Fast, DNA-sequence independent translocation by FtsK in a single-molecule experiment (original) (raw)
Abstract
Escherichia coli FtsK is an essential cell division protein, which is thought to pump chromosomal DNA through the closing septum in an oriented manner by following DNA sequence polarity. Here, we perform single-molecule measurements of translocation by FtsK50C, a derivative that functions as a DNA translocase in vitro. FtsK50C translocation follows Michaelis–Menten kinetics, with a maximum speed of ∼6.7 kbp/s. We present results on the effect of applied force on the speed, distance translocated, and the mean times during and between protein activity. Surprisingly, we observe that FtsK50C can spontaneously reverse its translocation direction on a fragment of E. coli chromosomal DNA, indicating that DNA sequence is not the sole determinant of translocation direction. We conclude that in vivo polarization of FtsK translocation could require the presence of cofactors; alternatively, we propose a model in which tension in the DNA directs FtsK translocation.
Keywords: chromosome segregation, DNA translocation, FtsK, magnetic tweezers, molecular motor
Introduction
Active transportation of DNA from one cellular compartment to another is central to many biological processes, such as viral DNA packaging, conjugation, sporulation, and cell division. In bacteria, a new class of proteins was recently identified that mediate the transport of double-stranded DNA through cell membranes and cell walls: the FtsK/SpoIIIE/TraSA family (Bath et al, 2000; Possoz et al, 2001; Aussel et al, 2002). These proteins belong to the AAA+ (ATPase Associated with various Activities) superfamily. As such, they are thought to form hexameric motors, which are responsible for the active translocation process. They are involved in a wide range of functions: TraSA is encoded by the plasmid pSAM2 of Streptomyces ambofaciens, and is responsible for the transfer of the plasmid from donor cells into acceptor cells during conjugation (Possoz et al, 2001). SpoIIIE is responsible for the transport of a chromosome into the small polar prespore compartment during sporulation in Bacillus subtilis (Bath et al, 2000). FtsK is an essential Escherichia coli cell division protein (Begg et al, 1995). Its N-terminal membrane domain is localized to the division septum (Wang and Lutkenhaus, 1998; Yu et al, 1998a), and is necessary for its formation (Draper et al, 1998; Chen and Beckwith, 2001). Its C-terminal motor domain (FtsKC) is implicated in chromosome segregation (Liu et al, 1998; Yu et al, 1998b). Indeed, FtsKC has two essential roles in chromosome dimer resolution (see Aussel et al, 2002; Capiaux et al, 2002; Corre and Louarn, 2002, and references therein for review). Chromosome dimers, formed by homologous recombination in organisms with circular chromosomes, are a threat to normal repartitioning of genetic information at cell division. In E. coli, chromosome dimers are resolved into monomers by the addition of a crossover at a specific 28 bp site, dif, by two tyrosine recombinases, XerC and XerD. Chromosome dimer resolution first requires colocalization of the dif sites; this is accomplished by translocation of the chromosomes by FtsKC (Capiaux et al, 2002; Corre and Louarn, 2002). Secondly, the recombination reaction itself is activated by FtsKC (Aussel et al, 2002), probably through a direct interaction with XerC/D (Yates et al, 2003; Massey et al, 2004). In addition, DNA translocation by FtsK could participate in segregation of normal chromosomes (Yu et al, 1998b; Donachie, 2002; Lau et al, 2003).
Coupled to these processes, there must be a mechanism of translocation polarity; that is, an orienting mechanism that ensures the DNA is transported in the correct direction. In B. subtilis, it has been argued that preferential assembly of SpoIIIE in one daughter cell establishes polarity in chromosome partitioning during sporulation (Sharp and Pogliano, 2002). In normal vegetative growth in E. coli, in contrast to sporulating B. subtilis, there is no obvious morphological nor protein expression difference between the two daughter cells that could explain asymmetric assembly of FtsK. Instead, it has been postulated that FtsK translocation polarity is defined by chromosomal sequence polarity, that is, a high skew of oligomeric sequences along the two replichores that inverts at dif (Salzberg et al, 1998; Lobry and Louarn, 2003). Chromosome dimer resolution is effective only when dif is located within a narrow zone of the chromosome (Cornet et al, 1996), functionally defined as the junction between DNA segments of opposite sequence polarity (Corre et al, 2000; Perals et al, 2000). Sequence polarity can be perturbed by the introduction, close to dif, of exogenous DNA, such as the genome of phage lambda (Corre et al, 2000). This change inactivates chromosome dimer resolution, and augments the probability of endogenous recombination in an FtsK-dependent manner (Corre and Louarn, 2002). This has led to the idea that polarization of the chromosome sequence could direct FtsK's translocation so as to juxtapose the chromosomal dif sites; this mechanism could also assist in clearing the septum of any residual DNA.
In vitro evidence for DNA translocation by the ATPase-motor domains of SpoIIIE and FtsK has been obtained, but all the assays employed were indirect, precluding detailed analysis of the translocation reaction (Bath et al, 2000; Aussel et al, 2002; Ip et al, 2003). Here, we directly monitor the DNA translocase activity of FtsK50C, an active, oligomeric C-terminal truncation of FtsK (Aussel et al, 2002), at the single-molecule level using magnetic tweezers (Strick et al, 1996). We measure, in various conditions of applied force and ATP concentration, the velocity, length, and duration of individual translocation events. Using these data, we deduce basic mechanochemical parameters of the protein's translocation reaction.
Surprisingly, we find that FtsK50C can spontaneously reverse its translocation direction, and thus travel both ways on the same segment of DNA. We test translocation on both lambda genomic DNA and an E. coli chromosomal fragment that has been shown to be polarized in vivo (Perals et al, 2000). No bias in translocation is observed on either of these substrates, suggesting that DNA sequence polarity does not directly affect FtsK50C activity. We conclude that in vivo polarization of FtsK translocation could require additional cofactors, such as domains of the full protein not included in the truncation used here. Alternatively, we propose a mechanism in which force, rather than DNA sequence, directs the protein's in vivo motion.
Results
FtsK50C causes transient decreases of DNA extension
Magnetic beads (4.5 μm) are tethered to a glass capillary by a section of DNA (see Figure 1A), as described in Materials and methods. We use a portion of lambda DNA in all experiments presented, with one exception (noted below) in which we use a fragment of E. coli chromosomal DNA. We search for beads that are tethered by a single, nicked DNA molecule; while we are able to identify and utilize unnicked DNA molecules, we concentrate here on nicked molecules so as to avoid the topological complications of supercoiling. Once a suitable bead is found, we flush the capillary with ATP-laden buffer, add purified FtsK50C monomers to a final concentration of 5–10 nM, and begin tracking the DNA extension with time. After a short time, many transient decreases in DNA extension are observed, as shown in Figure 1B. Preliminary data on unnicked molecules (OA Saleh, S Bigot, FX Barre and JF Allemand, in preparation) are in general very similar to nicked-molecule data; thus, nicks appear not to affect the protein activity. The typical waiting time between events (∼5 s) is greater than the typical event timespan (∼0.2 s), indicating that the measured activity is due to the action of a single protein complex. Note that the structure of FtsK50C in solution is unknown, and apparently multimeric (Aussel et al, 2002); throughout the text, we refer to the active protein unit as a complex. The events continue to occur for a long time without need of additional proteins or ATP. We detect no events in the absence of either ATP or FtsK50C, and we find that existing activity stops upon addition of EDTA to a final concentration of 20 mM. From these observations, we conclude that the events are caused by the ATP-dependent DNA translocation activity of FtsK50C.
