RNase-sensitive DNA modification(s) initiates S. pombe mating-type switching (original) (raw)
Abstract
Mating-type switching in fission yeast depends on an imprint at the mat1 locus. Previous data showed that the imprint is made in the DNA strand replicated as lagging. We now identify this imprint as an RNase-sensitive modification and suggest that it consists of one or two RNA residues incorporated into the mat1 DNA. Formation of the imprint requires _swi1_- and _swi3_-dependent pausing of the replication fork. Interestingly, swi1 and swi3 mutations that abolish pausing do not affect the use of lagging-strand priming site during replication. We show that the pausing of replication and subsequent formation of the imprint occur after the leading-strand replication complex has passed the site of the imprint and after lagging-strand synthesis has initiated at this proximal priming site. We propose a model in which a _swi1_- and _swi3_-dependent signal during lagging-strand synthesis leads to pausing of leading-strand replication and the introduction of the imprint.
Keywords: DNA modification, imprint, replication pausing, mating-type switching, Schizosaccharomyces pombe
Cellular differentiation mediated by DNA recombination has been described in several biological systems including Saccharomyces cerevisiae, Schizosaccharomyces pombe, and mammalian B-cell and T-cell differentiation. Several different mechanisms are used for initiation of these DNA recombination events. In S. cerevisiae, the site-specific endonuclease HO introduces a double-stranded break (DSB) at the MAT locus (Kostriken et al. 1983; for review, see Haber 1998). In B and T cells, V(D)J recombination is initiated by DSBs, introduced in the target DNA sequence by RAG1 and RAG2 proteins (McBlane et al. 1995; for review, see Gellert 2002). The combined action of the cytidine deaminase and uracil-DNA glycosylase is thought to generate abasic sites, leading to class switch recombination in B cells (Di Noia and Neuberger 2002; Rada et al. 2002). Here we show that S. pombe relies on RNase-sensitive modifications in the DNA double helix for the initiation of the replication-coupled recombination event that leads to mating-type switching in this yeast.
The S. pombe mating-type region consists of three gene cassettes, mat1, mat2P, and mat3M, each flanked by the homology boxes H1 and H2 (Fig. 1A). The donor loci mat2P and mat3M, containing P or M information, respectively, are transcriptionally silenced (Kelly et al. 1988). The mat1, which contains either P or M information, is transcriptionally active and determines the mating type of the cell. Switching between mating types occurs via a recombination event between mat1M and mat2P or mat1P and mat3M cassettes, which replaces the information at mat1 with the information of the opposite mating type (Fig. 1A; Beach 1983; Beach and Klar 1984; Arcangioli and de Lahondes 2000; for review, see Dalgaard and Klar 2001b).
Figure 1.

mat1 locus and the direction-of-replication model. (A) The mating-type region of S. pombe. mat1, containing either P or M information; mat2P and mat3M cassettes are shown. Each cassette is flanked by homology domains H1 and H2 (boxes). The positions of the centromere (circle) and the _cis_-acting sequences SAS1 and SAS2 (bracket) relative to mat1 are indicated. The recombination event, which replaces the cassette at mat1 with the information of the opposite mating type, is shown by black arrows. The silencing of mat2 and mat3 is indicated by the gray line. (B) The direction-of-replication model. The mating-type pedigree of a newly switched cell is shown. The cells are shown in yellow, and the cell generation is indicated to the right. Next to each cell is a diagram of the sister chromatids, present in the corresponding cell type. The strands, replicated as leading and lagging, are shown as red and blue arrows, respectively. The imprint is indicated by a yellow circle, and the diagram of initiation of the recombination event is displayed in brackets. (C) Replication at the mat1 locus. mat1 replication by a distal origin is secured by a polar replication terminator RTS1, shown as a gray triangle. Nascent leading and lagging strands are indicated by continuous red and discontinuous blue arrows, respectively. Homology domains H1 and H2 are indicated by gray boxes. The white arrows below represent the direction of replication at mat1. The _swi1_- and _swi3_-dependent replication pause is shown as a gray shadow.
Pedigree analysis of a mitotically dividing newly switched cell shows that one of the two daughter cells has a potential to switch (cells labeled ′ in Fig. 1B), and one of four granddaughter cells switches mating type (M in Fig. 1B; Miyata and Miyata 1981). Switchable cells carry a strand-specific imprint at the mat1 (Egel and Eie 1987; Klar 1987; Klar and Bonaduce 1993). Later studies showed that the imprint is converted into a DSB during standard methods of DNA purification (Arcangioli 1998; Dalgaard and Klar 1999). This break was mapped to a position close to the border of the homology domain H1 within the mat1 cassette on the strand, which contains the imprint (Fig. 1B; Nielsen and Egel 1989). The imprint was characterized either as an alkali-labile modification (Dalgaard and Klar 1999) or a nick (Arcangioli 1998). Genetic experiments show that imprinting occurs only when mat1 is replicated in a centromere-proximal direction (Fig. 1C; Dalgaard and Klar 1999, 2000, 2001a). A polar terminator of replication, RTS1, is located proximal to mat1 (Fig. 1C). This element ensures, by direction-specific replication termination, that the mat1 locus is always replicated in the correct orientation for the imprinting and mating-type switching (Dalgaard and Klar 2000, 2001a).
We previously proposed a model, termed the direction-of-replication model, which explains the switching pattern of S. pombe (Dalgaard and Klar 1999; line drawing, Fig. 1B). The mat1 locus is replicated by a distal origin, and the imprint is made only in the sister chromatid replicated as the lagging strand. The imprint is maintained in the DNA during the cell cycle until the next S-phase, when the imprinted strand is a template for leading-strand replication. There, it is thought that the replication fork encounters the modification and a DSB is formed for the initiation of the DNA recombination event, leading to mating-type switching (Dalgaard and Klar 2001b).
Imprinting depends on the _trans_-acting factors swi1, swi3, and swi7 (Egel et al. 1984). 2D-gel analysis of replication intermediates detects a pause of the replication fork in the proximity of the mat1 imprint. Interestingly, _trans_-acting factors swi1 and swi3 are required for this replication pause (Dalgaard and Klar 2000). swi1p is a homolog of S. cerevisiae Tof1p and TIM1/Timeless proteins in higher eukaryotes (Park and Sternglanz 1999; Lakin-Thomas 2000). Tof1p and swi1p have been implicated in _MEC1_- and _cds1_-dependent replication checkpoints in S. cerevisiae and S. pombe, respectively (Foss 2001; Noguchi et al. 2003). In addition, increased recombination is observed at hydroxyurea-stalled replication forks in swi1 mutants (Noguchi et al. 2003). The identification of swi3 gene has not yet been published. Importantly, swi7 was shown to be polymerase α which is predominantly involved in lagging-strand replication (Singh and Klar 1993). In addition, two _cis_-acting sequences, SAS1 and SAS2, which are located distal to mat1 (Fig. 1A), are essential for imprinting. SAS1 and SAS2 are thought to play a role in maintenance of the imprint (Arcangioli and Klar 1991). The DNA-binding protein sap1p was shown to bind to SAS1 and SAS2 in vitro (Arcangioli and Klar 1991). A 263-bp deletion, named smt-0 (Figs. 1A, 3A, below), has been identified. This deletion removes SAS1 and SAS2 and abolishes the imprint and switching (Styrkarsdottir et al. 1993). Interestingly, neither the swi7 nor the smt-0 mutations affect replication pausing at mat1 (Dalgaard and Klar 2000). Thus, replication pausing at mat1 is necessary but not sufficient for the formation and/or the maintenance of the imprint.
