Mechanistic Independence of Nef and Cyclophilin A Enhancement of Human Immunodeficiency Virus Type 1 Infectivity (original) (raw)
. Author manuscript; available in PMC: 2014 Feb 27.
Published in final edited form as: Virology. 1998 Aug 15;248(1):139–147. doi: 10.1006/viro.1998.9254
Abstract
Optimal HIV-1 infectivity requires the presence of both the viral factor Nef and the cellular protein cyclophilin A (CyPA) during virion assembly. These two proteins are integral components of HIV-1 particles. Both CyPA and Nef facilitate a step in the viral life cycle occurring between penetration and reverse transcription, suggesting a common mechanism of action. Experiments were performed to test the potential interplay of Nef- and CyPA-mediated enhancement of HIV-1 infectivity. In single-cycle infection assays, _nef_-defective virions were partially resistant to cyclosporin A (CsA), a drug which inhibits the binding of CyPA to the HIV-1 Gag precursor and CyPA incorporation into virions. Genetic dissection of the relative contributions of Nef and the cyclophilin A-Gag interaction to HIV-1 infectivity demonstrated the independence of these two effects. Nef was not required for incorporation of CyPA into HIV-1 virions, and vice-versa. Surprisingly, CyPA-deficient virions remained sensitive to inhibition by CsA, in a manner that depended strongly on the presence of a functional nef gene. These results demonstrate that Nef and CyPA act independently to render HIV-1 particles fully infectious. They further suggest that in addition to blocking the CyPA-Gag interaction, CsA can also inhibit HIV-1 replication through a novel mechanism involving suppression of Nef-directed enhancement of virus infectivity.
Introduction
HIV-1 Nef is required for efficient viral replication in primary CD4+ T cells and macrophages, as well as in many T cell lines (de Ronde et al., 1992; Miller et al., 1994; Spina et al., 1994; Zazopoulos and Haseltine, 1993). Several groups have previously shown that _nef_-defective virions are approximately five- to ten-fold less infectious than wild-type HIV-1 when tested in single-cycle infection assays using CD4-expressing Hela cells or the CEM human T cell line as targets (Aiken and Trono, 1995; Chowers et al., 1994; Miller et al., 1994; Schwartz et al., 1995). Although it is unclear what precise step in the HIV-1 life cycle Nef facilitates, two lines of evidence implicate an early post-entry event. First, measurements of virus internalization by cells reveal no differences between wild-type and _nef_-defective HIV-1. Second, pseudotyping by the envelope glycoproteins of the amphotropic murine leukemia virus (MuLV) fails to complement the Nef defect, suggesting that Nef does not facilitate CD4-dependent entry (Aiken and Trono, 1995; Miller et al., 1995). This conclusion is further supported by the observation that Nef enhances the infectivity of HIV-1(MuLV) pseudotypes irrespective of the presence or absence of CD4 on the target cells (Aiken and Trono, 1995). Although Nef does not appear to enhance HIV-1 entry, _nef_-defective HIV-1 undergoes inefficient reverse transcription in target cells, even though the virions themselves exhibit normal levels of reverse transcriptase activity (Aiken and Trono, 1995; Chowers et al., 1995; Schwartz et al., 1995). _Nef_-defective virions are impaired for synthesis of even the earliest products of reverse transcription in target cells (Aiken and Trono, 1995). It is therefore plausible that Nef facilitates a step of the HIV-1 life cycle between entry and initiation of reverse transcription—namely, virus uncoating. However, little is known regarding the molecular events associated with retroviral uncoating. The observation that Nef is incorporated into HIV-1 particles and is specifically cleaved by the viral protease (Pandori et al., 1996; Welker et al., 1996) suggests that Nef may enhance HIV-1 infection by acting directly in virions.
In addition to Nef, much attention has been focused on a human cellular protein that is also required for efficient HIV-1 infection. Cyclophilin A (CyPA), a peptidyl proline isomerase, is the target of the immunosuppressive drug cyclosporin A (CsA). CsA inhibits HIV-l replication in primary T cells and established T cell lines (Franke et al., 1994; Karpas et al., 1992; Rosenwirth et al., 1994; Thali et al., 1994). This activity is independent of the inhibitory effects of CsA on T cell activation, however, since nonimmunosuppressive analogs of CsA also block HIV-l replication (Billich et al., 1995; Rosenwirth et al., 1994). CyPA binds to the capsid region of the HIV-1 Gag precursor (Luban et al., 1993), and is incorporated into HIV-1 particles through an interaction with a proline-rich region of capsid (Franke et al., 1994; Thali et al., 1994). Mutations in this region which block CyPA binding render HIV-l particles poorly infectious. Importantly, the mechanisms by which Nef and CyPA enhance HIV-l infectivity appear intriguingly similar. Both Nef and CyPA are required at the time of HIV-1 particle formation to render HIV-1 virions fully infectious, and both proteins are components of HIV-l particles. The requirement for CyPA, like Nef, is not relieved by pseudotyping with the amphotropic MuLV envelope glycoprotein, indicating that the requirement for Gag-CyPA binding is independent of CD4-mediated HIV-l entry. Furthermore, both CyPA-deficient and _nef_-defective HIV-1 particles contain wild-type levels of endogenous reverse transcriptase activity but undergo inefficient reverse transcription in target cells. Recent observations from this laboratory have also demonstrated that pseudotyping HIV-1 particles by the envelope glycoprotein of vesicular stomatitis virus (VSV-G) suppressed the requirement for Nef (Aiken, 1997). Infection by HIV-1(VSV) pseudotypes was blocked by preventing endosomal acidification, indicating an endocytic route of entry. Inhibition of HIV-1 infectivity by CsA was also relieved in these pseudotypes; however, pseudotyping by VSV-G had no effect on the phenotype of HIV-l gag mutants defective for CyPA binding. These results suggested a possible mechanistic overlap between Nef-mediated HIV-1 infectivity enhancement and inhibition of HIV-1 infectivity by CsA.
Due to the similarities between Nef- and CyPA-mediated HIV-1 infectivity enhancement, experiments were designed to test whether Nef and CyPA employ a common mechanism to enhance HIV-1 infectivity. Viruses containing mutations in gag inhibiting binding of CyPA, in nef, or in both genes were produced by transfection of 293T cells in order to circumvent problems associated with production of replication-defective virions. Quantitative single-cycle infection assays were performed to determine the relative infectivity of mutant virions. Using this assay, the inhibitory effects of CsA on the infectivity of the mutants were also quantified. The results demonstrate that Nef and the CyPA-Gag interaction are independently required to enhance HIV-1 infectivity, and suggest that Nef-directed HIV-1 infectivity enhancement represents an additional viral function that is inhibited by CsA.