Figure 1.

(A) Schematic representation of the measurement apparatus. A magnetic bead is tethered to a glass capillary by a single, nicked DNA molecule. Magnets above the capillary create a field gradient that pulls on the bead with a force F, which is determined by measuring the bead's lateral fluctuations d_x_. By optically tracking the bead, we measure the extension L of the DNA with time. (B) Typical measurement of DNA extension, both with and without FtsK50C in the solution. The plotted data were measured at _F_=10.7 pN and with 5 mM ATP; the data sets have been offset for clarity. We attribute the transient decreases in DNA extension to the creation of loops of DNA by single complexes of FtsK50C. (C) A typical individual event extracted from the data shown in (B). All events begin with a constant-velocity decrease in DNA extension; we attribute the slope of the descent to the translocation velocity of the complex. The velocity, distance travelled d, and on-time are easily measured because of the well-defined event shape. As sketched, loop formation requires the complex to bind the DNA in two locations; see Figure 6 and Discussion for details. (D) FtsK50C activity versus ATP concentration for both bulk (rate of ATP consumption; red curve) and single molecule (translocation velocity; green points) assays. Both data sets are well fit by the Michaelis–Menten equation _V_max[ATP]/([ATP]+Km), with, respectively, _V_max=30±2 ATP/s and 6.7±0.1 kbp/s, and _K_m=270±40 and 330±20 μM. The fit to the single-molecule data (blue curve) is shown; it is highly coincident with the bulk data fit, which is omitted for clarity. Single-molecule data are taken at _F_∼5 pN, and each point is the average of typically 100 events; plotted bars indicate standard error. Inset: Histogram of measured translocation velocities (open circles) with best-fit Gaussian curve (solid line); data taken at 5 mM ATP and _F_=6 pN.
Translocation velocity represents protein activity
Each event begins with a linear decrease in bead height (Figure 1C), from which we can extract both the translocation velocity and the total distance travelled by the protein. Very rarely, we observe events containing pauses that interrupt the constant-velocity decrease. The pauses have no correlation with position along the DNA, and only occur in situations where many protein complexes appear to be active. We attribute pausing to protein–protein interactions, and do not characterize them here; see Supplementary data for more information. Within a single measurement (i.e., on one DNA molecule, and at constant force and ATP concentration), the translocation velocity measured from all events is normally distributed (inset, Figure 1D). Infrequently, an event is observed with almost precisely double the mean translocation velocity of other events in the data set (see Supplementary data). Such events presumably arise from two-motor activity (see Discussion), and are removed from all the analysis presented. The width of the distribution is accounted for by measurement noise and variations due to the stochastic stepping motion of the motor protein (Svoboda et al, 1994); thus, within the experimental noise, we observe no protein-to-protein or sequence-based velocity variation.
To confirm that FtsK50C's ATPase activity causes its translocation, we compare, at various ATP concentrations, the single-molecule velocity with the bulk ATPase activity measured in an excess of DNA (insuring each functional protein is active). As shown in Figure 1D, the assays agree quite well: each independently indicates that FtsK50C follows simple Michaelis–Menten kinetics with _K_m∼0.3 mM ATP and respective maximum velocities of ∼6.7 kbp/s and ∼30 ATP/FtsK50C monomer/s. Note that the latter is only a relative measure since it ignores the presence of nonfunctional (misfolded) proteins. These results, in the light of previous data showing that ATPase activity is highly stimulated in the presence of DNA (Aussel et al, 2002), are consistent with a mechanism in which ATP consumption is restricted to DNA-bound FtsK50C, and leads directly to DNA translocation.
Reversal of the translocation direction
After the initial translocation, the recovery back to the original bead height occurs in one of two ways, as shown in Figure 2A. The first type of recovery is an abrupt jump that occurs at speeds >20 μm/s, which is too fast to be resolved in our system. This is consistent with the protein completely unbinding, and the bead rising at a rate determined by the applied force and hydrodynamic Stokes drag (e.g. a 2 pN force will move a 4.5 μm diameter bead through water at a terminal velocity of ∼50 μm/s). The second type of recovery is a slower, constant-velocity increase, with a slope (at low forces) nearly equivalent to that of the decrease. We believe that slow recoveries are caused by a single complex that, after decreasing the bead height, reverses its translocation direction and causes the bead to rise (see Discussion and Figure 2A sketches). Both types of recoveries occur at all forces; at a given force, events with slow and fast recoveries are randomly interspersed. However, it is clear that the frequency of occurrence of direction reversals depends on the applied force: the proportion of slow recoveries decreases from ∼50% at low forces to nearly zero at high forces (inset, Figure 2A). Rarely, we observe events with more complicated mixed recoveries, wherein the bead height first increases slowly, then abruptly, or vice versa (see Supplementary data).
Figure 2.