Figure 3.

2D-gel analysis of the NdeI fragment, containing replication intermediates of mat1. (A) Analysis of the wild-type, SP835 (left), and smt-0, JZ108 (right), strains. The boxes above the autoradiographs show the magnification of the apex region of the Y-arc. The left and the middle correspond to the wild-type, and the right to the smt-0 2D gels. The arrows indicate the position of the cone signal. The line drawings below the autoradiographs show the position of the NdeI sites relative to mat1 and the distance from these sites to the imprint (circle) in wild-type and smt-0 DNA. The position and the length of smt-0 deletion are given. The diagram to the right displays the signals observed on a 2D gel of the wild type. The pause signal and the cone signal are shown, and the “chicken foot” structure, which constitutes the cone signal, is displayed above. The position of the 1N and 2N signals and the distance between them are indicated by the dotted lines and the bracket. The displacement of the cone signal relative to the pause signal is also shown by dotted lines and the small bracket below the diagram. (B) The quantification analysis of the 2D gels of wild-type and smt-0 strains. The diagram displays the signals quantified as well as the structure of corresponding replication intermediates. The intermediates present in switchable and unswitchable cells are shown. The intensities of the ascending (1, light gray) and descending (2, dark gray) parts of the Y-arc of wild type (JZ1) and smt-0 (JZ108) were quantified using Quantity One software from Bio-Rad on three 2D gels for each strain. The signal, labeled 1, was used to normalize the data. The average and standard deviation were calculated for value 2, as displayed on the histogram.
In this study, we first address how mating-type switching is initiated, when the leading-strand replication progression is blocked by the presence of the imprint in the template strand. A 2D gel of mat1 replication intermediates detects a signal, characteristic of “chicken foot” structure, formed when leading-strand replication encounters the imprint. Second, we show that the imprint is one or two RNase-sensitive nucleotides, incorporated into the mat1 DNA. Last, we characterize the spatial and temporal relationship between the replication pause necessary for imprinting and the formation of the imprint by mapping the ends of the nascent strands at the paused replication fork. The 3′-end of the leading strand and the 5′-end of the last Okazaki fragment map 354–369 and 342–345 nucleotides centromere-proximal to the imprint, respectively. Furthermore, the identified lagging-strand priming site is used with similar frequency in wild-type and swi7 strains, where replication pausing occurs, and in swi1 and swi3 strains, where replication pausing is abolished. Thus, the leading-strand replication pause at mat1 occurs after the synthesis of the lagging strand has initiated.
On the basis of these and previous observations, we propose the following additions to our earlier model for the mechanism of the imprinting: (1) The leading-strand replication complex replicates past the site of imprinting; (2) lagging-strand synthesis of the Okazaki fragment that is going to span over the site of the imprint is initiated at this proximal priming site; (3) a swi1p- and swi3p-dependent signal during lagging-strand elongation causes pausing of the leading-strand replication; and (4) the imprint is formed either via incorporation of the ribonucleotides during polymerization, or postreplicatively by an oxidation of the DNA backbone.
Results
Imprint in the template strand acts as a block for leading-strand replication
Mating-type switching has been proposed to initiate when the leading-strand replication complex encounters the imprint in the template strand (Arcangioli and de Lahondes 2000; Dalgaard and Klar 2001b; Fig. 1B, brackets). We analyzed the progression of the leading strand through the site of the imprint by applying to replication intermediates a high-resolution Southern blot technique (Nielsen and Egel 1989). The replication and switching intermediates were purified (Huberman et al. 1987) from JZ105 (wild-type mat1M) and SV1 (swi3) strains, each containing a deletion of the donor loci mat2P and mat3M. The deletion of mat2 and mat3 prevents the sequence homology at the donor cassettes from interfering with the analysis. In strains deleted for donor loci, the imprint at mat1 is still formed, but it is thought that instead of switching, homologous recombination occurs with the sister chromatid (Klar and Miglio 1986). In this experiment, the swi3 mutant strain acts as a control, as it lacks the mat1 imprint and therefore any switching-related intermediates. Replication intermediates were digested with SspI, purified from nonreplicating DNA by using benzoylated naphthoylated DEAE (BND) cellulose (Kiger and Sinsheimer 1969), and separated on a polyacrylamide gel. After subsequent electrotransfer, the filter was hybridized to the strand-specific probe that detects the lower strand. Using this technique, we mapped the 3′-end of the leading strand, formed when the replication fork encounters the imprint (Figs. 1B, 2B), exactly to the position where the imprint/break had been mapped on the other strand (Fig. 2A; Nielsen and Egel 1989). This result indicates that the leading-strand progression is blocked at the site of the imprint. Because recombination frequently occurs at stalled replication forks (Michel et al. 2001), the data support the model that the switching event is initiated when leading-strand replication encounters the imprint in the template strand.
Figure 2.