Results
_Nef_-defective HIV-1 is partially resistant to cyclosporin A
Treatment of cells producing HIV-1 with CsA blocks incorporation of CyPA into progeny virions and reduces viral infectivity (Franke et al., 1994; Thali et al., 1994). To dissect the effects of Nef and CyPA on HIV-1 infectivity, CsA was used to generate CyPA-deficient viruses. Wild-type and _nef_-defective HIV-1 stocks were produced by transfecting 293T cells with HIV-1 molecular clones and culturing in the presence of various concentrations of CsA. The resulting HIV-1-containing supernatants were assayed for reverse transcriptase activity and titrated by infecting the P4 indicator cell line. P4 cells express CD4, which renders them susceptible to HIV-1 infection, and contain an integrated E. coli lacZ gene driven by the HIV-1 LTR. Upon infection with HIV-1, Tat production from the integrated provirus leads to activation of the lacZ reporter, resulting in synthesis of b-galactosidase in these cells. Infected cells are identified by staining with X-gal 48 hours post-inoculation, allowing quantitation after a single round of HIV-1 infection. Using this single-cycle infection assay, the relative infectivity of a virus stock was calculated as the number of infected cells per unit of reverse transcriptase activity.
CsA caused a dose-dependent inhibition of wild-type HIV-1 infectivity reaching a level of 75% at 10 µM drug (Fig. 1, panel B). Virions produced in this concentration of CsA contained undetectable levels of CyPA (Fig. 2). Higher levels of CsA (20 (µM) were toxic to 293T cells and decreased virus production without further inhibiting viral infectivity. _Nef_-defective virions were less infectious than wild-type HIV-1 at all concentrations of CsA tested (Fig. 1, panel A). However, HIV-1Δnef was partially resistant to CsA, which caused 40% inhibition at 10 µM (Fig. 1, panel B).
Fig. 1.
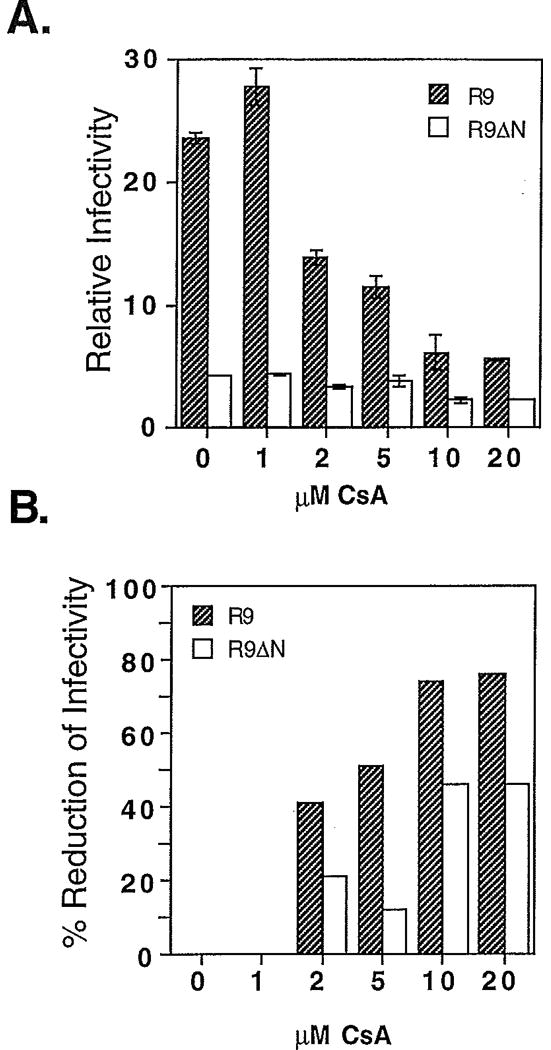
Effects of cyclosporin A on wild-type and _nef_-defective HIV-1 infectivity. Viruses were produced by culturing transfected 293T cells in the presence of the indicated concentrations of cyclosporin A. Culture supernatants were assayed for reverse transcriptase activity and their titers determined by infecting P4-2 indicator cells. Panel A: The ordinate values represent the number of infectious units per unit reverse transcriptase activity. R9: wild-type HIV-1; R9ΔN: _nef_-defective HIV-1. Shown are the mean values of triplicate infections, with error bars representing one standard deviation from the mean. The data are representative of two independent experiments. In Panel B, the data shown in Panel A are represented as the percentage by which viral infectivity is reduced by the indicated concentrations of CsA.
Fig. 2.
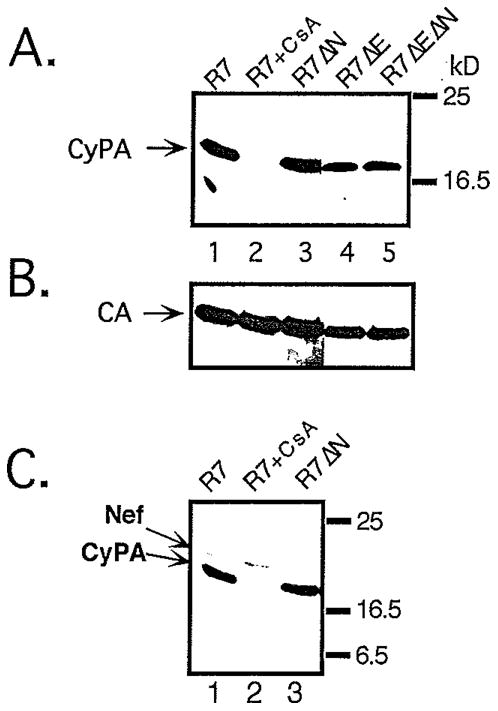
Levels of CyPA in wild-type and _nef_-defective HIV-1 particles. Virions produced by transfection of 293T cells were purified by centrifugation through 20% sucrose. The pellets were lysed and subjected to Western blot analysis using aliquots of cytoplasmic extracts normalized for p24 content by ELISA. Proteins were detected with (A) rabbit anti-cyclophilin A; (B) mouse anti-HIV-1 CA, and (C) rabbit anti-Nef; blots were developed using chemiluminiscent detection. Lane 1: wild-type HIV-1, lane 2: wild-type HIV-1 produced in the presence of 10 µM cyclosporin A; lane 3: _nef_-defective HIV-1, lane 4: _env_-defective HIV-1; lane 5: _env_-defective, _nef_-defective HIV-1.