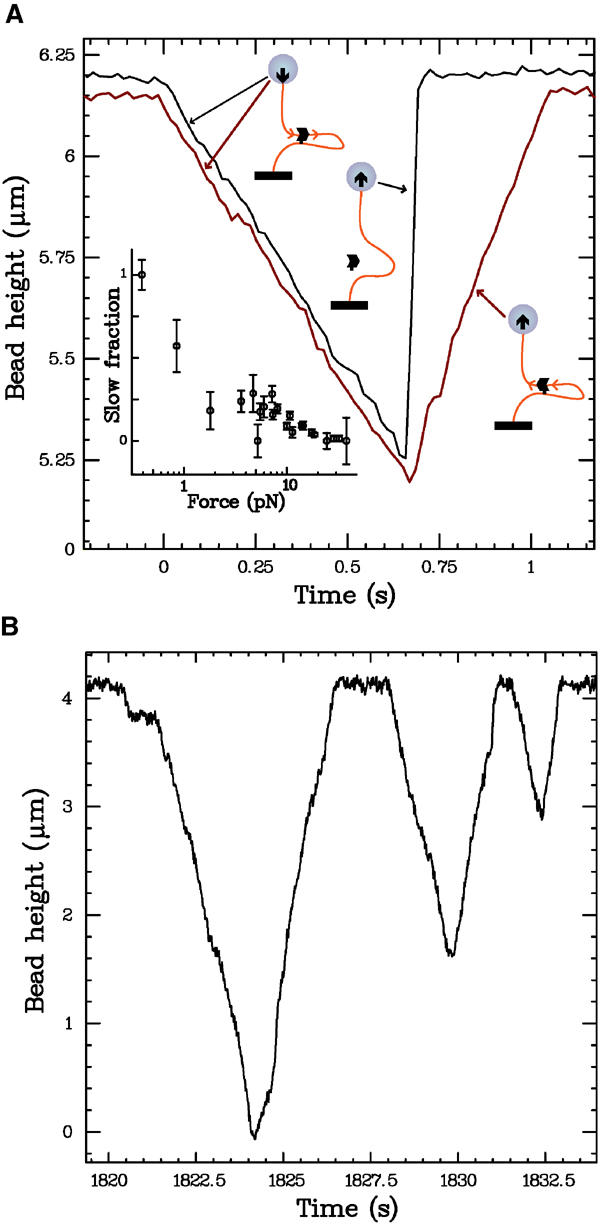
(A) Illustration of different types of recoveries. After decreasing, the DNA extension can recover its full length either abruptly (black curve) or slowly (red curve). As sketched, we attribute the former to protein unbinding, and the latter to a reversal of the translocation direction. The fraction of events exhibiting a slow recovery is force dependent, as plotted in the inset. The distance travelled before a reversal of direction is random, and consistent with the distribution of distances travelled before unbinding (see Figure 4). The events plotted were measured at 18 pN and 5 mM ATP, and are offset for clarity. The inset includes data taken at ATP concentrations from 0.5 to 5 mM; we observe no variation in the slow fraction with ATP. (B) Translocation of a fragment of E. coli chromosomal DNA that has been shown to have in vivo polar activity (Perals et al, 2000); data taken at 5 mM ATP and _F_=1 pN. All the three plotted events have slow, direction-reversal recoveries. In particular, in the first event, FtsK50C translocates the entire length of the DNA in both directions, indicating that no portion of the DNA sequence has a strong effect in biasing the translocation direction.
Direction reversal has potential implications for the in vivo mechanism of FtsK translocation polarity, as it shows that, on the section of lambda DNA utilized, the protein can travel in both directions on the same DNA sequence. To further explore this effect, we perform measurements on a fragment of E. coli chromosomal DNA with a proven in vivo polarity effect (Perals et al, 2000). As shown in Figure 2B, we still observe direction reversal events on the E. coli DNA, with no difference in either the shape of individual events, or the frequency of occurrence of slow recoveries. Furthermore, full-length direction reversal events occur (wherein the bead is brought from full height down to the capillary surface, then back up again, all at constant speed; see Figure 2B), indicating that no subsection of the E. coli DNA fragment has a definitive effect on the translocation direction of the complex.
Force dependence of the translocation velocity
In our assay, the applied force F depends only on the distance of the magnets to the capillary; we can easily adjust this distance to study the dependence of translocation velocity on force. During initial translocation, the protein lowers the bead, and must perform work against the applied force. In contrast, during direction-reversed translocation, the bead rises, so the protein works with the (upward) force. Thus, we can use initial translocation to measure the velocity with forces hindering the protein's motion, and reversed translocation to measure the velocity with forces assisting the motion (following previous practice, we define a hindering force as positive). In Figure 3, we show the force–velocity relationships measured in the presence of 1 and 5 mM ATP. At both concentrations, there is a clear plateau of constant velocity for small forces, along with a transition to increasingly higher velocities at large assisting forces; this transition occurs, respectively, at −4 and −11 pN for 1 and 5 mM ATP. At 5 mM ATP, the velocity begins to clearly decrease for _F_>15 pN; no such transition is seen in the 1 mM data up to _F=_29 pN.
Figure 3.
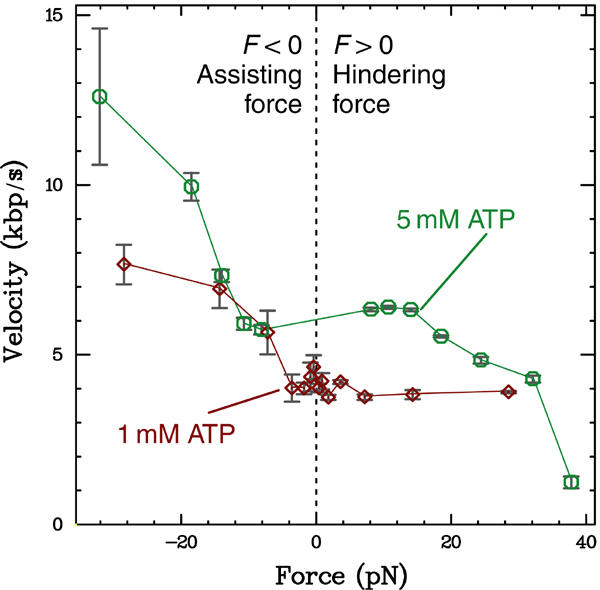
Force–velocity relations for FtsK50C measured at 1 and 5 mM ATP. At both ATP concentrations, there is a plateau of constant velocity for small forces. The velocity measured at 5 mM ATP begins to decrease above a hindering force of 15 pN, while no decrease is seen in the 1 mM data up to 29 pN. The hindering-force data indicate that the FtsK50C reaction pathway has force-independent biochemical step(s) in series with force-dependent mechanical step(s) (see Discussion). Plotted bars indicate standard error; large error estimates indicate regimes where the event frequency and size are small (see Figure 4).
Mean event length and on-time decrease with force
We are unable to measure the velocity above ∼35 pN because of a strong decrease in the size of each event, and in the frequency of event occurrence. The length and duration (i.e., on-time) of each event correspond, respectively, to the distance travelled by the protein, and the time spent bound to the DNA, before unbinding or reversing direction (see Figure 1C). Within a measurement at a given force and ATP concentration, both distance and on-time vary from event to event. We find that the distribution of each is always exponential (see inset, Figure 4B), indicating a constant unbinding probability with time (and thus distance, given the nearly constant velocity within a given measurement; see inset, Figure 1D); we can then fit an exponential curve to each distribution and extract the mean distance and on-time for the given conditions. In this way, we measure, at constant 5 mM ATP, the variation of mean distance and on-time with applied force. As shown in Figure 4A, the mean on-time decreases strongly, and exponentially, with force. Fitting the data to _t_0 exp(−F/_F_0) gives a decay constant _F_0=11.3±0.9 pN and a mean on-time at zero force of _t_0=1.6±0.3 s. The mean distance travelled also decreases strongly with force, as shown in Figure 4B. At low forces, the translocation velocity is fairly constant (Figure 3), so we expect the mean distance to vary in the same manner as the mean on-time. This is indeed the case: an exponential fit to the low-force points gives a decay constant _F_0=11.6±3.2 pN, nearly equivalent to the decay constant of the mean on-time. Above 20 pN, the mean distance drops off more quickly due to the decrease in velocity at high forces.