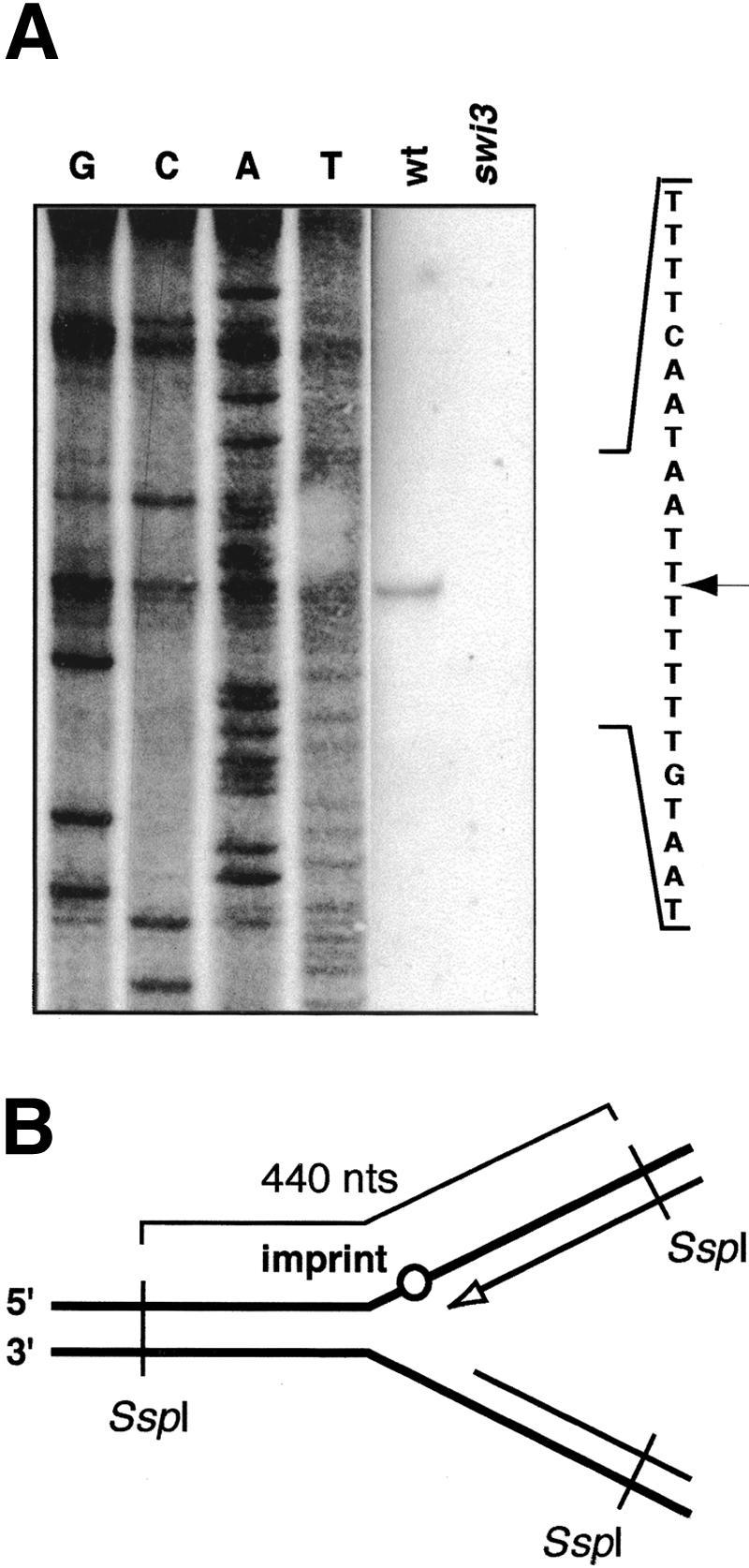
Leading-strand replication is blocked by the presence of the imprint in the template strand. (A) Mapping of the end of the nascent leading strand at the imprint using high-resolution Southern blot technique. Wild-type (JZ105) and a nonswitching swi3 (SV1) strains are analyzed in parallel. The sequence of the region analyzed is given to the right. The position of the band is indicated by the arrow. (B) Schematic diagram of the analyzed intermediates. The line drawing displays the position of the SspI sites and the size of the analyzed fragment. The imprint is indicated by a circle. The 3′-end of the nascent leading strand, mapped in A, is shown as an empty arrowhead.
There are fewer replication forks proximal to the site of the imprint in the strain that contains the imprint
According to the direction-of-replication model (Fig. 1B), approximately half of the cells in the population should have an imprint at mat1. Wild-type mat1 is replicated in the centromere-proximal direction. Therefore, on a 2D gel of mat1 replication intermediates, the ascending part of the Y-arc (Fig. 3B, left panel, region 1) will contain the forks approaching the imprint from the distal side, and the descending part of the Y-arc (Fig. 3B, left panel, region 2) will represent the forks leaving the site of the imprint on the proximal side (Fig. 3A,B). In the wild-type cells that contain the imprint, replication forks are converted into recombination intermediates at the site of the imprint, and these intermediates run at a different position on 2D gels (data not shown). We quantified the signal of the ascending and descending parts of the Y-arc (Fig. 3B, left panel) of the wild-type strain and the nonswitching smt-0 strain, and, as predicted, observed an approximately twofold decrease in signal of the descending part of the arc in the wild type compared with the smt-0 mutant (Fig. 3B, right panel, signal 2). Therefore, in the switching wild-type strain, the abundance of the forks leaving the site of the imprint on the proximal side is lower than in the nonswitching strain. This observation provides an additional support to the model that the recombination and switching are initiated when the leading-strand replication complex encounters the imprint in the template strand.
A `chicken foot' is formed when the leading-strand replication encounters the imprint in the template strand
Our data, published earlier, suggest that replication pausing at mat1 is prerequisite to imprinting (Dalgaard and Klar 2000). However, mat1 pausing and imprinting can be genetically separated: both swi7 and smt-0 mutants display a wild-type level of replication pausing at mat1, but lack the imprint (Dalgaard and Klar 2000). When we compared mat1 replication intermediates of wild-type and smt-0 strains by means of 2D gels, we detected a cone-shaped signal at the apex of the wild-type arc (Fig. 3A, wt). Importantly, this cone signal is absent on a 2D gel of the smt-0 strain (Fig. 3A, smt-0), suggesting that it represents the structure formed when the replication fork encounters the imprint in the template strand. These structures display a mobility characteristic of regressing replication forks similar to those formed when the leading-strand replication encounters DNA damage (Sogo et al. 2002; Courcelle et al. 2003). Indeed, both the replication fork that is blocked at the site of the imprint and the reversed fork generated from the blocked fork (Fig. 4B, right diagram) have the same molecular weight. Therefore, both fork structures should not separate in the first-dimension gel, but only in the second, where the regressed forks are retarded due to a more branched structure. Thus, on a 2D gel, the cone signal representing the regressing forks from the site of the imprint can only be positioned above the region of the Y-arc that corresponds to the forks blocked at the imprint.
Figure 4.

2D-gel analysis of the rqh1 mutant. (A) 2D gels of the NdeI-digested DNA from wild-type (JZ217) and rqh1 (JZ477) strains. The arrows show the position of the cone intermediates. (B) Interpretation of the autoradiographs. The diagram displays the position of the pause signal and the cone signal on the Y-arc, which represents mat1. The “chicken foot” structure, which constitutes the cone signal, is shown. The second Y-arc, which is visualized by the M-specific probe, represents mat3M. The line drawing below shows the position of the NdeI sites relative to mat1 and the distance from these sites to the imprint (circle). The line drawing to the right displays the process of fork reversal at the site of the imprint and the role of rqh1 in resetting reversed forks.
The “chicken foot” structures accumulate in the rqh1 mutant
Fork reversal is well characterized in bacterial systems. There, the helicase RecG can actively drive fork reversal, while another helicase, RecQ, is thought to counteract it, converting the X-shaped intermediate back into the Y-shaped replication fork. However, fork reversal can also happen spontaneously, without involvement of enzymes (for review, see Michel et al. 2001). Although the S. pombe homolog of RecG has not yet been identified, S. pombe rqh1p is thought to possess activities similar to RecQ (Fig. 4B; Murray et al. 1997; Doe et al. 2000, 2002). To confirm that the cone signal observed on 2D gels of mat1 represents fork regression, we analyzed the rqh1 mutant strain. Because rqh1 is involved in resetting reversed replication forks, we expected to see an increase in intensity of the cone signal, due to accumulation of the reversed forks. Indeed, the cone signal is more prominent on the 2D gel of the rqh1 mutant than on the gel of the wild-type strain (Fig. 4A). This result supports the suggestion that the observed signal represents fork regression.