Nef and CyPA are independently incorporated into HIV-1 particles
The decreased sensitivity of _nef_-defective HIV-1 to inhibition by CsA suggested that Nef and CyPA may act through a common pathway to enhance HIV-1 infectivity. To determine whether Nef influences virion incorporation of CyPA, purified wild-type and _nef_-defective HIV-1 particles were analyzed for their CyPA content by immunoblotting. Samples of viral lysates containing 100 ng of p24, as determined by ELISA, were subjected to electrophoresis on polyacrylamide gels containing SDS. After transfer of proteins to nitrocellulose, the relative levels of CyPA present in the particles were assessed by probing the blot with a rabbit antiserum specific for human CyPA. No significant differences in CyPA content were observed between wild-type and _nef_-defective virions (Fig. 2, panel A, lanes 1 and 3), or in their corresponding envelope-defective variants (lanes 4 and 5). As previously reported (Franke, Yuan, and Luban, 1994; Thali et al., 1994), wild-type HIV-1 virions produced in the presence of 10 µM CsA contained undetectable levels of CyPA (lane 2). To assess whether equal quantities of viral lysates were loaded, the blot was reprobed with a CA-specific monoclonal antibody (Fig. 2, panel B). As expected, the results confirmed that similar amounts of capsid protein were present in each lane.
In addition to CyPA, Nef is also a component of HIV-1 virions, and is cleaved in virions by the HIV-1 protease (Pandori et al., 1996; Welker et al., 1996). To ask whether CsA influences virion incorporation of Nef, the blot shown in Fig. 2 was reprobed with a rabbit antiserum specific for HIV-1 Nef. No significant reduction of proteolytically processed Nef was observed in virions produced in the presence of CsA (panel C, lanes 1 and 2). This result indicates that CsA does not modulate Nef function by influencing incorporation of Nef into HIV-1 particles or its subsequent cleavage by the viral protease.
Nef and the cyclophilin A-Gag interaction independently enhance HIV-1 infectivity
Previous reports have demonstrated that mutations of capsid residues glycine 221 or proline 222 to alanine inhibit CyPA incorporation into HIV-1 virions and reduce HIV-1 infectivity (Braaten et al., 1996; Franke et al., 1994). To genetically dissect the relative contributions of Nef and CyPA to HIV-1 infectivity, the G221A and P222A mutations were introduced into HIV-1 proviral DNA constructs carrying intact and disrupted nef genes. Viruses containing the G221A or P222A mutations were produced in parallel with wild-type and HIV-1Δnef by transfection of 293T cells, and the resulting virus supernatants assayed for reverse transcriptase activity and titers determined by infecting P4 cells. The infectivities of the G221A or P222A viruses were reduced to 18% and 33% of that of wild-type HIV-1 (Fig. 3). These values correlated well with the relative levels of CyPA present in the virions, as shown by immunoblot analysis of detergent lysates of purified virions (Fig. 4). Furthermore, the effects of gag mutations on HIV-1 infectivity were strictly independent of the presence of a functional nef gene. For example, the GagP222A mutation decreased HIV-1 infectivity by three-fold regardless of the nef genotype, and Nef enhanced HIV-1 infectivity by approximately nine-fold regardless of the presence of either gag mutation (Fig. 3). Thus, the effects of the Gag-CyPA interaction and Nef on HIV-1 infectivity are functionally distinct. This result contrasts with the results of previous experiments, in which HIV-1Δnef exhibited a decreased sensitivity to CsA (Fig. 1). This apparent contradiction suggests that inhibition of HIV-1 infectivity by CsA may be pleiotropic, perhaps involving a Nef-dependent component.
Fig. 3.
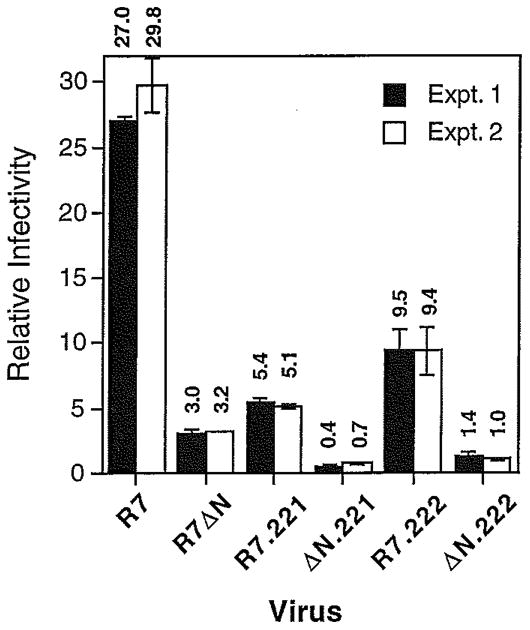
Nef and the CyPA-Gag interaction independently enhance HIV-1 infectivity. Viruses were produced by transfection of 293T cells and were assayed for infectivity using P4-2 cells as targets. Shown are the mean values of triplicate infections, with error bars representing one standard deviation. The paired solid and open bars are the results of independent virus stocks. R7:wild-type HIV-1; R7ΔN:_nef_-defective HIV-1. R7.221 and ΔN.221 contain a mutation of Gly221 of capsid to Ala; R7.222 and ΔN.222 contain a mutation of Pro222 to Ala.
Fig. 4.
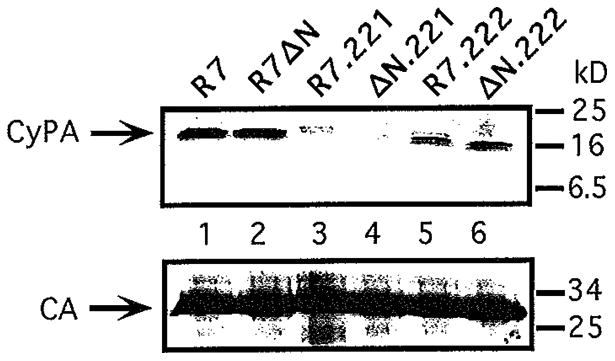
Effects of gag mutations of HIV-1 packaging of CyPA. Virions produced by transfection of 293T cells with proviral DNA constructs and were purified by ultracentrifugation through 20% sucrose. Viral pellets were dissolved in lysis buffer, assayed for p24 content by ELISA, and subjected to immunoblot analyses. The blots were probed sequentially with rabbit anti-CyPA (panel A) and mouse anti-CA (panel B), and protein bands were visualized using chemiluminescent detection.