Figure 4.
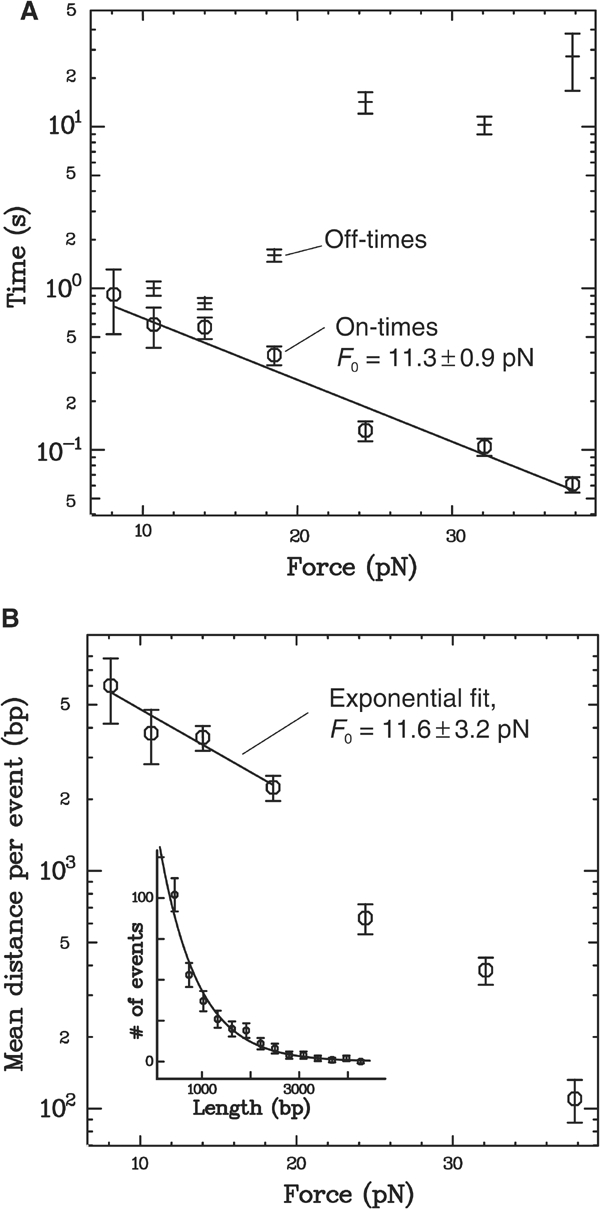
(A) Force dependence of the on- and off-times at 5 mM ATP. As with the distance travelled (see inset, B), the distribution of on-times from many events within a measurement is exponential (data not shown), indicating a constant unbinding probability with time. We extract a mean on-time from each distribution through fitting an exponential function. Here, we plot the variation with force of the mean on-time (circles), and fit it with an exponential function _t_0 exp(−F/_F_0). The mean on-time decreases with force with a decay constant _F_0=11.3±0.9 pN and an on-time at zero force of _t_0=1.6±0.3 s. This exponential force dependence can be simply explained if we assume that binding of the protein causes a distortion that decreases the DNA length by at least 0.36±0.03 nm (see Discussion). (B) Dependence of the mean distance travelled per event on force at 5 mM ATP. The mean distance travelled at a given force should follow directly from the on-times (A) and the velocity (Figure 3). Indeed, at low forces, where the velocity is constant, we observe an exponential decay constant, _F_0=11.6±3.2 pN, nearly identical to that of the on-times. For higher forces, the decrease in velocity causes the measured mean distance to drop off even more quickly. Inset: A typical distribution of distances, and exponential fit; data taken from ∼300 events at _F_=24 pN and 5 mM ATP.
Force dependence of the off-times
Along with the distances and on-times, we extract from each measurement distributions of the off-times, that is, the time duration between events (see Figure 1B). The off-time distributions from a given measurement clearly do not follow a single exponential curve, but rather a curve that is the sum of (at least) two exponentials with, respectively, fast and slow time constants (see Supplementary data). The presence of fast and slow rebinding rates is presumably due to the varying timescales of processes that contribute to protein–DNA binding (see Discussion). We find strong variations in the mean off-time for different measurements, which we attribute to variations in the amount and active percentage of the added protein. However, since a single addition of protein to the capillary results in long-lasting activity, we can vary the force and confidently measure the response of the off-times at constant protein concentration. As shown in Figure 4A, we find that the mean values of the off-time increase with force, indicating that binding rate of the protein to the DNA is decreased by the application of higher forces.
Noise analysis reveals FtsK50C step size
The completion of an enzymatic cycle results in a single forward step of a motor protein. If the step size is larger than the measurement noise, and the average time to complete an enzymatic cycle is larger than the time resolution of the measurement, it would be possible to observe individual steps directly. This is not the case here: we observe no discrete steps in the traces of bead position versus time. However, it is still possible to estimate the step size by considering the fluctuations in the measurement. In the absence of protein activity, the resolution of our measurement is limited by the bead's Brownian fluctuations, which, at low frequency f, create a frequency-independent ‘white' noise. During protein activity (i.e., during the linear decrease in bead height), we find that the measurement noise increases at low frequencies as 1/_f_2, and thus cannot be attributed only to Brownian fluctuations (see Figure 5). Such a low-frequency increase in noise has been observed in previous single-molecule measurements (Svoboda et al, 1994), and has been shown to be proportional to the enzymatic step size (Svoboda et al, 1994; Charvin et al, 2002); it is caused by the random distribution of the times between individual steps (Svoboda et al, 1994).
Figure 5.

Power spectrum analysis of the variance in bead height during (circles) and in the absence of (squares) protein activity. Each set of points is the average of 26 segments of a data trace acquired at 5 mM ATP and _F_=19 pN. The spectrum of noise during protein activity shows a clear 1/_f_2 increase over the spectrum of the measurement noise alone. Note that the latter spectrum is calculated from quiescent segments between the bursts of activity that give rise to the former spectrum, and is thus a nearly simultaneous measure of the background noise. This increase is due to stochastic variations of the motion of the protein, and is proportional to the protein's step size δ. The line is a fit to the predicted increase (Charvin et al, 2002): _y=_δ〈_v_〉/2π2_f_2+b, where 〈_v_〉 is the mean velocity and b accounts for the (white) measurement noise. From many such fits, we find that, if the FtsK50C enzymatic cycle has a single rate-determining step, it advances 12±2 bp per cycle.
