Clinical spectrum of 4H leukodystrophy caused by POLR3A and POLR3B mutations (original) (raw)
Abstract
Objective:
To study the clinical and radiologic spectrum and genotype–phenotype correlation of 4H (hypomyelination, hypodontia, hypogonadotropic hypogonadism) leukodystrophy caused by mutations in POLR3A or POLR3B.
Methods:
We performed a multinational cross-sectional observational study of the clinical, radiologic, and molecular characteristics of 105 mutation-proven cases.
Results:
The majority of patients presented before 6 years with gross motor delay or regression. Ten percent had an onset beyond 10 years. The disease course was milder in patients with POLR3B than in patients with POLR3A mutations. Other than the typical neurologic, dental, and endocrine features, myopia was seen in almost all and short stature in 50%. Dental and hormonal findings were not invariably present. Mutations in POLR3A and POLR3B were distributed throughout the genes. Except for French Canadian patients, patients from European backgrounds were more likely to have POLR3B mutations than other populations. Most patients carried the common c.1568T>A POLR3B mutation on one allele, homozygosity for which causes a mild phenotype. Systematic MRI review revealed that the combination of hypomyelination with relative T2 hypointensity of the ventrolateral thalamus, optic radiation, globus pallidus, and dentate nucleus, cerebellar atrophy, and thinning of the corpus callosum suggests the diagnosis.
Conclusions:
4H is a well-recognizable clinical entity if all features are present. Mutations in POLR3A are associated with a more severe clinical course. MRI characteristics are helpful in addressing the diagnosis, especially if patients lack the cardinal non-neurologic features.
4H leukodystrophy (4H) (HLD7, OMIM 607694 and HLD8, OMIM 614381) is typically characterized by the triad of hypomyelination, hypodontia, and hypogonadotropic hypogonadism. It was first identified in 4 children too young for the assessment of pubertal development. Clinical hallmarks were early-onset ataxia, delayed dentition, and hypomyelination (ADDH).1 Four adult patients with milder neurologic signs and hypogonadotropic hypogonadism were described shortly thereafter,2 and it soon became clear that they all had the same entity.3 Additional signs such as severe myopia and small stature emerged, but patient numbers were too small to confirm that they are typical of 4H.3 Several years later, tremor-ataxia with central hypomyelination (TACH) was described in French Canadian patients4 and mapped to 10q22.3–10q23.31 with subsequent identification of the responsible gene, POLR3A, coding for the largest subunit of RNA polymerase III.5 Mutations in POLR3A and POLR3B, encoding another subunit of this polymerase, were then shown to also cause 4H, as well as 2 other entities: “hypomyelination with cerebellar atrophy and hypoplasia of the corpus callosum,” and “leukodystrophy with oligodontia.”6–8 The name Pol III-related leukodystrophies has been suggested in order to include all the variations described above. As the term 4H encompasses the non-neurologic symptoms and is therefore clinically helpful for the diagnosis, we chose to refer to the condition as 4H leukodystrophy in this article.
We report the clinical, molecular, and radiologic characteristics of 105 patients with mutation-proven 4H leukodystrophy.
METHODS
Patients.
A cross-sectional observational study was performed including 105 patients (table 1) with 4H leukodystrophy present in the white matter disorders database of the Centre for Childhood White Matter Disorders, VU University Medical Centre, Amsterdam, the Netherlands; the Montreal Children's Hospital of the McGill University Health Center, Canada; and the Myelin Disorders Bioregistry Project at Children's National Medical Center, Washington, DC. Clinical questionnaires were completed by the referring physician or family, supplemented by information from medical records. Thirty-six patients were seen by N.I.W. (19), A.V. (8), or G.B. (9). Some of the patients had already been published when delineating the clinical entity or describing the genetic basis of the disease (table e-1 on the _Neurology_® Web site at Neurology.org). Patients were initially recruited when noted to have hypomyelination and dental abnormalities, later on the basis of typical MRI abnormalities.