The analyzed strains contain the S. cerevisiae LEU2 marker inserted distal to mat1. This insertion does not affect pausing or imprinting (data not shown), but because of the different length of the analyzed fragment, the pause site and the site of the imprint are displaced along the Y-arc compared with that shown in Figure 3A. Importantly, the cone signal is also displaced, indicating that the “chicken foot” is linked to the position of the imprint in the DNA.
Imprint is one or two ribonucleotides
Because the imprint is an alkali-labile modification present in the strand that was replicated as lagging, it was suggested earlier that it might be an RNA primer remaining after Okazaki-fragment processing (Dalgaard and Klar 1999). To directly test whether the imprint is an RNA, we used RNase treatment of the imprinted DNA, followed by a primer extension assay. It was shown earlier that some DNA preparation methods convert the imprint into a DSB, whereas others may maintain it intact (Arcangioli 1998; Dalgaard and Klar 1999). When DNA is purified as for 2D gels (Huberman et al. 1987), the imprint is converted into a DSB at only a low level. Surprisingly, when the primer extension was performed on double-stranded, nonreplicating DNA purified by this method, none or very little signal at the site of the imprint was detected (Fig. 5A, lane 1). This observation suggests that the VentR(exo–) polymerase used in the assay for primer extension is able to read through the imprint. We repeated the experiment, first converting the imprint into a break by using sodium hydroxide treatment before the primer extension (Fig. 5A, line drawing). As expected, the treatment generated a signal at the site where the imprint has been mapped previously and an additional weaker band could be detected one nucleotide below (Fig. 5A, lane 3; Nielsen and Egel 1989). These data show that the VentR(exo–) polymerase is able to read through the imprint under the conditions used in this assay, possibly due to its lack of proofreading activity. To test whether the imprint is an RNA, we treated imprinted DNA with the unspecific single-stranded RNase T2. Indeed, the treatment generated a break, showing that the imprint might be one, or several, RNA residue(s) (Fig. 5A, lane 2). To verify that RNase T2 is able to cleave single-stranded DNA that contains only one ribonucleotide, we used an oligo with one ribonucleotide in a sequence context that corresponded to that of the mat1 imprint. The treatment of the 5′ end-labeled oligo verified that RNase T2 can cleave such a substrate (Fig. 5B). To estimate how many RNA residues comprise the imprint, we used our observation that the VentR(exo–) DNA polymerase can read through the intact imprint. We used synthetic templates with one, two, three, or eight ribonucleotide residues in a sequence context that corresponded to that of the mat1 imprint (Fig. 5D). Primer extension using one cycle showed that the VentR(exo–) polymerase is partly able to read through one and two residues, and that this read-through is almost complete if, as in the primer extension assay, 30 cycles of extension are performed (Fig. 5C, left panel). However, if the number of ribonucleotides is increased to three or eight residues, they act as a complete block for the polymerase (Fig. 5C, right panel). These data suggest that the imprint is one or two ribonucleotides incorporated into the DNA duplex.
Figure 5.

Characterization of the mat1 imprint. (A) Primer extension on the intact mat1 imprint allows read-through of the VentR(exo–) polymerase, and the RNase T2 treatment converts the imprint into a break. Nonreplicating DNA from JZ105 strain was treated with RNase T2 or NaOH and analyzed by primer extension. The sequence of the region analyzed is given to the right, where the position of the bands is indicated by the arrows. The line drawing below shows the position of the primer used for the analysis relative to the site of the imprint and the interpretation of the obtained result. (B) RNase T2 can recognize one ribonucleotide present in the single-stranded DNA. End-labeled oligonucleotides 1 (without ribonucleotide) and 2 (with one ribonucleotide), shown in D, were treated with RNase T2. The arrow indicates the position of the band generated by RNase treatment. (C) Primer extension using VentR(exo–) polymerase on different synthetic templates. The templates, given in D, and the number of cycles used are shown above the autoradiographs. (D) Sequences of the single-stranded templates for primer extension used in B and C. The position where primer priSV2 anneals is shown. The sequence of homology domain H1 is outlined by a gray box. (E) Analysis of the imprint by primer extension on broken DNA, treated with different nucleases. The enzymes used are shown above the respective lanes. The drawing to the right displays the conversion of the imprint into a break by ribonucleases.
Last, we analyzed the properties of the imprint by using different ribonucleases. As mentioned earlier, some DNA preparations contain a break at the site of the imprint, and this break generates a single band in a primer extension assay. When such DNA is treated with single-stranded RNases T1 or T2, a second band is generated just below (Fig. 5E). The bond between the last deoxyribonucleotide and the first ribonucleotide on the 3′-end of the proximal fragment is not recognized by single-stranded RNases, because it is identical to the bonds between deoxyribonucleotides. Therefore, hydrolysis of the single-stranded RNA–DNA substrate by RNase will leave an RNA residue on the 3′-end of the proximal fragment. The next bond, between the first and the second ribonucleotides, is an RNA bond, and it appears to be most prone to hydrolysis during DNA purification, giving a band after primer extension on otherwise intact DNA. The appearance of the second band on a gel upon RNase treatment can be explained by removal of one ribonucleotide by cleaving the RNA bond between the 3′-end of the ribonucleotide and the 5′-end of the following deoxyribonucleotide on the distal fragment (Fig. 5E, line drawing). As expected, treatment with RNase H fails to generate the second band, because RNase H recognizes a minimum of four to six ribonucleotides (Fig. 5E). However, neither alkali treatment nor the digest with single-stranded RNases hydrolyzes the imprinted DNA so that only one, shorter extension product is observed (Fig. 5A,E). Together, these data strongly suggest that the wild-type DNA contains a population of imprinted molecules with one or two ribonucleotides, incorporated into the DNA duplex.
No RNA primers, synthesized during lagging-strand replication, map to the site of the imprint
Because swi7/polymerase α forms a complex with primase, which synthesizes RNA primers during replication, it was proposed that the imprint at mat1 is a part of such an RNA primer, left after Okazaki-fragment processing (Dalgaard and Klar 1999). To test this hypothesis, we mapped the position of RNA primers, formed during lagging-strand synthesis at mat1, by using the method for replication initiation point (RIP) mapping (Gerbi and Bielinsky 1997). The replication intermediates were purified as for 2D gels, phosphorylated using T4 polynucleotide kinase, and subsequently digested with λ-exonuclease. This enzyme recognizes 5′-phosphorylated DNA ends; thus the exonuclease digest leaves only nascent DNA molecules, protected by RNA primers on the 5′-end (Fig. 6A, right line drawing, solid black lines). The nascent DNA is then used as a template for primer extension. Extensive analysis with different primers revealed problems with unspecific annealing. To circumvent this problem, we analyzed wild-type mat1 and smt-0 DNA in parallel. The smt-0 deletion is located distal to mat1 (Fig. 3A, line drawing). By using a primer within the region, covered by the smt-0 deletion, we could identify bands formed due to specific annealing, as they should appear only in the lane with wild-type DNA. Primer priSV3 was selected as giving the highest specificity. Surprisingly, no specific band can be detected at the position of the imprint (Fig. 6A, left panel). This result suggests that the imprint is not formed from an RNA primer left after Okazaki-fragment processing and raises the possibilities that either the imprint is made during elongation of an Okazaki fragment, or the modification is introduced after the replication of the region is completed.