To estimate the step size, we select the active segment of a suitable event and compute, at each time point, the difference between the bead's measured position and its mean position (as predicted by the mean velocity). We then find the power spectrum of that difference, average the spectra over many events from a single measurement, and fit this average with the theoretically predicted curve (Charvin et al, 2002); an example is shown in Figure 5. By performing this analysis, we estimate that the step size of FtsK50C is 12±2 bp. In our application of the model, we assume that the entire enzymatic cycle has only one rate-determining step. Relaxation of this assumption (i.e., if the rates of two or more substeps of the cycle are comparable) would decrease the randomness of the distribution of times between steps (Svoboda et al, 1994); in turn, the estimated step size would necessarily increase to account for the measured level of noise. Thus, our estimate of 12±2 bp is technically a lower bound. We see no significant variation in the step size with ATP concentrations of 1 or 5 mM or forces between 5 and 19 pN. If we assume the active motor is a hexamer (Aussel et al, 2002), and that 100% of the added protein in the bulk ATPase measurement were active, we can use the maximum velocities of translocation (∼6.7 kbp/s) and ATP hydrolysis (∼30 ATP/monomer/s) to estimate that the FtsK50C motor moves ∼37 bp per hydrolyzed ATP. Given the step size, this leads to the impossible result that less than one ATP is hydrolyzed per enzymatic cycle. It is important to note that this does not invalidate our result for the lower bound of the step size: a much more likely explanation is that a significant fraction of the protein in the ATPase assay was inactive; this would not affect the single-molecule measurement.
Discussion
FtsK50C forms loops of DNA
We attribute the observed transient decreases in DNA extension (Figure 1B) to the extrusion of loops of DNA by translocating protein complexes: a protein solely moving along a DNA molecule will cause no change in extension in our assay, and since each molecule is nicked we rule out shortening due to an accumulation of supercoils. Formation of DNA loops has been postulated before to explain the topological modification of DNA substrates by FtsK50C complexes (Aussel et al, 2002; Ip et al, 2003). A recurrent tentative hypothesis was that loop extrusion was due to the activity of two connected motors, with each translocating, but in opposite directions (Ip et al, 2003). Since FtsK is an AAA+ protein, it is probable that each motor is a hexamer encircling the DNA; thus two connected motors would form a double ring. We cannot rule out that each complex contains more than two motors; on the contrary, the characteristics of direction-reversal events are best explained by an FtsK50C complex containing three or more motors (see below). The formation of such higher order complexes is compatible with previously published electron microscopy and gel filtration data on FtsK50C (Aussel et al, 2002).
A single motor is active during loop extrusion
The range of measured timespans indicates that each event is due to only a single complex of FtsK50C. As mentioned, each complex might be composed of several functional hexameric motors, two of which could bind to the DNA and form a loop. However, we argue that only one motor is translocating during the observed events: at low forces, the processivity increases to the extent that we see full-length, constant-velocity events (in which the bead starts at full height and is brought smoothly to the capillary surface). If loops were extruded by two motors simultaneously, a full-length, constant-speed event would only occur if each motor bound exactly to the middle of the DNA and worked in opposite directions until each hit a surface (the bead or the capillary). Such a precise starting position should be rare, yet we observe this type of event frequently at low force. Further evidence disfavoring multiple active motors is the appearance of direction-reversal events, in which the velocities of descent and ascent are nearly equivalent (Figure 2A). This process is difficult to explain if two motors are acting, since both motors would have to switch directions simultaneously. For these reasons, we conclude that during each measured event, only one motor is translocating.
Localizing the protein to a DNA extremity
Although only one motor is active, loop formation requires the FtsK50C complex to contact the DNA in two locations. In what manner could the complex form a second, nontranslocating contact? The protein could possibly have a second (immobile) type of DNA-binding domain, but this is unlikely for an aforementioned reason: full-length, constant-velocity events require that the protein begins translocation at one of the extremities of the DNA, and random DNA binding would not efficiently localize the protein to an extremity. Instead, we suggest that the second binding location is also a motor bound to the DNA, but stalled at the bead or capillary surface, as diagrammed in Figure 6. In this scenario, a diffusing complex first binds to the DNA through one motor, which translocates (without loop formation or a change in bead height) and transports the complex to a surface, where the motor stalls but remains bound to the DNA. Other motors in the complex are then free to bind; translocation of these motors causes loop formation and reduces the bead height. The complex is localized to the extremity of the DNA, as required to explain the measured full-length events. The rare events with twice the normal velocity (see Supplementary data) occur when a second motor binds before the first has stalled, resulting in a change in bead height at twice the single-motor rate. Finally, this model explains the multiple timescales in the off-time distributions (see Supplementary data): short off-times occur when only one of the two motors unbinds, leaving the complex in close proximity to the DNA and thus enabling fast rebinding, while long off-times occur when both motors unbind, and rebinding is limited by free diffusion of the protein complexes.
Figure 6.

Diagram of proposed steps in loop formation by FtsK50C complexes. (A) Free complexes, containing several identical (probably hexameric) motors, diffuse into the vicinity of the DNA. (B) The complexes bind the DNA through a single motor, which translocates and carries the complex toward an extremity of the DNA without changing the bead height. (C) Upon reaching the surface, the motor stalls but does not unbind, allowing other motors in the complex to bind and translocate. (D) Translocation of the second motor extrudes a loop of DNA, and decreases the bead height at the single-motor velocity. The decrease can be halted by (E) unbinding of either motor, leading to a fast recovery of the bead height, or (F) reversal of the translocation direction, and a slow recovery of bead height. We believe that direction reversal proceeds through unbinding of the translocating motor, followed by binding and translocation by an oppositely directed motor from the same complex (see Discussion).
Brief motor unbinding accompanies direction reversal
As shown in the inset to Figure 2A, direction reversal is less likely at higher forces. This can be explained by assuming that direction reversal first involves unbinding of the translocating motor, followed immediately by binding of an oppositely directed motor: the observed decrease in binding rate with force (i.e., the increase in off-time, see Figure 4A) would then explain the decrease in the probability of direction reversal with force (inset, Figure 2A). The oppositely directed motor could, in principle, be an inverted configuration of the first motor, but we consider it more likely that it is a different (but nearby) motor within the same complex.