Table 1.
Demographic data on our patient cohort

Standard protocol approvals, registrations, and patient consents.
We received approval from the ethical standards committee for our research on patients with leukoencephalopathies, and written informed consent was obtained from the guardians.
Mutation analysis.
Mutation analysis of POLR3A and POLR3B was performed in Amsterdam and Montreal, except for 5 patients, who were diagnosed in clinical laboratories (4H-41, 4H-42, 4H-43, 4H-104, and 4H-105), and 2 patients (4H-37 and 4H-38) using whole exome sequencing, followed by Sanger sequencing.
MRI.
The latest available MRI was analyzed for this study. MRI was unavailable for 4 patients and in 5 patients, not all items could be scored due to insufficient quality of the MRI. MRIs were evaluated according to a previously published protocol9 by either N.I.W. or G.B. For hypomyelination, previously defined MRI criteria were applied.10 Studies typically included sagittal T1-weighted and axial T1-weighted, T2-weighted, and fluid-attenuated inversion recovery images.
Pathology.
Six-micrometer-thick formalin-fixed paraffin-embedded tissue sections of the brain of patient 4H-92, who had died of pneumonia at age 14 years, were deparaffinized and stained according to standard protocols (supplemental data).
RESULTS
Molecular data.
Of the 105 patients, 43 had mutations in POLR3A and 62 in POLR3B (table e-1 and figure e-1). Most patients of French Canadian descent carried the c.2015G>A mutation in POLR3A in a homozygous or heterozygous state. Only one patient without French Canadian descent was heterozygous for this mutation. Patients with Mediterranean ancestry mostly carried mutations in POLR3A. With the exception of French Canadian patients, patients of European descent were much more likely to have mutations in POLR3B (53/62 cases). A total of 51 of 62 patients were compound heterozygous for c.1568T>A in POLR3B. Surprisingly, only one sibling pair was homozygous for this mutation, although the carrier frequency is estimated to be 0.5%.8 The combination c.1568T>A and c.2084-6A>G was present in 5 patients, c.1568T>A and c.2570+1G>A in 4. Nonsense mutations were rare and never homozygous. In 4 families, a second mutation could not be ascertained. Six additional patients with typical clinical and radiologic features of 4H syndrome were negative for mutations in both genes and were not included in the further analysis.
Neurologic signs.
In about half of the patients (52%; 54/103), development was delayed. This was usually noted between the age of 1 and 2 years. Nineteen children (17%) were never able to walk independently (4 with POLR3A, 15 with POLR3B mutations). If unsupported walking was possible, it was usually achieved before age 2 years. There were 10/105 (10%) patients presenting late, after age 10 years. Only one patient, a 21-year-old woman diagnosed at age 16 years when she developed optic neuritis, had no cerebellar signs. Many patients had severe intention tremor and dysmetria; in others, gait ataxia was evident, whereas appendicular ataxia was mild. Abnormal smooth pursuit and gaze-evoked nystagmus were present in most patients. Limitation of vertical gaze was present in 18/91 (20%) patients for whom the information was available, and this was often an early sign. Pyramidal signs were usually absent in young children and developed slowly in older patients. Extrapyramidal signs, typically dystonia, were prominent in only a few patients. Wheelchair dependence was usually delayed and occurred from the end of the first decade (mean age of 14 years), but half of the patients (48%, 26/54) were ambulatory as adults. The main limitation to walking was ataxia. Expressive language and swallowing deteriorated over time as well.
Cognition varied widely, from normal cognitive abilities in some patients to learning difficulties or mild to moderate intellectual disability in most. Cognition usually deteriorated slowly in the second decade, but language comprehension and nonverbal communication were present until late. Epilepsy was infrequent (present in 19/99 [19%] patients for whom the information is available), and usually well-controlled with medication. Neurologic deterioration with infections was present in half (42/37, 53%), with not all children regaining their previous level. Eight patients, only one of whom carried mutations in POLR3B, died due to disease progression, the youngest at age 8 years, the oldest at age 36 years.