Cyclosporin A sensitivity correlates with nef expression
To distinguish the effects of CsA on the Gag-CyPA interaction from possible inhibition of Nef-enhanced HIV-1 infectivity, wild-type and _nef_-defective HIV-1 particles containing mutations in gag were produced in the presence and absence of CsA and assayed for infectivity using P4 cells. Interestingly, mutant virions containing decreased CyPA levels were significantly inhibited by CsA (Fig. 5, panel A). Furthermore, CyPA-deficient HIV-1 particles containing a functional nef gene were inhibited by CsA to a degree similar to that of wild-type HIV-1. In contrast, _nef_-defective viruses were relatively resistant to the inhibitory effects of CsA irrespective of the ability of Gag to bind CyPA (Fig. 5, panel B). This result suggests that a significant component of the inhibitory effect of CsA on HIV-1 replication is due to inhibition of Nef-mediated infectivity enhancement.
Fig. 5.
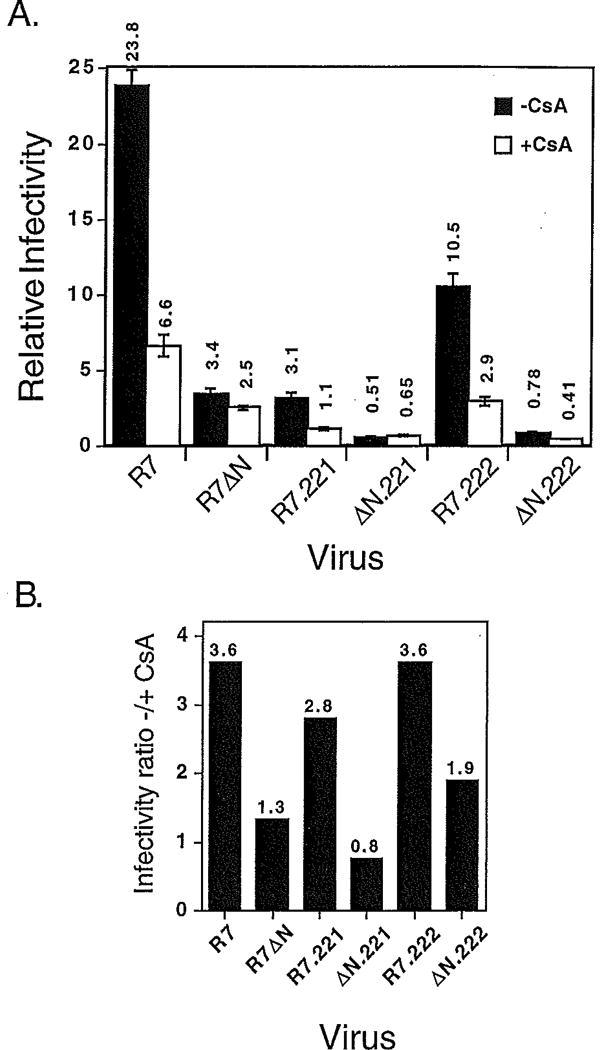
Inhibition of HIV-1 infectivity by CsA correlates with the expression of Nef. Viruses were produced by culturing transfected 293T cells for two days in the presence or absence of 10 µM CsA. Virus stocks were assayed for reverse transcriptase activity and their titers determined by infecting P4 cells. Panel A: shown are the mean values of triplicate infections, with error bars representing one standard deviation. The results are representative of two experiments. Shown in panel B are the ratios of the infectivity values obtained for viruses produced in the absence of CsA to those of viruses produced in the presence of CsA.
To determine whether these effects extended to HIV-1 replication, the growth kinetics of wild-type and _nef_-defective viruses containing the G221A and P222A mutations were monitored in CEM cells cultured in the presence and absence of CsA (Fig. 6). The growth of both gag mutant viruses was delayed relative to wild-type HIV-1 (Fig. 6, panels C and E), to an extent that correlated with the CyPA levels in the virions (Fig. 4). Similar effects of CsA were also observed for the _nef_-defective variants (Fig. 6, panels B, D, and E). In most of the CsA-containing cultures, positive reverse transcriptase activity was detected by 40 days, possibly due to the acquisition of drug-resistant mutations, as previously described (Aberham et al., 1996). Exceptions include the poorly-replicating viruses ΔN.222, R7.221, and ΔN.221. The replication kinetics of _nef_-defective viruses were delayed relative to their nef+ counterparts (Fig. 6, panels D and F), consistent with the results from single-cycle assays that the effects of Nef and CyPA on HIV-1 infectivity are independent (Fig. 5). However, in contrast to the previous result that _nef_-defective viruses are relatively resistant to CsA when assayed on P4 cells (Fig. 1), the growth of both nef+ and _nef_-defective variants of the gag mutants in CEM cells was markedly inhibited by CsA (Fig. 6, panels C-F).
Fig. 6.
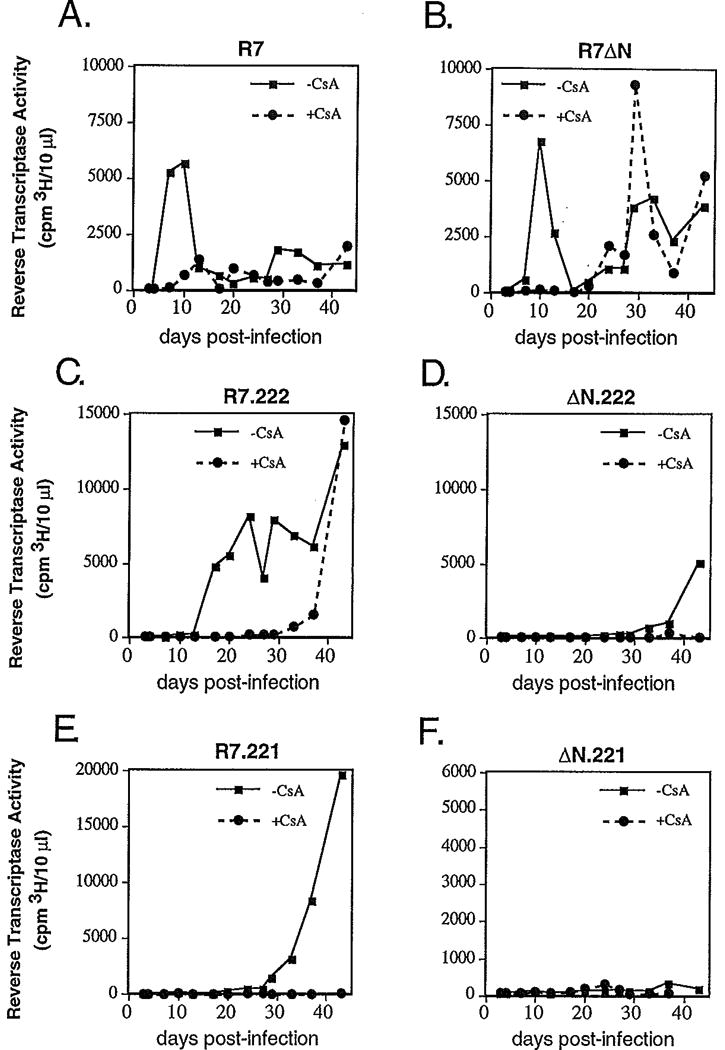
Cyclosporin A inhibits replication of mutant HIV-1 lacking CyPA. CEM cells were inoculated with aliquots of viruses containing 30,000 cpm reverse transcriptase activity. After overnight culturing, the cells were pelleted to remove input virus, and cultured in 2 ml media in the presence (dashed lines) and absence (solid lines) of 2.5 µM CsA. Virus production was monitored periodically by assaying for reverse transcriptase activity in the supernatants. Panel A: wild-type HIV-1; C: HIV-1 containing a mutation of Pro222 to Ala in capsid; E: HIV-1 containing a mutation of Gly221 to Ala in capsid. Replication of the _nef_-defective variants of R7, R7.222, and R7.221 are shown in panels B, D, and F, respectively.