Figure 6.

Analysis of replication intermediates at mat1. (A) Primer extension on Okazaki fragments from the wild-type (JZ105), _smt_-0 (JZ108), swi1 (SV5), swi3 (SV1), and swi7 (SP469) strains. The left panel shows the analysis of the wild-type and smt-0 strains. The position of the homology domain H1 and the site of the imprint are given to the right of the autoradiograph. The drawing below shows the annealing position of the primer used for the assay on the wild-type DNA and lack of specific annealing to smt-0 DNA. The right panel shows the analysis of swi1, swi3, and swi7 mutants. Black lines indicate the position of the analyzed region relative to the left panel. The drawing below indicates that in this experiment the parental DNA, shown by dotted lines, was digested away by λ-exonuclease before primer extension. (B) Mapping of the pause site on the lagging strand on gel-purified paused intermediates. Strains are given in A. The sequence of the region (upper strand) is shown to the right, and the arrows indicate the position of the bands observed. The drawing below shows that RNase H removes RNA primers at 5′-ends of pausing intermediates of the lagging strand. The drawing also indicates that in this experiment primer can anneal both to nascent and to parental DNA. (C) Mapping of the pause site on the leading strand. The nonswitching strain smt-0 (JZ108) was used for clarity of the analysis, to avoid detecting imprinting and switching intermediates. The sequence of the analyzed region is shown to the right. The position of the BstNI restriction site on the upper strand is labeled, and the region of pausing is designated by the brackets. The line drawing below displays the position of the BstNI and SfaNI sites and the size of the full-length fragment of the lower strand. The strands, visualized on the gel, are shown as thick lines, and the mapped 3′-end of the nascent leading strand is shown as an empty arrowhead.
The 5′-end of the paused lagging strand maps 342–345 nucleotides proximal to the imprint
Interestingly, the cone signal observed in Figures 3A and 4A, which represents reversal of the replication fork at the site of the imprint, is not protruding from the center of the pause signal, as would be expected if pausing occurred at the site of imprinting. Instead, it originates from a position that is displaced along the Y-arc (Fig. 3A, right diagram). This physical separation of the two signals suggests, surprisingly, that the replication fork pausing occurs at the position different from the imprinting site. Because mat1 is replicated in a unidirectional manner, the position of the cone signal on the apex and the pause signal on the descending part of the Y-arc shows that the pause occurs within the mat1 cassette at the centromere-proximal side of the imprint.
In the primer extension experiments described earlier, the most prominent band observed on a gel mapped ∼350 nucleotides upstream of the imprint, also on a centromere-proximal side (Fig. 6A, left panel). To test whether this band represents the mat1 pause site, we compared wild-type, swi1, swi3, and swi7 strains on the gel: in original 2D-gel analysis, the pause site was shown to be present in wild type and swi7 and absent in swi1 and swi3 mutants (Dalgaard and Klar 2000). Surprisingly, the RIP-mapping analysis detected this prominent band with equal intensity in all four strains (Fig. 6A, right panel). We therefore repeated the experiment with paused intermediates purified from the agarose gels, omitting the λ-exonuclease treatment, and here we observed a significant difference between wild-type and swi3 strains. The signal that appears as two bands at positions 342 and 345 is much stronger in the wild type than in swi3 and absent in smt-0 (Figs. 6B, 7A). We speculated that when the replication fork pauses, Okazaki-fragment processing takes place, and RNase H removes the RNA primers from the long-lived paused molecules detected by 2D gels. λ-Exonuclease, used in the first experiment, removes the processed paused intermediates, which are not protected by an RNA primer, in the wild-type DNA. Thus, there is no difference in frequency the lagging-strand priming site is used in the strains that lack pausing, but swi1p- and swi3p-dependent pausing allows accumulation of Okazaki fragments where the RNA primers have been processed by RNase H.
Figure 7.

Replication pausing at mat1. (A) The summary of the mapping data. The position of the homology domain H1 is indicated by the gray box, and the nucleotides that the imprint consists of are circled. The palindromic bases around the imprint are labeled by arrows, indicating the center of the palindrome (Nielsen and Egel 1989). The start of the smt-0 deletion, extending into the sequence distal to mat1, is shown (Styrkarsdottir et al. 1993). The restriction sites, used in this study for the leading-strand mapping, are labeled, and the positions of the mapped replication intermediates are indicated by horizontal arrows and brackets. (B) The three-step model for the role of pausing in the formation of the imprint (see text). (C) Model for how the imprint can be introduced postreplicatively by oxidation.
The 3′-end of the paused leading strand maps close to the 5′-end of the lagging strand
To map the end of the leading strand in the paused replication fork, we used the high-resolution Southern blot method described earlier. A similar approach was used previously to map the blocked replication fork in the rDNA repeats of S. cerevisiae (Gruber et al. 2000), where 150 copies of rDNA repeats per genome are present. However, for a single-copy mat1 locus, it proved to be very difficult. The balance between analyzing enough DNA to detect the signal and using a thin enough gel to maintain resolution allows mapping of the paused leading strand within a 10- to 20-nucleotide region. The DNA was digested to generate a 265-nucleotide full-length fragment of the bottom strand (Fig. 6C, line drawing). The probe, specific to the leading (bottom) strand, detected two signals in a range of 354–361 and 362–369 nucleotides upstream of the site of the imprint, which are absent in the swi1 mutant (Figs. 6C, 7A; Supplementary Fig. 1). This signal is close to the 5′-end, mapped by primer extension on the lagging-strand paused intermediates.