Considerations on the mechanochemistry of the FtsK50C motor
The force–velocity relationships of FtsK50C (Figure 3) show a clear plateau for small forces of either orientation, along with a decrease in velocity (at saturating 5 mM ATP) for large hindering forces. This behavior is qualitatively similar to both theoretical predictions (Keller and Bustamante, 2000) and previous results obtained on RNA polymerase (Wang et al, 1998; Neuman et al, 2003). The behavior is indicative of two types of sequential steps in the reaction that causes forward movement of the protein: a force-dependent mechanical step (involving motion along the DNA), and one or more force-independent biochemical steps (involving no motion along the DNA, but rather, for example, ATP binding or a change in the protein's internal conformation). The mechanical step requires the protein to change the height of the bead by a distance δ and perform a work against the force of _F_δ; thus, the kinetic barrier of that step will increase at high hindering forces, decreasing the rate. If, at zero force, the mechanical step is much faster than the biochemical step, the zero-force velocity _v_0 will depend only on the step size and the biochemical step rate. As the force increases, the mechanical step rate slows until it becomes comparable to the biochemical step rate. For small F, the biochemical step determines the rate (and the velocity equals _v_0, independent of the force), while for large F the mechanical step determines the rate (and the velocity decreases with force). This description qualitatively matches our data at 5 mM ATP. Furthermore, it is consistent with the 1 mM ATP data, where no velocity decrease is seen up to _F=_29 pN. The decrease to 1 mM ATP slows the ATP-binding process, thus slowing the rate of the biochemical steps. To make the mechanical step rate comparable to this lowered biochemical rate (i.e., to see a velocity decrease) will thus require a larger force than was needed at 5 mM ATP; we apparently do not reach this regime at the maximum force of 29 pN utilized in the 1 mM data.
For assisting forces, our data notably deviate from the above model and the measurements on RNA polymerase (Neuman et al, 2003); in both, there is a continuation of the constant-velocity plateau. Instead, we observe a clear increase in velocity for large assisting forces (Figure 3). The velocity increase is not likely due to a change in the enzymatic turnover rate (since it is unlikely that assisting force would quicken the limiting biochemical processes), but rather due to an increase in the mechanical step size. Supporting this, we note that the velocity is still dependent on ATP in this regime; thus, the biochemical processes are still rate-determining. However, since our data are sparse at large negative forces due to the rarity of direction reversals (inset, Figure 2A) and the decrease in event size (Figure 4A), we cannot rule out the existence of a completely separate mechanism of forward motion.
Force dependence of binding statistics indicates a distortion of the DNA
The mean value of the on-time decreases exponentially with force (see Figure 4A) until, at high hindering force, protein activity is limited by this parameter. The exponential dependence can be accounted for by assuming that the DNA length when bound by FtsK50C is shorter than when free. We model this effect as a two-state system (Evans and Ritchie, 1997; Rief et al, 1998), where the bound and unbound states are separated by a transition state with a higher free energy. Any DNA length change l between the bound and transition states would require unbinding to perform a work −Fl against the applied force. This would affect the mean on-time (the inverse of the mean unbinding rate) by a factor exp(−Fl/kT), consistent with our observations of an exponential dependence on force. Based on the fitted exponential decay constant of 11.3±0.9 pN, we estimate that unbinding of FtsK50C causes the DNA length to increase by _l_⩾0.36±0.03 nm (the estimate is a lower bound since the measurement is not sensitive to any further change in DNA length between the transition and unbound state; see Supplementary data). This length change is much smaller than the minimal 80 bp binding site required to stimulate FtsK50C's ATPase activity (Massey et al, 2004). Therefore, it is consistent only with a small bend in the DNA induced at the protein's binding site.
Implications of direction reversal for models of FtsK polarity
It has been suggested that some chromosomal oligomeric sequences, with a high skew that inverts at dif, could direct the FtsK translocation process (Perals et al, 2000; Corre and Louarn, 2002), just like Chi sequences alter the enzymatic properties of RecBCD complexes (Spies et al, 2003). In this regard, direction reversal is surprising because it indicates that FtsK50C can move in both directions on the same segment of DNA. Furthermore, we observe full-length direction-reversal translocation events on a lambda DNA fragment of approximately half the size of the phage genome; introduction of a complete phage lambda genome has been shown to perturb chromosome polarity in vivo (Corre et al, 2000). To confirm this finding, we perform experiments on a fragment of the E. coli genome that was shown to be polarized (Perals et al, 2000). Full-length translocation events after direction reversal are again observed (Figure 2B). We conclude that the oligomeric DNA motifs that polarize the E. coli and lambda genomes do not constitute absolute blocks to DNA translocation by FtsK50C. We cannot rule out the possibilities that their action is probabilistic, or that it requires the presence of the N-terminal or linker domains of FtsK. However, our data raise the possibility that DNA sequence information plays only an indirect role on polarity of translocation by FtsK. For example, an additional protein could bind to specific DNA sequences and block FtsK translocation, just like Tus binding to Ter sites can stop replication forks (Kamada et al, 1996), or, rather than acting on FtsK, the DNA sequence could direct progressive, oriented condensation of the nucleoids, thereby imposing that the dif region is the last chromosomal section to be moved by FtsK.
A force-rectified translocation model could impose directionality
Based on the observed sensitivity of the travelled distance to force (Figure 4B), we propose an alternate mechanism that could account for in vivo FtsK directionality. The parameters of the exponential curve fit of mean on-time versus force (Figure 4A), along with the maximum speed, indicate that at zero force the FtsK50C complex will travel, on average, ∼11 kbp per binding event. Since DNA translocation most likely involves the binding of two identical motor units (Figure 6), the zero-force mean distance for each motor is then twice that of the complex, or ∼22 kbp. This is much less than the total length of the E. coli chromosome (4.5 Mbp). However, the chromosome is compacted in the cell and it is highly probable that to clear the septum, FtsK works on DNA loops of sizes similar to the zero-force mean distance.
We propose that a septum-bound FtsK motor, upon binding a chromosomal loop, can translocate in either direction. However, if pumping in the wrong direction, it will pull against the large fraction of condensed DNA and work against other proteins in the nascent daughter cell that maintain the chromosome position, such as MukB and active next-generation DNA replisomes (Sherratt, 2003). The counteracting activities of FtsK and these other proteins will create a tension in the DNA analogous to the force we apply to the bead in our assay. Just as we observe (Figure 4B), this tension will decrease the distance FtsK can travel before unbinding. FtsK motors that pump in the wrong direction will thus quickly fall off, allowing other, perhaps correctly oriented, motors to bind. Although the lack of structural data on FtsK precludes affirmation, the formation of higher order complexes by FtsK50C and the resulting in vitro direction-reversal activity might indicate that higher order complexes are also formed in vivo by FtsK, which would facilitate this process. Motors pumping in the correct direction will not create tension in the DNA, and will translocate a correspondingly greater distance. In this way, FtsK would clear any misplaced chromosomal loops that pass through the septum. Interestingly, this model could be applied to sporulation in B. subtilis, assuming that condensation of DNA in the prespore generates sufficient tension.
This force-rectified translocation model applies to segregation of normal daughter chromosomes and to resolution of chromosome dimers. For normal chromosome segregation, FtsK would simply accelerate the clearing process; both genetic and cellular biology data indicate that FtsK is not absolutely required for cohesion and mid-cell positioning of the large terminus region of the E. coli chromosome (Capiaux et al, 2002; Corre and Louarn, 2002). In the presence of chromosome dimers, misplaced loops could still occur; FtsK would clear these loops from the septum until there remained only the two chromosome-connecting strands that contain the dif sites required for XerC/D recombination.