Discussion
Results of experiments presented here demonstrate that Nef enhances HIV-1 infectivity independently of the CyPA-Gag interaction. The infectivity of HIV-1 virions containing reduced levels of CyPA by virtue of mutations in capsid was stimulated by Nef to the same degree as wild-type virus. This was shown by quantitative single-cycle infection assays using Hela-CD4 cells as targets, and confirmed by measuring viral replication in CEM human T cells. _nef_-defective HIV-1 contained normal levels of CyPA, and virions produced in the presence of CsA exhibited normal amounts of proteolytically cleaved Nef protein. Thus, virion incorporation of CyPA is not regulated by Nef, and vice-versa.
The observation that Nef and the CyPA-Gag interaction independently enhance HIV-1 infectivity has implications for the mechanisms by which these two proteins function. Both Nef and CyPA appear to act at a step of the virus infection pathway that follows entry into the target cell but precedes reverse transcription (Aiken and Trono, 1995; Braaten et al., 1996; Chowers et al., 1995; Schwartz et al., 1995; Steinkasserer et al., 1995). Genetic disruption of Nef, or of the CyPA-Gag interaction, results in the formation of normal amounts of virus particles that appear normal both structurally and biochemically (Aiken, unpublished observations; Braaten et al., 1996; Miller et al., 1995). Both proteins are significant components of virions (Franke et al., 1994; Pandori et al., 1996; Thali et al., 1994; Welker et al., 1996). Although the subviral localization of Nef is unknown, CyPA is probably associated with the viral core through its interaction with capsid. Thus, it was not difficult to imagine that both Nef and CyPA might utilize a common mechanism to facilitate virus uncoating—a step that follows entry but precedes reverse transcription. If this were the case, one might have expected some overlap between the effects of mutations abolishing nef expression and those preventing CyPA incorporation. Since the effects were strictly independent, it is likely that these two proteins enhance HIV-1 infectivity by separate mechanisms.
In the single-cycle infectious assays employed here, a four-fold reduction of wild-type HIV-1 infectivity was observed when the virus is produced in the presence of 10 µM CsA. This concentration was sufficient to virtually abolish CyPA incorporation into HIV-1 particles. The fact that CsA and mutations in gag only partially inhibited infection in this assay allowed dissection of the relative contributions of Nef and CyPA to viral infectivity. _Nef_-defective virions were relatively resistant to CsA, as their infectivity was inhibited only two-fold by 10 µM CsA. Other workers reported a 9-to 14-fold reduction of HIV-1 infectivity by CsA using a _nef_-defective HIV-1 carrying a chloramphenicol acetyl transferase (CAT) gene (Thali et al., 1994). Although the cause of the decreased effects of CsA in the present experiments is not clear, one possibility may be the different assay systems employed: the previous results were obtained using bulk assays of CAT activity in cell lysates, while the assay employed in the present study involved counting individual infected cells. In this system, some variation in the intensity of blue staining of infected cells is always evident, but very dark and very light stained cells are scored as single infectious units. If CsA-treatment results in a shift in the relative proportion in the number of light or dark blue cells, a greater or lesser effect would be observed by assaying enzyme activity in cell lysates. The signal from bulk assays of reporter activity may thus be amplified by differences in reporter expression arising from delays in the time of integration, as well as effects on post-integrational processes such as transcriptional activation or virus-induced cytotoxicity. In support of this, preliminary results from this laboratory suggest that the apparent effects of CsA are markedly enhanced by assaying bulk lysates of infected cells for soluble β-galactosidase activity (C. Aiken, unpublished observations). Another possible explanation is that the cell lines used to produce the viruses may differ in CyPA concentrations, resulting in modulation of the phenotype of viruses produced in the presence of CsA. Cell-specific effects related to intracellular CyPA concentrations have recently been reported for the replication of the HIV-1 GagP222A mutant (Ackerson et al., 1998).
HIV-1 particles produced in the presence of 10 µM CsA are devoid of CyPA, and it has generally been concluded that disruption of the CyPA-Gag interaction is the mechanism through which CsA suppresses HIV-1 replication. Evidence for this comes from the fact that mutations conferring resistance to cyclosporins map to the capsid region of Gag (Aberham et al., 1996; Braaten et al., 1996). In addition, replacement of the CA-p2 region of the simian immunodeficiency virus (SIV), a virus that is resistant to CsA, by the corresponding region from HIV-1 renders SIV sensitive to the drug (Dorfman and Gottlinger, 1996). These findings argue strongly for the CyPA-Gag interaction as the principal target for CsA inhibition of HIV-1 replication.
Evidence presented here suggests that in addition to preventing virion incorporation of CyPA, CsA may block HIV-1 replication by inhibiting an additional viral function. First, single-cycle infection assays revealed that _nef_-defective virions exhibited a two- to three-fold reduced sensitivity to CsA relative to wild-type HIV-1. Secondly, CsA also reduced the infectivity of HIV-1 mutants deficient in CyPA by a similar degree as that of the wild-type virus and delayed the replication of these mutant viruses in a human T cell line. Together, these results suggest that CsA may target another viral function besides the CyPA-Gag interaction. The observation that CsA sensitivity correlates with the presence of a functional nef gene strongly suggests that CsA can inhibit the ability of Nef to enhance HIV-1 infectivity. In support of this interpretation, recent results from this laboratory have demonstrated that pseudotyping HIV-1 by the glycoprotein of vesicular stomatitis virus suppresses both the requirement for Nef and the sensitivity to CsA but does not relieve the infectivity impairment resulting from a gag mutation that inhibits CyPA binding (Aiken, 1997). This finding further demonstrates a functional distinction between CyPA-Gag binding and inhibition of HIV-1 infectivity by CsA. Nevertheless, CsA effectively inhibited the growth of both wild-type and _nef_-defective HIV-1 in CEM cells. This apparent discrepancy may be due to the differing dynamics of virus replication in continuous culture versus those in single-cycle infection assays. The two- to three-fold higher sensitivity of nef+ HIV-1 to CsA may not be apparent in viral growth curves where the mode of transmission can switch from cell-free infection to a cell-to-cell mode after the first round. Thus, it is probable that the CyPA-Gag interaction is the major target for CsA inhibition of HIV-1 replication in T cells. Nevertheless, CsA inhibition of Nef-dependent HIV-1 infectivity enhancement represents an effect of the drug that may be relevant in some experimental systems, including those measuring virus infection during a single round.