Non-neurologic manifestations.
Dental abnormalities were present in 88/101 patients for which the information was available (87%). Natal teeth were present in 18/94 (19%). Delayed dentition with abnormal order of deciduous tooth eruption was reported in 65/91 (71%), with the upper median incisors erupting late or not at all. Hypodontia was present in 71/98 (72%) (figure 1).
Figure 1. Typical dental abnormalities.
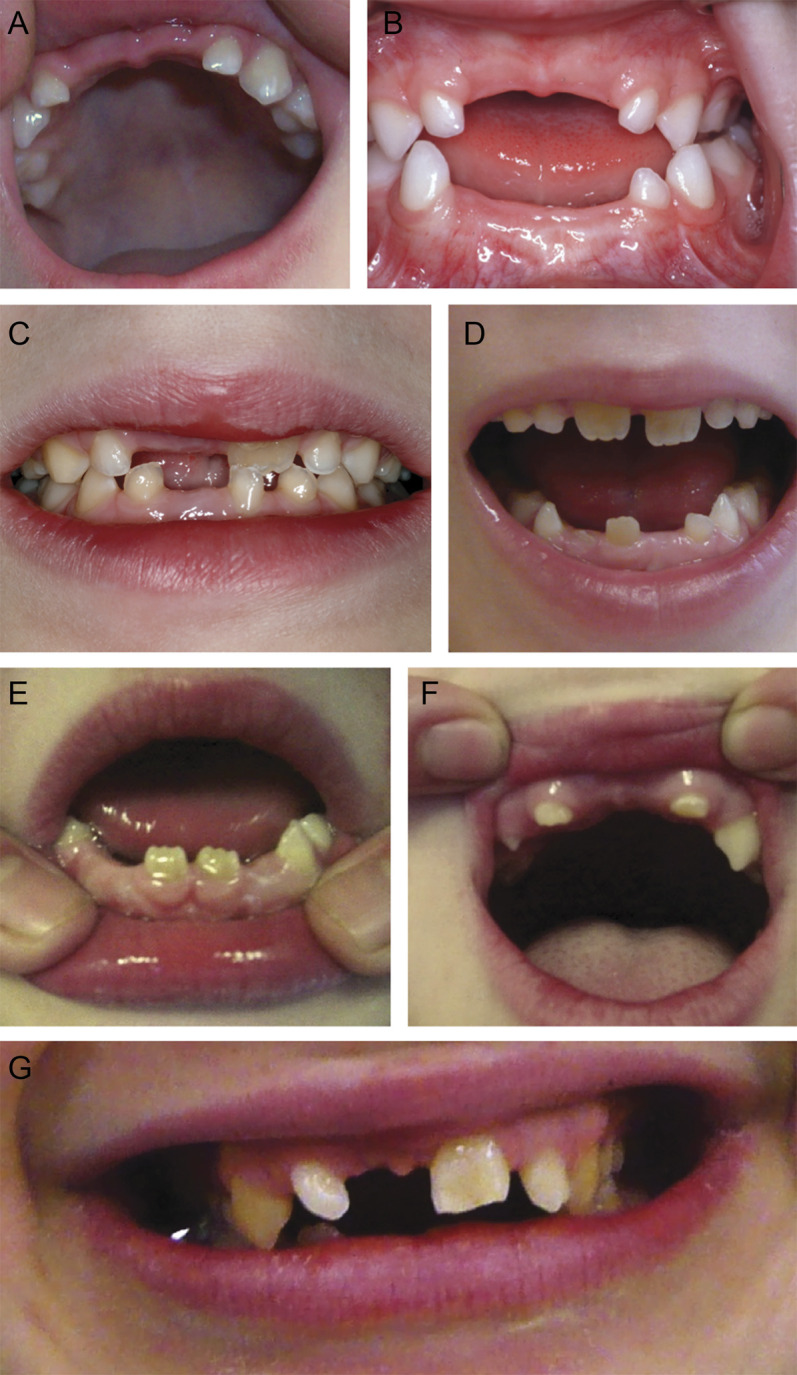
In the 2 youngest children (A: 4H-70, age 3 years; B: 4H-51, age 4 years), the most typical abnormality is the lack of the upper median deciduous incisors. In patient 4H-47 (age 7 years [C] and age 8 years [D]), the deciduous maxillary median incisors never erupted, but the permanent incisors did. In patient 4H-55, age 9 years (E, F), these teeth are still absent, and the permanent lower median incisors have only recently erupted. (G) In patient 4H-93 (age 19 years), the right upper median incisor never erupted. The right lateral median incisor is still a deciduous tooth, as is the left maxillary canine.
