Scholar]
- Takayanagi H. (2012). New developments in osteoimmunology. Nat Rev Rheumatol 8:684-689. [DOI] [PubMed] [Google Scholar]
- Takayanagi H, Kim S, Koga T, Nishina H, Isshiki M, Yoshida H, et al. (2002). Induction and activation of the transcription factor NFATc1 (NFAT2) integrate RANKL signaling in terminal differentiation of osteoclasts. Dev Cell 3:889-901. [DOI] [PubMed] [Google Scholar]
- Tanaka S, Miyazaki T, Fukuda A, Akiyama T, Kadono Y, Wakeyama H, et al. (2006). Molecular mechanism of the life and death of the osteoclast. Ann NY Acad Sci 1068:180-186. [DOI] [PubMed] [Google Scholar]
- Tanaka S, Wakeyama H, Akiyama T, Takahashi K, Amano H, Nakayama KI, et al. (2010). Regulation of osteoclast apoptosis by Bcl-2 family protein Bim and Caspase-3. Adv Exp Med Biol 658:111-116. [DOI] [PubMed] [Google Scholar]
- Tang Y, Wu X, Lei W, Pang L, Wan C, Shi Z, et al. (2009). TGF-beta1-induced migration of bone mesenchymal stem cells couples bone resorption with formation. Nat Med 15:757-765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teitelbaum SL. (2011). The osteoclast and its unique cytoskeleton. Ann NY Acad Sci 1240:14-17. [DOI] [PubMed] [Google Scholar]
- Vaira S, Alhawagri M, Anwisye I, Kitaura H, Faccio R, Novack DV. (2008). RelA/p65 promotes osteoclast differentiation by blocking a RANKL-induced apoptotic JNK pathway in mice. J Clin Invest 118:2088-2097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wada T, Nakashima T, Hiroshi N, Penninger JM. (2006). RANKL-RANK signaling in osteoclastogenesis and bone disease. Trends Mol Med 12:17-25. [DOI] [PubMed] [Google Scholar]
- Wang Z, McCauley LK. (2011). Osteoclasts and odontoclasts: signaling pathways to development and disease. Oral Dis 17:129-142. [DOI] [PubMed] [Google Scholar]
- Whyte MP, Obrecht SE, Finnegan PM, Jones JL, Podgornik MN, McAlister WH, et al. (2002). Osteoprotegerin deficiency and juvenile Paget’s disease. N Engl J Med 347:175-184. [DOI] [PubMed] [Google Scholar]
- Xiong J, O’Brien CA. (2012). Osteocyte RANKL: new insights into the control of bone remodeling. J Bone Miner Res 27:499-505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yagi M, Ninomiya K, Fujita N, Suzuki T, Iwasaki R, Morita K, et al. (2007). Induction of DC-STAMP by alternative activation and downstream signaling mechanisms. J Bone Miner Res 22:992-1001. [DOI] [PubMed] [Google Scholar]
- Yao Z, Xing L, Boyce BF. (2009). NF-kappaB p100 limits TNF-induced bone resorption in mice by a TRAF3-dependent mechanism. J Clin Invest 119:3024-3034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yasui T, Hirose J, Tsutsumi S, Nakamura K, Aburatani H, Tanaka S. (2011). Epigenetic regulation of osteoclast differentiation: possible involvement of Jmjd3 in the histone demethylation of Nfatc1. J Bone Miner Res 26:2665-2671. [DOI] [PubMed] [Google Scholar]
- Zhao B, Ivashkiv LB. (2011). Negative regulation of osteoclastogenesis and bone resorption by cytokines and transcriptional repressors. Arthritis Res Ther 13:234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao C, Irie N, Takada Y, Shimoda K, Miyamoto T, Nishiwaki T, et al. (2006). Bidirectional ephrinB2-EphB4 signaling controls bone homeostasis. Cell Metab 4:111-121. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material