How might CsA inhibit Nef function? CsA binds to CyPA, promoting the formation of a ternary complex with calcineurin (CN) and blocking the activity of this protein phosphatase essential for efficient T-cell activation. However, _nef_-defective HIV-1 was also resistant to analogs of CsA that are incapable of mediating complex formation with CN (C. Aiken, unpublished observations). This activity of CsA is therefore unlikely to mediate the apparent inhibition of Nef. Nef contains a highly conserved PGPG sequence element that forms part of a turn on the protein surface (Lee et al., 1996; Shugars et al., 1993) that is reminiscent of the CyPA-binding domain of capsid (Gamble et al., 1996). Binding of CyPA to Nef has been observed in vitro (Billich et al., 1995), and CyPA peptidylprolyl isomerase activity may be required for the proper folding or intracellular transport of Nef. CsA does not appear to affect Nef synthesis or stability, since the levels of Nef protein in transfected cells are not reduced by CsA (C. Aiken, unpublished observations). Furthermore, preliminary results indicate that CsA treatment of _nef_-expressing T cells does not significantly inhibit another activity of Nef, CD4 downregulation. CD4 downregulation and HIV-1 infectivity enhancement are genetically distinct activities of the viral protein (Goldsmith et al., 1995; Saksela, Cheng, and Baltimore, 1995), and CyPA binding to Nef may be required for one activity and not the other. Alternatively, CsA treatment of cells could interfere with a downstream effector molecule, such as a Nef-associated protein kinase, through which Nef may mediate HIV-1 infectivity enhancement (Saksela, Cheng, and Baltimore, 1995; Sawai et al., 1994). Resolution of these issues will require a more detailed understanding of the biochemical mechanism by which Nef enhances HIV-1 infectivity.
Materials and Methods
Cells and viruses
293T cells, and the Hela-CD4/LTR-lacZ indicator cell line P4 (Charneau et al., 1992) were cultured in Dulbecco's modified Eagle's medium (DMEM, Cellgro) supplemented with 10% fetal bovine serum (FBS, Atlanta Biologicals) and penicillin plus streptomycin (Cellgro). The human T cell lines CEM, SupT1, and Jurkat were cultured in RPMI 1640 containing 10% FBS and penicillin plus streptomycin. The viruses used in this study were derived from R7, a full-length clone of HIV-1 HXB2 containing a complete nef open-reading frame (Kim et al., 1989). R7ΔN contains a frameshift mutation at the _Xho_I site in nef, and has been characterized previously (Aiken et al., 1994). Capsid mutations were created by oligonucleotide-directed mutagenesis using the polymerase chain reaction to amplify a _Bss_HII-to-_Spe_I fragment. PCR-amplified regions were subjected to DNA sequencing using Sequenase (Amersham), and cloned into R7. R9 and R9ΔN are full-length plasmid clones of HIV-1 that were created by replacing a _Bss_HII -to- _Bam_HI fragments of R7 or R7ΔN, respectively, with the corresponding fragment from pNL4-3 (Adachi et al., 1986), as previously described (Gallay et al., 1995). In these viruses, the gag, pol, and gpl20 region of env are derived from NL4-3, while the LTRs and nef are from HXB2. These viruses therefore encode functional vpu and vpr genes. HIV-1 stocks were produced by transfection of 293T cells using the calcium phosphate-BBS method (Chen and Okayama, 1987). After overnight incubation with the precipitates in an incubator set at 35°C and 3% CO2, the cells were washed once with phosphate buffered saline (PBS) and the medium replaced. In experiments involving cyclosporin A (Sigma) the drug was added as a 1 mM stock solution in absolute ethanol to achieve the desired final concentrations in culture media (1-20 µM). Control viruses were produced in the presence of equivalent concentrations of ethanol. Two days after transfection, virus-containing supernatants were harvested, passed through 0.45 µm cellulose acetate syringe filters, and assayed for reverse transcriptase activity and virus infectivity prior to storage at −75°C.
Infections
For single-cycle assays of HIV-1 infectivity, P4 cells (4 × 104) were plated in 12-mm wells and infected in triplicate one day later with 0.25 ml of virus dilutions containing DEAE-dextran (20 µg/ml). Two hours later, complete medium (2 ml) was added. Cells were subsequently cultured for two days, and were fixed and stained with 5-bromo-4-chloro-3-indolyl-galactopyranoside (X-gal) as described (Kimpton and Emerman, 1992). To determine the number of infectious units per ml of a virus stock, wells containing betweeen 20 and 500 blue cells were counted under the light microscope, and the mean values calculated and multiplied by the virus dilution. Values obtained for serially-diluted virus samples behaved linearly within this range. Uninfected wells consistently exhibited fewer than 5 cells per well. The infectivity of a given virus was calculated as the number of infectious units per ml virus stock divided by the cpm of reverse transcriptase activity contained in 10 µl supernatant.
Infections of T cell lines were performed by inoculating cells (5 × 105) in 1-ml volumes with amounts of virus stocks normalized for reverse transcriptase activity. After overnight incubation, the cells were pelleted to remove the virus inocula and resuspended in 2 ml fresh medium containing 2.5 µM cyclosporin A (Sigma). Supernatants were sampled every three days for virus production, and the cultures were split 1:5 or 1:10 as necessary into fresh medium with or without cyclosporin A.
Reverse transcriptase assays were performed in duplicate as previously described (Aiken and Trono, 1995). Values obtained were all within the linear range of the assay, and varied between 2000 and 80,000 cpm 3H, with background values between 100 and 200 cpm. The values of reverse transcriptase activity measured using this assay correlated well with p24 concentrations of the virus stocks determined by ELISA.