Delayed puberty was defined as absence of pubertal development after age 14 (girls) or 16 years (boys). Thirty-seven patients were too young to assess pubertal development. Delayed puberty or primary amenorrhea was present in 27/33 patients with POLR3A (81%) and 20/29 patients (69%) with POLR3B mutations.
The majority of patients (73/84, 87%) had myopia, often pronounced, and usually progressive. Optic atrophy was present in older patients and is probably more frequent than reported here, as not all patients were assessed for it. Cataracts were seen in only 3 patients, including 1 sibling pair. About half of the patients had short stature (47/91, 51%); this was slightly more frequent in children with onset before 3 years. Growth hormone deficiency was confirmed in 5 of 10 patients tested and might be more common, as many patients were not investigated for this. Several patients were tested for hypothyroidism with normal results. Three patients had mild osteopetrosis detected by chance on routine hip X-rays.11 In one of these patients, hip X-ray results had normalized at age 18 years.
MRI findings.
Hypomyelination was invariably present with myelination of the optic radiation seen in almost all patients (93/97; 95%). A small T2 hypointense dot in the posterior limb of the internal capsule was seen in 42/60 (70%) patients with POLR3B, but only 5/37 (13%) patients with POLR3A mutations. Relative T2 hypointensities (relative myelin preservation) of the ventrolateral thalamus (88/97; 91%) and the dentate nucleus (90/97; 93%) were also characteristic (figure 2). Supratentorial atrophy was invariably present in the adult patients but rarely seen before age 10 years. A thin corpus callosum was present in all patients examined beyond 17 years, reflecting generalized atrophy and white matter loss. In children younger than 10 years, thinning of the corpus callosum was more frequent with POLR3A (10/12) than POLR3B (14/26) mutations. In 8 patients, a small cyst was seen in the splenium. Cerebellar atrophy was present in all POLR3B patients except for one 3-year-old girl; it was absent in 8 patients with mutations in POLR3A (2–12 years at MRI) (figure e-2).
Figure 2. MRI changes with age.

MRI of 4 patients with 4H syndrome at 6 (A–D, patient 4H-52, row 1), 12 (E–H, patient 4H-53, row 2), 23 (I–L, patient 4H-58, row 3), and 40 (M–P, patient 4H-94, row 4) years of age. The axial T2-weighted images all show diffusely elevated white matter signal, less hyperintense than CSF, consistent with hypomyelination. The signal of the optic radiations is hypointense, indicating myelination. In the youngest patient, there is a small hypointense dot in the posterior limb of the internal capsule (B). All patients show a relatively hypointense signal of the ventrolateral thalamus. The corpus callosum becomes thinner with age (sagittal T1 images, A, L, P, and sagittal T2 image, H). The sagittal images also demonstrate cerebellar atrophy. White matter signal of the middle cerebellar peduncles and the cerebellar white matter is too high on the T2-weighted images; the dentate nucleus appears hypointense as well as the dorsal tegmentum (C, G, K, O). (Q–T) MRI of patient 4H-45 homozygous for c.1586T>A (POLR3B) at age 16 years. Myelination of the perirolandic cortex and the parieto-occipital white matter is relatively good on these axial T2-weighted images, compared to the frontal white matter (Q, R, S). The splenium and the anterior limb of the internal capsule (R) as well as the optic radiations (S) are well-myelinated. There is a small lesion in the right optic radiation (R). No cerebellar atrophy is seen on the sagittal T1-weighted image (T).