Discussion
We test here key aspects of the imprinting mechanism that leads to mating-type switching in S. pombe. First, we show that the imprint constitutes a barrier for the replication fork. According to the “one-in-four” rule, there are two populations of cells in the wild-type culture: “unswitchable” and “switchable”. The unswitchable population carries a chromatid that is intact at the mat1 locus, and the switchable population—imprinted (Fig. 1B). This means that the pattern observed on 2D gels represents two events: replication fork pausing and the formation of the imprint in one population, and replication fork encountering the imprint in the template strand with initiation of mating-type switching in the other. We observed a cone signal, which is likely to represent fork regression and formation of a transient “chicken foot” structure (Figs. 3A, 4) when replication encounters the imprint in the second population. Fork regression is known to occur when the leading-strand replication complex encounters an obstacle in the template DNA, such as DNA damage (Courcelle et al. 2003). At mat1, this signal can be seen in the strains of different genotypes, where imprinting is not affected (Dalgaard and Klar 2000). We map the end of the nascent leading strand, formed when the replication fork encounters the imprint, to the position of the imprint. Together, these data support our earlier proposal that switching is initiated when the leading-strand replication complex encounters the imprint in the template strand and is unable to read through. Thus, the biological function of the imprint is to create an appropriate 3′-end of the invading strand, necessary for the subsequent recombination between mat1 and one of the donor loci, mat2P or mat3M. This can occur either through a replication fork collapse, where fork regression has to occur before the formation of a break in the imprinted template strand, or directly by using the protruding 3′-end of the newly synthesized leading strand, exposed in the “chicken foot”.
Second, we demonstrate that the site of the mat1 imprint/”chicken foot” structure and the site of replication pause, prerequisite to imprinting and mating-type switching, are separated on a 2D gel (Figs. 3A, 4). We also map the 3′-end of the nascent leading strand in the paused replication complex 354–369 nucleotides upstream of the site of the imprint, using high-resolution Southern blot, and the 5′-end of the nascent lagging strand 342–345 nucleotides upstream of the site of the imprint, using primer extension (Fig. 7A). This means that the leading-strand replication complex has already passed the region where the imprint is to be introduced (Fig. 7B, Step 1). Primer extension on Okazaki fragments shows that wild type, swi1, swi3, and swi7 display a similar amount of fragments, the DNA part of which starts 342–345 nucleotides from the imprint. However, when primer extension is performed on purified paused intermediates in the wild type, the signal is greatly increased. These data suggest that pausing of lagging-strand replication occurs after polymerase α has initiated the synthesis of the last Okazaki fragment, and that RNase H is able to remove the RNA primers at the stalled fork (Fig. 7B, Step 2).
We do not know what the signal for the imprinting event is. Because the 5′-end of the lagging-strand template becomes exposed earlier than the 3′-end, it would remain single-stranded for a longer period of time. There is evidence from experiments with trinucleotide repeat tracts in S. cerevisiae that this allows formation of the secondary structure in the DNA, thus affecting lagging-but not leading-strand synthesis (Maurer et al. 1998). At the mat1 locus of S. pombe, the sequence around the imprint has symmetric properties (Nielsen and Egel 1989) and the homology domain H1 (Fig. 1A) is important for replication pausing as well as for the formation of the imprint (Styrkarsdottir et al. 1993; Kaykov et al. 2004). Alternatively, a _trans_-acting protein might bind the exposed single-stranded DNA at the implicated H1 sequence. Therefore, the lagging-strand replication might encounter difficulties during elongation through this region, either due to the secondary DNA structure, or due to involvement of a _trans_-acting factor. Such barriers can constitute the signal that leads to imprinting (Fig. 7B, Step 2).
Last, we show that the mat1 imprint is sensitive to RNases, and we estimate that it consists of one or two ribonucleotides. The assay used here could not detect an RNA primer at the position of the mat1 imprint, which suggests that the imprint is not a part of a primer that is normally synthesized to initiate a new Okazaki fragment. We see at least three possible mechanisms for imprint introduction (Fig. 7B, Step 3). (1) It could be that mistakes happen during lagging-strand synthesis, when the lagging-strand replication complex attempts to escape from the stall site. In S. cerevisiae trinucleotide repeat tracts, this leads to tract expansion (Pelletier et al. 2003). Ribonucleotide(s) at mat1 can be introduced when the lagging-strand replication is overcoming the barrier present in the template DNA. (2) The studies using the SV40 DNA replication system showed that synthesis of the first ∼30 nucleotides of an Okazaki fragment by polymerase α is followed by polymerase switch, and the rest of the fragment is completed by polymerase δ (Waga and Stillman 1998). Because of the signal during lagging-strand progression through the site of the imprint, a reverse change of polymerases may take place, and the ribonucleotides are incorporated in a swi7/polα-dependent manner. (3) Alternatively, such ribonucleotides could be formed postreplicatively by an oxidation of the DNA backbone (Fig. 7C). The difference between ribose and deoxyribose is one oxygen atom at the 2′ position. The enzyme ribonucleotide-reductase converts ribonucleotides into deoxyribonucleotides using NADPH as a source of energy. We imagine that the reverse process can be catalyzed by an oxidating enzyme that has a structure to bind the DNA duplex and access the 2′ position of the deoxyribose, and the signal from the lagging-strand synthesis might designate the site for the modification.
The presence of ribonucleotides in the newly replicated DNA was previously shown by RNase sensitivity assays in mitochondrial DNA, where ribonucleotides are present in both nascent strands, but are much more abundant in the newly synthesized lagging strand (Yang et al. 2002). The data presented here is the first evidence of a site-specific RNase-sensitive modification in the nuclear eukaryotic DNA. Such imprints could act as a mark for other cellular processes. Indeed, the imprinting function of swi1, swi3, and swi7 is important for alkylation damage repair in S. pombe (E. Sommariva, T. Pellny, S. Vengrova, T. Eydmann and J. Dalgaard, in prep.). There, such imprints are proposed to mark the sites where the replication forks were stalled by damage to the template DNA. swi1p, swi3p, and swi7p proteins, involved in imprinting in S. pombe, are conserved from yeast to metazoans, suggesting that similar mechanisms may play a role in other organisms.
Materials and methods
S. pombe strains
Strains were constructed as described elsewhere (Moreno et al. 1991). The genotypes of the strains used are as follows: JZ105, h90 mat1M Δ_mat 2,3_::LEU2, ade6-210, leu1-32, ura4-D18, his2; JZ108, mat1M smt-0 Δ_mat 2,3_::LEU2, ade6-210, leu1-32, his2; JZ217, h90 mat1 Δ_1.5 Kb distal_::LEU2, ade6-210, leu1-32, ura4-D18, swi6 mod+; JZ477, h90 mat1 Δ_1.5 Kb distal_::LEU2, ade6-210, leu1-32, ura4-D18, swi6 mod+, rqh1_Δ::ura4; SV1, h90 mat1M Δ_mat 2,3::LEU2, ade6-216, his2, swi3; SV5, h90 mat1M Δ_mat 2,3_::LEU2, ade6-210, leu1-32, ura4-D18, his2, swi1_Δ::ura4; SP469, h90 mat1M Δ_mat 2,3::LEU2, ade6-216, leu1-32, his2, swi7 (A. Klar, National Cancer Institute, Frederick, MD); SP835, h90 mat1P Δ_mat2,3_::LEU2 ade6-216 leu1-32 ura4-D18 his2 (A. Klar); SP976, h90 ade6-210, leu1-32, ura4-D18, swi6 mod+ (A. Klar).