Protein Analyses
HIV-1 virions were pelleted by ultracentrifugation of virus supernatants (8 ml) through a 2 ml layer of 20% sucrose in PBS for 2 hours at 100,000 x_g_ in a Beckman SW41 rotor. Pellets were resuspended in 1 ml PBS and recentrifuged at 100,000 x_g_ for 30 min in a Sorvall RPA45 rotor. The resulting pellet was resuspended in lysis buffer (20 mM Tris-HCl pH 7.5, 100 mM NaCl, 0.5% NP40) and assayed for p24 by ELISA (Biotech Research Laboratories). Immunoblotting of viral proteins was performed by subjecting samples of viral lysates containing 100 ng p24 to electrophoresis on 4-20% gradient acrylamide gels containing SDS. Proteins were transferred to nitrocellulose, and blocked overnight in TBST (10 mM Tris-HCl pH 8.0, 150 mM NaCl, 0.05% Tween 20) containing 5% nonfat dry milk. Blots were probed with rabbit anti-Nef (Aiken and Trono, 1995), rabbit anti-CyPA (the generous gift of Louis Henderson), and mouse anti-CA (obtained from the NIH AIDS Research and Reference Reagent Program). Protein bands were revealed using chemiluminescent detection (ECL, Amersham Corp.).
Acknowledgments
I thank C. Hicks for technical assistance, F. Clavel for P4 cells, L. Henderson for antiserum to cyclophilin A, P. Spearman for the use of his BL-3 laboratory, and D. Ballard, T. Dermody, P. Green, J. Hawiger, and E. Ruley for helpful comments. Antiserum to HIV-1 p25/24 Gag from Dr. Kathelyn Steimer, Chiron Corporation, was obtained through the AIDS Research and Reference Reagent Program, Division of AIDS Program, NIAID, NIH.
This work was supported by NIH grant AI40364. I was supported in part by an AmFAR/Genentech, Inc. Scholar Award.
References
- Aberham C, Weber S, Phares W. Spontaneous mutations in the human immunodeficiency virus type 1 gag gene that affect viral replication in the presence of cyclosporins. J Virol. 1996;70:3536–3544. doi: 10.1128/jvi.70.6.3536-3544.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ackerson B, Rey O, Canon J, Krogstad P. Cells with high cyclophilin A content support replication of human immunodeficiency virus type 1 Gag mutants with decreased ability to incorporate cyclophilin A. J Virol. 1998;72:303–308. doi: 10.1128/jvi.72.1.303-308.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adachi A, Gendelman HE, Koenig S, Folks T, Willey R, Rabson A, Martin MA. Production of acquired immunodeficiency syndrome-associated retrovirus in human and nonhuman cells transfected with an infectious molecular clone. J Virol. 1986;59:284–291. doi: 10.1128/jvi.59.2.284-291.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aiken C. Pseudotyping human immunodeficiency virus type 1 (HIV-1) by the glycoprotein of vesicular stomatitis virus targets HIV-1 entry to an endocytic pathway and suppresses both the requirement for Nef and the sensitivity to cyclosporin A. J Virol. 1997;71:5871–5877. doi: 10.1128/jvi.71.8.5871-5877.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aiken C, Konner J, Landau NR, Lenburg ME, Trono D. Nef induces CD4 endocytosis: requirement for a critical dileucine motif in the membrane-proximal CD4 cytoplasmic domain. Cell. 1994;76:853–864. doi: 10.1016/0092-8674(94)90360-3. [DOI] [PubMed] [Google Scholar]
- Aiken C, Trono D. Nef stimulates human immunodeficiency virus type 1 proviral DNA synthesis. J Virol. 1995;69:5048–5056. doi: 10.1128/jvi.69.8.5048-5056.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Billich A, Hammerschmid F, Peichl P, Wenger R, Zenke G, Quesniaux V, Rosenwirth B. Mode of action of SDZ NIM 811, a nonimmunosuppressive cyclosporin A analog with activity against human immunodeficiency virus (HIV) type 1: interference with HIV protein-cyclophilin A interactions. J Virol. 1995;69:2451–2461. doi: 10.1128/jvi.69.4.2451-2461.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braaten D, Aberham C, Franke EK, Yin L, Phares W, Luban J. Cyclosporine A-resistant human immunodeficiency virus type 1 mutants demonstrate that Gag encodes the functional target of cyclophilin A. J Virol. 1996;70:5170–5176. doi: 10.1128/jvi.70.8.5170-5176.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braaten D, Franke EK, Luban J. Cyclophilin A is required for an early step in the life cycle of human immunodeficiency virus type 1 before the initiation of reverse transcription. J Virol. 1996;70:3551–3560. doi: 10.1128/jvi.70.6.3551-3560.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charneau P, Alizon M, Clavel F. A second origin of DNA plus-strand synthesis is required for optimal human immunodeficiency virus replication. J Virol. 1992;66:2814–2820. doi: 10.1128/jvi.66.5.2814-2820.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen C, Okayama H. High-efficiency tranformation of mammalian cells by plasmid DNA. Mol Cell Biol. 1987;7:2745–2752. doi: 10.1128/mcb.7.8.2745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chowers MY, Pandori MW, Spina CA, Richman DD, Guatelli JC. The growth advantage conferred by HIV-1 nef is determined at the level of viral DNA formation and is independent of CD4 downregulation. Virology. 1995;212:451–457. doi: 10.1006/viro.1995.1502. [DOI] [PubMed] [Google Scholar]
- Chowers MY, Spina CA, Kwoh TJ, Fitch NJS, Richman DD, Guatelli JC. Optimal infectivity in vitro of human immunodeficiency virus type 1 requires an intact nef gene. J Virol. 1994;68:2906–2914. doi: 10.1128/jvi.68.5.2906-2914.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Ronde A, Klaver B, Keulen W, Smit L, Goudsmit J. Natural HIV-1 Nef accelerates virus replication in primary human lymphocytes. Virology. 1992;188:391–395. doi: 10.1016/0042-6822(92)90772-h. [DOI] [PubMed] [Google Scholar]
- Dorfman T, Gottlinger HG. The human immunodeficiency virus type 1 capsid p2 domain confers sensitivity to the cyclophilin-binding drug SDZ NIM 811. J Virol. 1996;70:5751–5757. doi: 10.1128/jvi.70.9.5751-5757.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franke EK, Yuan HEH, Luban J. Specific incorporation of cyclophilin A into HIV-1 virions. Nature. 1994;372:359–362. doi: 10.1038/372359a0. [DOI] [PubMed] [Google Scholar]