Relation between genotype and phenotype.
Patients with POLR3A had, in general, more severe disease than patients with POLR3B mutations, despite the fact that disease onset was slightly earlier in the latter group (4.3 years vs 3.4 years). This is reflected in both age at loss of supported walking and survival (figure 3).
Figure 3. Disease progression.
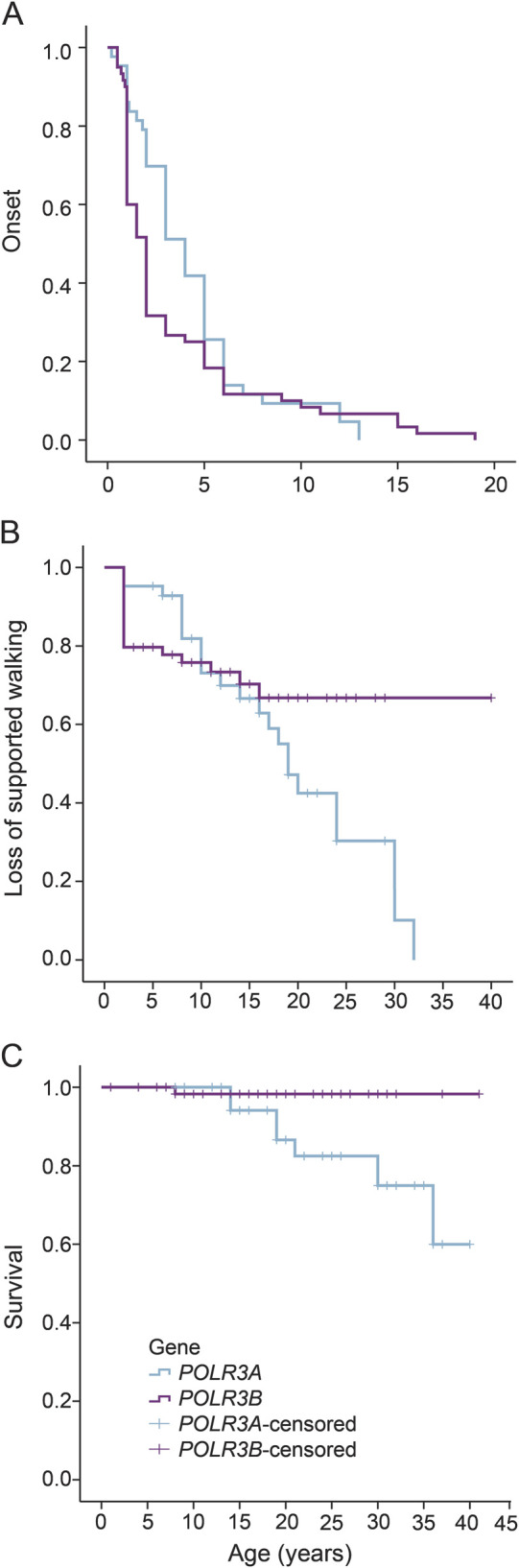
Cumulative probability of age at onset, age at wheelchair dependence, and age at death in our patients' cohort. (A) Age at onset of the patients according to the mutated gene. Patients with POLR3B mutations have an earlier disease onset than patients with POLR3A mutations. Cumulative probability of (B) age at wheelchair dependence and (C) death in our patients' cohort.