DNA preparation
Yeast chromosomal DNA was purified form logarithmically growing culture as described by Huberman et al. (1987). Replication intermediates were separated from nonreplicating DNA by BND-cellulose chromatography (Kiger and Sinsheimer 1969). 2D gels were performed as described by Brewer and Fangman (1987), except that the first- and second-dimension gels contained 0.5% and 1.2% agarose, respectively.
High-resolution Southern blot
Replication intermediates were digested with SspI or BstNI/SfaNI and enriched using BND-cellulose. The DNA was precipitated overnight with isopropanol, redissolved in loading buffer, and loaded on a 6% denaturing polyacrylamide gel. Sequencing reactions, made using primers _mat1M_-SspI (GCACTCCCTAC CATAATATACTCACTAAT) or _mat1M_-SfaNI (CTAAAACAG TATGGCATTAC) and a bacterial plasmid containing the EcoRI fragment of mat1M (pBZ85), were run in parallel as a marker. The DNA was electroblotted on a GeneScreen membrane (NEN). For Figure 2A, the membrane was probed with a strand-specific RNA probe (Nielsen and Egel 1989). For Figure 6C, the probe was synthesized by linear primer extension, using primer _mat1M_-BstNI (TGGTTATAAGTACCAGCCTAG), on a 268-bp BstNI/SfaNI fragment (Ruven et al. 1994).
Primer extension
NaOH treatment of nonreplicating DNA was performed as follows: DNA was incubated in 40 mM NaOH, 2 mM EDTA at 25°C for 2 h, neutralized with equimolar HCl, and precipitated before primer extension. RNase treatment of nonreplicating DNA was performed by incubating heat-denatured DNA with 10 units of RNase T2 or 1000 units of RNase T1 (both from Life Technologies) for 30 min to 2 h at 37°C, followed by phenol/chloroform extraction and precipitation before primer extension. For RNase H (Life Technologies) digest, 4 units of the enzyme were used and the DNA was not denatured. Nascent DNA was purified using λ-exonuclease as for RIP mapping (Gerbi and Bielinsky 1997). For Figure 6B, the replication intermediates were digested with NdeI and separated as for 2D gels on a first-dimension gel. The region from 3.5 to 6 kb was excised, and the DNA was purified using a Qiagen gel-purification kit. Primer extension was performed using primers priSV1 (ATC TCGTTAGAGGGAAGGGGAAGGT) and priSV3 (AATATT AGTGAGTATATTATGGT) and VentR(exo–) DNA polymerase (New England Biolabs). Sequencing reactions using corresponding primers and a bacterial pBZ85 plasmid were run in parallel as a marker. Synthetic templates were obtained from Helena Biosciences. Primer priSV2 and template 1 were end-labeled with γ32P-ATP (Amersham Pharmacia) and T4 polynucleotide kinase (New England Biolabs). Extension reactions were performed as for RIP mapping.
Acknowledgments
We thank our colleagues at MCRI for helpful suggestions and discussions. Special thanks to Dr. Robert Cross for suggestions and help with the manuscript. This research was sponsored by the Association of International Cancer Research (J.Z.D.) and the Marie Curie Cancer Care.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
Supplemental material is available at http://www.genesdev.org.
Article published ahead of print. Article and publication date are at http://www.genesdev.org/cgi/doi/10.1101/gad.289404.
Corresponding author.
References
- Arcangioli B. 1998. A siteand strand-specific DNA break confers asymmetric switching potential in fission yeast. EMBO J. 17**:** 4503-4510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arcangioli B. and de Lahondes, R. 2000. Fission yeast switches mating type by a replication-recombination coupled process. EMBO J. 19**:** 1389-1396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arcangioli B. and Klar, A.J. 1991. A novel switch-activating site (SAS1) and its cognate binding factor (SAP1) required for efficient mat1 switching in Schizosaccharomyces pombe. EMBO J. 10**:** 3025-3032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beach D.H. 1983. Cell type switching by DNA transposition in fission yeast. Nature 305**:** 682-687. [Google Scholar]
- Beach D.H. and Klar, A.J. 1984. Rearrangements of the transposable mating-type cassettes of fission yeast. EMBO J. 3**:** 603-610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brewer B.J. and Fangman, W.L. 1987. The localization of replication origins on ARS plasmids in S. cerevisiae. Cell 51**:** 463-471. [DOI] [PubMed] [Google Scholar]
- Courcelle J., Donaldson, J.R., Chow, K.H., and Courcelle, C.T. 2003. DNA damage-induced replication fork regression and processing in Escherichia coli. Science 299**:** 1064-1067. [DOI] [PubMed] [Google Scholar]
- Dalgaard J.Z. and Klar, A.J. 1999. Orientation of DNA replication establishes mating-type switching pattern in S. pombe. Nature 400**:** 181-184. [DOI] [PubMed] [Google Scholar]
- ____. 2000. swi1 and swi3 perform imprinting, pausing, and termination of DNA replication in S. pombe. Cell 102**:** 745-751. [DOI] [PubMed] [Google Scholar]
- ____. 2001a. A DNA replication-arrest site RTS1 regulates imprinting by determining the direction of replication at mat1 in S. pombe. Genes & Dev. 15**:** 2060-2068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ____. 2001b. Does S. pombe exploit the intrinsic asymmetry of DNA synthesis to imprint daughter cells for mating-type switching? Trends Genet. 17**:** 153-157. [DOI] [PubMed] [Google Scholar]
- Di Noia J. and Neuberger, M.S. 2002. Altering the pathway of immunoglobulin hypermutation by inhibiting uracil-DNA glycosylase. Nature 419**:** 43-48. [DOI] [PubMed] [Google Scholar]
- Doe C.L., Dixon, J., Osman, F., and Whitby, M.C. 2000. Partial suppression of the fission yeast rqh1(-) phenotype by expression of a bacterial Holliday junction resolvase. EMBO J. 19**:** 2751-2762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doe C.L., Ahn, J.S., Dixon, J., and Whitby, M.C. 2002. Mus81-Eme1 and Rqh1 involvement in processing stalled and collapsed replication forks. J. Biol. Chem. 277**:** 32753-32759. [DOI] [PubMed] [Google Scholar]
- Egel R. and Eie, B. 1987. Cell lineage asymmetry for Schizosaccharomyces pombe: Unilateral transmission of a high-frequency state of mating-type switching in diploid pedigrees. Curr. Genet. 12**:** 429-433. [Google Scholar]