FtsK50C's high velocity enables fast processing of misaligned DNA
FtsK50C's maximum velocity of ∼6.7 kbp/s makes it the fastest DNA-based motor protein yet measured. Speeds similar to our measurements have been estimated for the total transfer rate of the B. subtilis genome into a nascent spore by SpoIIIE (Errington et al, 2001). As mentioned above, we do not expect FtsK to mobilize such a large fraction of the E. coli genome, but rather a tangle of DNA loops. Unravelling such a tangle could require loops to be successively processed several times, as has been reported in vitro for replicative catenanes (Ip et al, 2003); thus FtsK50C's high speed could be needed to complete multiple processing of misaligned loops quickly.
Materials and methods
Protein, substrate DNA, and slide preparation
FtsK50C is purified as described in Aussel et al (2002). λ DNA (bp 25 882–45 679) is amplified by long-range PCR. E. coli chromosomal DNA (bp 8877–22 422 after the XerC-binding site of dif) is purified from plasmid pFC94 (Perals et al, 2000). DNA substrates are ligated to biotin- and digoxigenin-modified DNA fragments, which are prepared by PCR using cognate modified nucleotides. The resulting constructs are incubated with 4.5 μm diameter paramagnetic streptavidin beads and added to 1-mm square cross-section glass capillaries, which had been washed with 0.1 M NaOH, coated with SigmaCote, and sequentially incubated with solutions of anti-digoxigenin and bovine serum albumin (BSA). We insure that the beads we use are bound to the capillary by a single, nicked DNA molecule, and not a single unnicked, molecule or multiple molecules, as described in Strick et al (1996, 1998).
Measurement of applied force and bead height
The image of each bead is captured by a CCD camera at 60 Hz and processed, using custom-written software, to give the 3D bead position. Before protein addition, we calibrate the apparatus for each bead by measuring the applied force as a function of magnet/capillary separation distance. The applied force is measured by monitoring the mean squared transverse fluctuations 〈d_x_2〉 of the bead (see Figure 1A), and applying the equipartition theorem: ½_k_T = ½〈d_x_2〉F/L, where L is the measured bead height (Strick et al, 1996). Microscope drift is removed by simultaneously tracking a reference bead stuck to the capillary surface, and subtracting its height from that of the experimental bead.
Addition of protein to the capillary
Experiments are performed in a buffer containing 10 mM MgCl2, 10 mM Tris pH 7.9, 50 mM NaCl, 1 mM DTT, 0.01% Triton X-100, and ATP. In all, 5–10 μl of 200 nM FtsK50C monomers is added to 200 μl of buffer in the capillary, and gently mixed.
Long data acquisitions
Activity resulting from a single addition of protein can last for hours. To control for possible degradation of the protein or ATP in that time, we measure the activity at a reference force multiple times throughout the experiment. We never observe a decrease in velocity in those reference measurements. In some long measurements, we do observe an increase in the mean off-time; we attribute this to a decrease in the active protein concentration through degradation or nonspecific adsorption to the capillary. Data sets containing such a decrease are not used to construct the off-time plot in Figure 4A.
Data analysis
Measured traces of bead height correspond directly to DNA extension; no filtering has been applied to any of the plots shown. Translocation velocities, lengths, and on- and off-times are extracted by analyzing each data set with custom-written software that applies a filter (Chung and Kennedy, 1991) in order to identify events automatically, while still measuring each parameter from the unfiltered data. Data quoted in units of base pairs have been corrected for the difference between measured DNA extension and the true contour length by applying the worm-like chain model for DNA elasticity (Bouchiat et al, 1999). To calculate the fraction of slow recoveries (inset, Figure 2A), the number of slow recovery segments of a certain minimum size (typically 0.5 μm; less at higher forces) are counted, and divided by the total number of events of that size; the threshold is needed due to the difficulty in characterizing very short recoveries. For the fluctuation analysis, within a data set, only long (>0.75 μm) events are analyzed. A straight line is fit to, and subtracted from, each event to acquire the difference at each time point. The mean variance from the fit line is computed for each event; typically, the mean variance across all events forms a compact distribution. Outliers from this distribution (more than 2 standard deviations larger than the mean) sometimes occur, and are considered to contain anomalous noise; thus, the corresponding events are discarded. Power spectra are then calculated from the suitable events using standard algorithms.
Bulk ATPase assay
Reactions are performed with 100 nM of FtsK50C (monomer), 5 nM of 3 kb supercoiled plasmid DNA, 1 nM [α32P]ATP and cold ATP in 10 μl of 10 mM Tris–HCl, pH 7.9, 10 mM MgCl2, 50 mM NaCl, and 1 mM DTT. Reactions are incubated for 3 min before being stopped with EDTA and excess ATP. The ratio of ATP to ADP is analyzed by thin layer chromatography. We check that our measurements correspond to initial rates and that DNA is in a 10-fold excess in the reaction (data not shown).
Supplementary Material
Supplementary Data
Acknowledgments
We thank D Bensimon, V Croquette, J-M Louarn, and F Cornet for constant support and helpful discussions. We thank V Croquette for sharing analysis software, and J-Y Bouet, G Charvin, K Neuman, and T Lionnet for critical reading of the manuscript. Research was funded by the CNRS and the Ecole Normale Supérieure. Research in Paris was supported by grants from the French Research Ministry ACI Jeune Chercheur program and from the EU MolSwitch program. Research in Toulouse was supported by grants from the CNRS ATIP program and from the French Research Ministry Fundamental Microbiology ACI program.