In French Canadian patients with homozygosity for c.2015G>A in POLR3A, age at onset was usually in infancy or early childhood, except for one patient with a milder disease course and onset at 5 years. There were several patients with a similar combination of POLR3B mutations: c.1568T>A with c.2084-6A>G and c.1568T>A with c.2570+1 G>A. One patient compound heterozygous for c.2084-6A>G and c.1568T>A presented late (at 15 years with delayed puberty), while the presentation of the other patients with this combination was early. The 2 siblings homozygous for c.1568T>A had an exceptionally mild clinical course, the older of the 2 having no neurologic signs at age 21 years. On brain MRI, myelination was much better in the latter 2 patients, especially in the parieto-occipital white matter, than in the other patients (figure 2, Q–S). Only 1 out of 12 sibpairs showed significant intrafamilial variability, with a very mildly affected and a severely affected sibling.
Pathology.
Gross examination of the brain of a 14-year-old girl (4H-92; homozygous for c.1797 G>C/p.Gln599His in POLR3A) showed diffuse white matter atrophy with thin corpus callosum and enlarged lateral ventricles. The white matter was nonhomogeneously discolored, also involving U-fibers, commissures, and capsules. Histologic analysis revealed variable white matter rarefaction, lack of myelin, and reduced numbers of oligodendrocytes with better preservation of the perivascular myelin in the centrum semiovale (figure e-3). The degree of axonal loss was proportional to the myelin involvement. Foamy macrophages were seen clustering around small blood vessels. Microglial activation was mild. Neurons showed neither mineralization in the cortex (as seen in another patient12) nor iron accumulation in the globus pallidus. The cerebellar folia were mildly atrophic with some loss of granular neurons and, in places, of Purkinje cells. There was nonhomogeneous lack of myelin in the deeper cerebellar white matter, with relative sparing of the hilus of the dentate nucleus.
Analysis of the sural nerve of the same patient showed mild lack of myelin. The ovaries appeared normal apart from a slightly reduced number of oocytes. Immunohistochemical analysis of the pituitary gland for follicle-stimulating hormone, luteinizing hormone, growth hormone, and prolactin showed no abnormalities.
DISCUSSION
From our dataset concerning a cohort of 105 mutation-proven cases, it is evident that 4H is an insidiously progressive disorder with declining motor function due to increasing ataxia, sometimes with episodes of faster deterioration triggered by minor infections. Albeit not formally evaluated, cognition remains relatively spared until late. 4H leukodystrophy comes with a spectrum of disease severity. At the severe end, affected children do not achieve independent walking and have mild to moderate intellectual disability. At the milder end of the spectrum, patients present after age 5 years with learning difficulties and motor clumsiness. Exceptionally, patients become symptomatic only in late adolescence or early adulthood. It is likely that this late-onset group represents an underestimation, since clinical presentation of this age group is mild, with one family even diagnosed by chance.
In general, patients with POLR3A mutations are more severely affected than patients with POLR3B mutations with faster regression and shorter life expectancy. Strikingly, the disease starts slightly later in _POLR3A_-mutated patients and, in contrast to _POLR3B_-mutated patients, most _POLR3A_-mutated patients achieve independent walking. Homozygosity for the common c.1568T>A POLR3B mutation, which we found much less often than expected given the estimated carrier frequency,8 causes a mild clinical presentation. Considerable intrafamilial variability is rare, seen in only 1 of the 12 sibling pairs. All these findings suggest that there is a genotype–phenotype correlation, but due to the numerous private mutations we could not confirm such a relationship.
The characteristic dental abnormalities—natal teeth, delayed dentition, abnormal order of teeth eruption, hypodontia—are present in two-thirds of the patients and as such are not obligatory for the diagnosis. The same is true for hypogonadotropic hypogonadism, which is absent in a quarter of the patients. When all features are present, the likelihood for finding mutations in either POLR3A or POLR3B is high, although the small number of mutation-negative cases opens the possibility of additional responsible genes. As our study initially only included patients with all features present, the proportion of patients with neither abnormal puberty nor dental abnormalities might be higher. Our study has made clear that the disturbance of pubertal development itself is also variable. Several female patients had normal breast development but primary amenorrhea. One patient did not respond to pulsatile GnRH, suggesting that delayed puberty might be secondary to a defect in the GnRH receptor or in gonadotropin bioactivity.13 Endocrine abnormalities not only include hypogonadotropic hypogonadism, but in a small number of patients also growth hormone deficiency. The latter might be more common as the majority of patients, even in the large group with short stature, were never evaluated for growth hormone deficiency. Almost all patients have myopia, usually progressive and severe.