- Gallay P, Swingler S, Song J, Bushman F, Trono D. HIV nuclear import is governed by the phosphotyrosine-mediated binding of matrix to the core domain of integrase. Cell. 1995;83:569–576. doi: 10.1016/0092-8674(95)90097-7. [DOI] [PubMed] [Google Scholar]
- Gamble TR, Vajdos FF, Yoo S, Worthylake DK, Houseweart M, Sundquist WI, Hill CP. Crystal structure of human cyclophilin A bound to the amino-terminal domain of HIV-1 capsid. Cell. 1996;87:1285–1294. doi: 10.1016/s0092-8674(00)81823-1. [DOI] [PubMed] [Google Scholar]
- Goldsmith MA, Warmerdam MT, Atchison RE, Miller MD, Greene WC. Dissociation of the CD4 downregulation and viral infectivity enhancement functions of human immunodeficiency virus type 1 Nef. J Virol. 1995;69:4112–4121. doi: 10.1128/jvi.69.7.4112-4121.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karpas A, Lowdell M, Jacobson S, Hill F. Inhibition of human immunodeficiency virus and growth of infected T cells by the immunosuppressive drugs cyclosporin A and FK506. Proc Natl Acad Sci USA. 1992;89:8351–8355. doi: 10.1073/pnas.89.17.8351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim S, Byrn R, Groopman J, Baltimore D. Temporal aspects of DNA and RNA synthesis during human immunodeficiency virus infection: evidence for differential gene expression. J Virol. 1989;63:3708–3713. doi: 10.1128/jvi.63.9.3708-3713.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimpton J, Emerman M. Detection of replication-competent and pseudotyped human immunodeficiency virus with a sensitive cell line on the basis of activation of an integrated β-galactosidase gene. J Virol. 1992;66:2232–2239. doi: 10.1128/jvi.66.4.2232-2239.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee CH, Saksela K, Mirza UA, Chait BT, Kuriyan J. Crystal structure of the conserved core of HIV-1 Nef complexed with a Src family SH3 domain. Cell. 1996;85:931–942. doi: 10.1016/s0092-8674(00)81276-3. [DOI] [PubMed] [Google Scholar]
- Luban J, Bossolt KL, Franke EK, Kalpana GV, Goff SP. Human Immunodeficiency Virus type 1 Gag protein binds to cyclophilins A and B. Cell. 1993;73:1067–1078. doi: 10.1016/0092-8674(93)90637-6. [DOI] [PubMed] [Google Scholar]
- Miller MD, Warmerdam MT, Gaston I, Greene WC, Feinberg MB. The human immunodeficiency virus-1 nef gene product: a positive factor for viral infection and replication in primary lymphocytes and macrophages. J Exp Med. 1994;179:101–113. doi: 10.1084/jem.179.1.101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller MD, Warmerdam MT, Page KA, Feinberg MB, Greene WC. Expression of the human immunodeficiency virus type 1 (HIV-1) nef gene during HIV-1 production increases progeny particle infectivity independently of gpl60 or viral entry. J Virol. 1995;69:570–584. doi: 10.1128/jvi.69.1.579-584.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pandori MW, Fitch NJS, Craig HM, Richman DD, Spina CA, Guatelli JC. Producer-cell modification of human immunodeficiency virus type 1: Nef is a virion protein. J Virol. 1996;70:4283–4290. doi: 10.1128/jvi.70.7.4283-4290.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenwirth B, Billich A, Datema R, Donatsch P, Hammerschmid F, Harrison R, Hiestand P, Jaksche H, Mayer P, Peichl P, Quesniaux V, Schatz F, Schuurman HJ, Traber R, Wenger R, Wolff B, Zenke G, Zurini M. Inhibition of human immunodeficiency virus type 1 replication by SDZ NIM 811, a nonimmunosuppressive cyclosporine analog. Antimicrob Agents Chemother. 1994;38:1763–1772. doi: 10.1128/aac.38.8.1763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saksela K, Cheng G, Baltimore D. Proline-rich (PxxP) motifs in HIV-1 Nef bind to SH3 domains of a subset of Src kinases and are required for the enhanced growth of Nef+ viruses but not down-regulation of CD4. EMBO J. 1995;14:484–491. doi: 10.1002/j.1460-2075.1995.tb07024.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sawai ET, Baur A, Struble H, Peterlin BM, Levy JA, Cheng-Mayer C. Human immunodeficiency virus type 1 Nef associates with a cellular serine kinase in T lymphocytes. Proc Natl Acad Sci USA. 1994;91:1539–1543. doi: 10.1073/pnas.91.4.1539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwartz O, Marechal V, Danos O, Heard JM. Human immunodeficiency virus type 1 Nef increases the efficiency of reverse transcription in the infected cell. J Virol. 1995;69:4053–4059. doi: 10.1128/jvi.69.7.4053-4059.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shugars DC, Smith MS, Glueck DH, Nantermet PV, Seillier-Moiseiwitsch F, Swanstrom R. Analysis of human immunodeficiency virus type 1 nef gene sequences present in vivo. J Virol. 1993;67:4639–4650. doi: 10.1128/jvi.67.8.4639-4650.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spina CA, Kwoh TJ, Chowers MY, Guatelli JC, Richman DD. The importance of nef in the induction of human immunodeficiency virus type 1 replication from primary quiescent CD4 lymphocytes. J Exp Med. 1994;179:115–123. doi: 10.1084/jem.179.1.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steinkasserer A, Harrison R, Billich A, Hammerschmid F, Werner G, Wolff B, Peichl P, Palfi G, Schnitzel W, Mlynar E, Rosenwirth B. Mode of action of SDZ NIM 811, a nonimmunosuppressive cyclosporin A analog with activity against human immunodeficiency virus type 1 (HIV-1): interference with early and late events in HIV-1 replication. J Virol. 1995;69:814–824. doi: 10.1128/jvi.69.2.814-824.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thali M, Bukovsky A, Kondo E, Rosenwirth B, Walsh CT, Sodroski J, Gottlinger HG. Functional association of cyclophilin A with HIV-1 virions. Nature. 1994;372:363–365. doi: 10.1038/372363a0. [DOI] [PubMed] [Google Scholar]
- Welker R, Kottler H, Kalbitzer HR, Krausslich HG. Human immunodeficiency virus type 1 Nef protein is incorporated into virus particles and specifically cleaved by the viral proteinase. Virology. 1996;219:228–236. doi: 10.1006/viro.1996.0240. [DOI] [PubMed] [Google Scholar]
- Zazopoulos E, Haseltine WA. Effect of nef alleles on replication of human immunodeficiency virus type 1. Virology. 1993;194:20–27. doi: 10.1006/viro.1993.1230. [DOI] [PubMed] [Google Scholar]