- Egel R., Beach, D.H., and Klar, A.J. 1984. Genes required for initiation and resolution steps of mating-type switching in fission yeast. Proc. Natl. Acad. Sci. 81**:** 3481-3485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foss E.J. 2001. Tof1p regulates DNA damage responses during S phase in Saccharomyces cerevisiae. Genetics 157**:** 567-577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gellert M. 2002. V(D)J recombination: RAG proteins, repair factors, and regulation. Annu. Rev. Biochem. 71**:** 101-132. [DOI] [PubMed] [Google Scholar]
- Gerbi S.A. and Bielinsky, A.K. 1997. Replication initiation point mapping. Methods 13**:** 271-280. [DOI] [PubMed] [Google Scholar]
- Gruber M., Wellinger, R.E., and Sogo, J.M. 2000. Architecture of the replication fork stalled at the 3′ end of yeast ribosomal genes. Mol. Cell. Biol. 20**:** 5777-5787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haber J.E. 1998. Mating-type gene switching in Saccharomyces cerevisiae. Annu. Rev. Genet. 32**:** 561-599. [DOI] [PubMed] [Google Scholar]
- Huberman J.A., Spotila, L.D., Nawotka, K.A., el-Assouli, S.M., and Davis, L.R. 1987. The in vivo replication origin of the yeast 2 microns plasmid. Cell 51**:** 473-481. [DOI] [PubMed] [Google Scholar]
- Kaykov A., Holmes, A.M., and Arcangioli, B. 2004. Formation, maintenance and consequences of the imprint at the mating-type locus in fission yeast. EMBO J 23**:** 930-938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelly M., Burke, J., Smith, M., Klar, A., and Beach, D. 1988. Four mating-type genes control sexual differentiation in the fission yeast. EMBO J. 7**:** 1537-1547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiger J.A. and Sinsheimer, R.L. 1969. Vegetative lambda DNA. IV. Fractionation of replicating lambda DNA on benzoylated-naphthoylated DEAE cellulose. J. Mol. Biol. 40**:** 467-490. [DOI] [PubMed] [Google Scholar]
- Klar A.J. 1987. Differentiated parental DNA strands confer developmental asymmetry on daughter cells in fission yeast. Nature 326**:** 466-470. [DOI] [PubMed] [Google Scholar]
- Klar A.J. and Bonaduce, M.J. 1993. The mechanism of fission yeast mating-type interconversion: Evidence for two types of epigenetically inherited chromosomal imprinted events. Cold Spring Harb. Symp. Quant. Biol. 58**:** 457-465. [DOI] [PubMed] [Google Scholar]
- Klar A.J. and Miglio, L.M. 1986. Initiation of meiotic recombination by double-strand DNA breaks in S. pombe. Cell 46**:** 725-731. [DOI] [PubMed] [Google Scholar]
- Kostriken R., Strathern, J.N., Klar, A.J., Hicks, J.B., and Heffron, F. 1983. A site-specific endonuclease essential for mating-type switching in Saccharomyces cerevisiae. Cell 35**:** 167-174. [DOI] [PubMed] [Google Scholar]
- Lakin-Thomas P.L. 2000. Circadian rhythms: New functions for old clock genes. Trends Genet. 16**:** 135-142. [DOI] [PubMed] [Google Scholar]
- Maurer D.J., O'Callaghan, B.L., and Livingston, D.M. 1998. Mapping the polarity of changes that occur in interrupted CAG repeat tracts in yeast. Mol. Cell. Biol. 18**:** 4597-4604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McBlane J.F., van Gent, D.C., Ramsden, D.A., Romeo, C., Cuomo, C.A., Gellert, M., and Oettinger, M.A. 1995. Cleavage at a V(D)J recombination signal requires only RAG1 and RAG2 proteins and occurs in two steps. Cell 83**:** 387-395. [DOI] [PubMed] [Google Scholar]
- Michel B., Flores, M.J., Viguera, E., Grompone, G., Seigneur, M., and Bidnenko, V. 2001. Rescue of arrested replication forks by homologous recombination. Proc. Natl. Acad. Sci. 98**:** 8181-8188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyata H. and Miyata, M. 1981. Mode of conjugation in homothallic cells of Schizosaccharomyces pombe. J. Gen. Appl. Microbiol. 27**:** 365-371. [Google Scholar]
- Moreno S., Klar, A., and Nurse, P. 1991. Molecular genetic analysis of fission yeast Schizosaccharomyces pombe. Methods Enzymol. 194**:** 795-823. [DOI] [PubMed] [Google Scholar]
- Murray J.M., Lindsay, H.D., Munday, C.A., and Carr, A.M. 1997. Role of Schizosaccharomyces pombe RecQ homolog, recombination, and checkpoint genes in UV damage tolerance. Mol. Cell. Biol. 17**:** 6868-6875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nielsen O. and Egel, R. 1989. Mapping the double-strand breaks at the mating-type locus in fission yeast by genomic sequencing. EMBO J. 8**:** 269-276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noguchi E., Noguchi, C., Du, L.L., and Russell, P. 2003. Swi1 prevents replication fork collapse and controls checkpoint kinase Cds1. Mol. Cell. Biol. 23**:** 7861-7874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park H. and Sternglanz, R. 1999. Identification and characterization of the genes for two topoisomerase I-interacting proteins from Saccharomyces cerevisiae. Yeast 15**:** 35-41. [DOI] [PubMed] [Google Scholar]
- Pelletier R., Krasilnikova, M.M., Samadashwily, G.M., Lahue, R., and Mirkin, S.M. 2003. Replication and expansion of trinucleotide repeats in yeast. Mol. Cell. Biol. 23**:** 1349-1357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rada C., Williams, G.T., Nilsen, H., Barnes, D.E., Lindahl, T., and Neuberger, M.S. 2002. Immunoglobulin isotype switching is inhibited and somatic hypermutation perturbed in UNG-deficient mice. Curr. Biol. 12**:** 1748-1755. [DOI] [PubMed] [Google Scholar]
- Ruven H.J., Seelen, C.M., Lohman, P.H., Mullenders, L.H., and van Zeeland, A.A. 1994. Efficient synthesis of 32P-labeled single-stranded DNA probes using linear PCR; application of the method for analysis of strand-specific DNA repair. Mutat. Res. 315**:** 189-195. [DOI] [PubMed] [Google Scholar]
- Singh J. and Klar, A.J. 1993. DNA polymerase-alpha is essential for mating-type switching in fission yeast. Nature 361**:** 271-273. [DOI] [PubMed] [Google Scholar]
- Sogo J.M., Lopes, M., and Foiani, M. 2002. Fork reversal and ssDNA accumulation at stalled replication forks owing to checkpoint defects. Science 297**:** 599-602. [DOI] [PubMed] [Google Scholar]
- Styrkarsdottir U., Egel, R., and Nielsen, O. 1993. The smt-0 mutation which abolishes mating-type switching in fission yeast is a deletion. Curr. Genet. 23**:** 184-186. [DOI] [PubMed] [Google Scholar]
- Waga S. and Stillman, B. 1998. The DNA replication fork in eukaryotic cells. Annu. Rev. Biochem. 67**:** 721-751. [DOI] [PubMed] [Google Scholar]
- Yang M.Y., Bowmaker, M., Reyes, A., Vergani, L., Angeli, P., Gringeri, E., Jacobs, H.T., and Holt, I.J. 2002. Biased incorporation of ribonucleotides on the mitochondrial L-strand accounts for apparent strand-asymmetric DNA replication. Cell 111**:** 495-505. [DOI] [PubMed] [Google Scholar]