An important question is how to reach a diagnosis in the absence of dental and endocrine abnormalities. Brain MRI pattern recognition proves an excellent means to facilitate this. Hypomyelination in combination with cerebellar atrophy, relative T2 hypointensity of the ventrolateral thalamus, and myelination of the pyramidal tracts within the posterior limb of the internal capsule, dentate nuclei, and the optic radiation are present in most patients and suggest the diagnosis.10,14 The corpus callosum is usually normal in young children, but invariably thin in adults, reflecting slowly progressive cerebral white matter volume loss. Interestingly, although it was considered as one of the essential radiologic findings,10 cerebellar atrophy is absent or mild in a substantial number of patients with POLR3A mutations, confirming the results from a previous small study on 6 patients.15
We are still far from understanding the pathogenesis of 4H leukodystrophy. The combination of hypomyelination with the peculiar dental anomalies remains enigmatic. As a few patients show signs of osteosclerosis,11 we hypothesize that a defect of osteoclasts is at the basis of the dentition abnormalities by preventing normal tooth eruption, which necessitates local bone resorption. Regarding the white matter abnormalities, it emerges that defects of proteins essential for RNA synthesis—RNA polymerase III in 4H leukodystrophy, cytoplasmic aspartyl tRNA synthetase in hypomyelination with brainstem involvement and leg spasticity16—cause hypomyelination, but the underlying mechanism is unresolved. Which cell type is primarily affected in 4H leukodystrophy, the oligodendrocytes providing the myelin sheath or the neurons whose axonal integrity is essential to induce myelination, remains unclear. Histopathology results with widespread axonal damage and, in the cerebellum, Purkinje cell loss and axonal and dendritic swellings, point to an important neuroaxonal involvement. However, all end-stage brain disorders, including white matter disorders, involve neurons without necessarily providing clues for the initial problem. This is true even for the prototypic oligodendrocyte disorder Pelizaeus-Merzbacher disease, in which the gene coding for proteolipid protein 1 is mutated, and where neuroaxonal degeneration is a feature of late disease.17 The fact that, in 4H leukodystrophy, clinical presentation is comparatively mild in many patients during the first years of life and the subsequent disease progression slow opens the possibility of intervention once its pathogenesis has been elucidated.
Supplementary Material
Data Supplement
Accompanying Editorial
ACKNOWLEDGMENT
The authors thank the patients and families; the Genome Quebec Innovation Center and McGill University; and Tiny Uithuisje, Jolanda Keek, Marian Muijs, Eline van der Vooren, Carola van Berkel, and Truus E.M. Abbink for help with sequencing.