References
- Aussel L, Barre FX, Aroyo M, Stasiak A, Stasiak AZ, Sherratt D (2002) FtsK is a DNA motor protein that activates chromosome dimer resolution by switching the catalytic state of the XerC and XerD recombinases. Cell 108: 195–205 [DOI] [PubMed] [Google Scholar]
- Bath J, Wu LJ, Errington J, Wang JC (2000) Role of Bacillus subtilis SpoIIIE in DNA transport across the mother cell-prespore division septum. Science 290: 995–997 [DOI] [PubMed] [Google Scholar]
- Begg KJ, Dewar SJ, Donachie WD (1995) A new Escherichia coli cell division gene, ftsK. J Bacteriol 177: 6211–6222 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bouchiat C, Wang MD, Allemand JF, Strick T, Block SM, Croquette V (1999) Estimating the persistence length of a worm-like chain molecule from force–extension measurements. Biophys J 76: 409–413 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Capiaux H, Lesterlin C, Perals K, Louarn JM, Cornet F (2002) A dual role for the FtsK protein in Escherichia coli chromosome segregation. EMBO Rep 3: 532–536 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charvin G, Bensimon D, Croquette V (2002) On the relation between noise spectra and the distribution of time between steps for single molecular motors. Single Molecules 3: 43–48 [Google Scholar]
- Chen JC, Beckwith J (2001) FtsQ, FtsL and FtsI require FtsK, but not FtsN, for co-localization with FtsZ during Escherichia coli cell division. Mol Microbiol 42: 395–413 [DOI] [PubMed] [Google Scholar]
- Chung SH, Kennedy RA (1991) Forward-backward nonlinear filtering technique for extracting small biological signals from noise. J Neurosci Methods 40: 71–86 [DOI] [PubMed] [Google Scholar]
- Cornet F, Louarn J, Patte J, Louarn JM (1996) Restriction of the activity of the recombination site dif to a small zone of the Escherichia coli chromosome. Genes Dev 10: 1152–1161 [DOI] [PubMed] [Google Scholar]
- Corre J, Louarn JM (2002) Evidence from terminal recombination gradients that FtsK uses replichore polarity to control chromosome terminus positioning at division in Escherichia coli. J Bacteriol 184: 3801–3807 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corre J, Patte J, Louarn JM (2000) Prophage lambda induces terminal recombination in Escherichia coli by inhibiting chromosome dimer resolution. An orientation-dependent _cis_-effect lending support to bipolarization of the terminus. Genetics 154: 39–48 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donachie WD (2002) FtsK: Maxwell's demon? Mol Cell 9: 206–207 [DOI] [PubMed] [Google Scholar]
- Draper GC, McLennan N, Begg K, Masters M, Donachie WD (1998) Only the N-terminal domain of FtsK functions in cell division. J Bacteriol 180: 4621–4627 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Errington J, Bath J, Wu LJ (2001) DNA transport in bacteria. Nat Rev Mol Cell Biol 2: 538–544 [DOI] [PubMed] [Google Scholar]
- Evans E, Ritchie K (1997) Dynamic strength of molecular adhesion bonds. Biophys J 72: 1541–1555 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ip SCY, Bregu M, Barre F-X, Sherratt DJ (2003) Decatenation of DNA circles by FtsK-dependent Xer site-specific recombination. EMBO J 22: 6399–6407 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamada K, Horiuchi T, Ohsumi K, Shimamoto N, Morikawa K (1996) Structure of a replication-terminator protein complexed with DNA. Nature 383: 598–603 [DOI] [PubMed] [Google Scholar]
- Keller D, Bustamante C (2000) The mechanochemistry of molecular motors. Biophys J 78: 541–556 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lau IF, Filipe SR, Soballe B, Okstad OA, Barre FX, Sherratt DJ (2003) Spatial and temporal organization of replicating Escherichia coli chromosomes. Mol Microbiol 49: 731–743 [DOI] [PubMed] [Google Scholar]
- Liu GW, Draper GC, Donachie WD (1998) FtsK is a bifunctional protein involved in cell division and chromosome localization in Escherichia coli. Mol Microbiol 29: 893–903 [DOI] [PubMed] [Google Scholar]
- Lobry JR, Louarn JM (2003) Polarisation of prokaryotic chromosomes. Curr Opin Microbiol 6: 101–108 [DOI] [PubMed] [Google Scholar]
- Massey TH, Aussel L, Barre F-X, Sherratt DJ (2004) Asymmetric activation of Xer site-specific recombination by FtsK. EMBO Rep 5: 399–404 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neuman KC, Abbondanzieri EA, Landick R, Gelles J, Block SM (2003) Ubiquitous transcriptional pausing is independent of RNA polymerase backtracking. Cell 115: 437–447 [DOI] [PubMed] [Google Scholar]
- Perals K, Cornet F, Merlet Y, Delon I, Louarn JM (2000) Functional polarization of the Escherichia coli chromosome terminus: the dif site acts in chromosome dimer resolution only when located between long stretches of opposite polarity. Mol Microbiol 36: 33–43 [DOI] [PubMed] [Google Scholar]
- Possoz C, Ribard C, Gagnat J, Pernodet JL, Guerineau M (2001) The integrative element pSAM2 from Streptomyces: kinetics and mode of conjugal transfer. Mol Microbiol 42: 159–166 [DOI] [PubMed] [Google Scholar]
- Rief M, Fernandez JM, Gaub HE (1998) Elastically coupled two-level systems as a model for biopolymer extensibility. Phys Rev Lett 81: 4764–4767 [Google Scholar]
- Salzberg SL, Salzberg AJ, Kerlavage AR, Tomb JF (1998) Skewed oligomers and origins of replication. Gene 217: 57–67 [DOI] [PubMed] [Google Scholar]
- Sharp MD, Pogliano K (2002) Rote of cell-specific SpoIIIE assembly in polarity of DNA transfer. Science 295: 137–139 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sherratt DJ (2003) Bacterial chromosome dynamics. Science 301: 780–785 [DOI] [PubMed] [Google Scholar]
- Spies M, Bianco PR, Dillingham MS, Handa N, Baskin RJ, Kowalczykowski SC (2003) A molecular throttle: the recombination hotspot chi controls DNA translocation by the RecBCD helicase. Cell 114: 647–654 [DOI] [PubMed] [Google Scholar]
- Strick TR, Allemand JF, Bensimon D, Bensimon A, Croquette V (1996) The elasticity of a single supercoiled DNA molecule. Science 271: 1835–1837 [DOI] [PubMed] [Google Scholar]
- Strick TR, Allemand JF, Bensimon D, Croquette V (1998) Behavior of supercoiled DNA. Biophys J 74: 2016–2028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Svoboda K, Mitra PP, Block SM (1994) Fluctuation analysis of motor protein movement and single enzyme-kinetics. Proc Natl Acad Sci USA 91: 11782–11786 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang LL, Lutkenhaus J (1998) FtsK is an essential cell division protein that is localized to the septum and induced as part of the SOS response. Mol Microbiol 29: 731–740 [DOI] [PubMed] [Google Scholar]
- Wang MD, Schnitzer MJ, Yin H, Landick R, Gelles J, Block SM (1998) Force and velocity measured for single molecules of RNA polymerase. Science 282: 902–907 [DOI] [PubMed] [Google Scholar]
- Yates J, Aroyo M, Sherratt DJ, Barre FX (2003) Species specificity in the activation of Xer recombination at dif by FtsK. Mol Microbiol 49: 241–249 [DOI] [PubMed] [Google Scholar]
- Yu XC, Tran AH, Sun Q, Margolin W (1998a) Localization of cell division protein FtsK to the Escherichia coli septum and identification of a potential N-terminal targeting domain. J Bacteriol 180: 1296–1304 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu XC, Weihe EK, Margolin W (1998b) Role of the C terminus of FtsK in Escherichia coli chromosome segregation. J Bacteriol 180: 6424–6428 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Data