GLOSSARY
4H
4H leukodystrophy
ADDH
ataxia, delayed dentition, and hypomyelination
TACH
tremor-ataxia with central hypomyelination
Footnotes
AUTHOR CONTRIBUTIONS
Nicole I. Wolf: designed and supervised the study, collected data, wrote the first draft of the article. Adeline Vanderver: collected patient and MRI data, reviewed the article. Rosalina M.L. van Spaendonk: responsible for molecular genetic analysis of a large part of the patients, reviewed the article. Raphael Schiffmann: collected patient and MRI data, reviewed the article. Bernard Brais: collected patient and MRI data, reviewed the article. Marianna Bugiani: performed additional histopathologic stainings, wrote part of the article. Erik Sistermans: collected mutation data, reviewed the article. Coriene Catsman-Berrevoets: collected patient and MRI data, reviewed the article. Johan M Kros: performed the autopsy on one patient, reviewed the article. Pedro Soares Pinto: collected patient and MRI data, reviewed the article. Daniela Pohl: collected patient and MRI data, reviewed the article. Sandya Tirupathi: collected patient and MRI data, reviewed the article. Petter Strømme: collected patient and MRI data, reviewed the article. Ton de Grauw: collected patient and MRI data, reviewed the article. Sébastien Fribourg: analyzed molecular data. Michelle Demos: collected patient and MRI data, reviewed the article. Amy Pizzino: collected patient and MRI data, reviewed the article. Sakkubai Naidu: collected patient and MRI data, reviewed the article. Kether Guerrero: responsible for molecular genetic analysis of a large part of the patients, reviewed the article. Marjo S van der Knaap: collected patient and MRI data, reviewed the article. Geneviève Bernard: designed and supervised the study, collected data, reviewed the article.
STUDY FUNDING
Supported by the Canadian Institute of Health Research (MOP-G-287547). Dr. Bernard has received a Research Scholar Junior 1 of the Fonds de Recherche du Québec en Santé (FRQS). Also supported by National High Technology Research and Development Program of China—863 Program (2012AA02A201), the Guangdong Enterprise Key Laboratory of Human Disease Genomics, National Gene Bank Project of China, Shenzhen Key Laboratory of Transomics Biotechnologies (CXB201108250096A), Shenzhen Engineering Laboratory for Clinical Molecular Diagnostics, and The Children's Hospital of Philadelphia Research Institute.
DISCLOSURE
N. Wolf serves as communicating editor of the Journal of Inherited Metabolic Disease and as editor for Neuropediatrics, both without payment. A. Vanderver has acted on an advisory board for Shire Pharmaceuticals as well as an unpaid consultant for Stem Cells Inc. R. van Spaendonk reports no disclosures relevant to the manuscript. R. Schiffmann has received honoraria and grant support from Shire and Amicus Therapeutics. B. Brais, M. Bugiani, E. Sistermans, C. Catsman-Berrevoets, J. Kros, and P.S. Pinto report no disclosures relevant to the manuscript. D. Pohl has received compensation from Biogen Idec, Merck Serono, Bayer Schering, Teva, and Sanofi-Aventis (speaker's honoraria and for serving on scientific advisory boards). S. Tirupathi, P. Strømme, T. de Grauw, S. Fribourg, M. Demos, A. Pizzino, S. Naidu, K. Guerrero, and M. van der Knaap report no disclosures relevant to the manuscript. G. Bernard has received compensation from Actelion Pharmaceuticals, Genzyme, Shire, and Santhera Pharmaceuticals (speaker's honoraria and for serving on scientific advisory boards). Go to Neurology.org for full disclosures.
REFERENCES
- 1.Wolf NI, Harting I, Boltshauser E, et al. Leukoencephalopathy with ataxia, hypodontia, and hypomyelination. Neurology 2005;64:1461–1464. [DOI] [PubMed] [Google Scholar]
- 2.Timmons M, Tsokos M, Asab MA, et al. Peripheral and central hypomyelination with hypogonadotropic hypogonadism and hypodontia. Neurology 2006;67:2066–2069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wolf NI, Harting I, Innes AM, et al. Ataxia, delayed dentition and hypomyelination: a novel leukoencephalopathy. Neuropediatrics 2007;38:64–70. [DOI] [PubMed] [Google Scholar]
- 4.Bernard G, Thiffault I, Tetreault M, et al. Tremor-ataxia with central hypomyelination (TACH) leukodystrophy maps to chromosome 10q22.3-10q23.31. Neurogenetics 2010;11:457–464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bernard G, Chouery E, Putorti ML, et al. Mutations of POLR3A encoding a catalytic subunit of RNA polymerase pol III cause a recessive hypomyelinating leukodystrophy. Am J Hum Genet 2011;89:415–423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Tetreault M, Choquet K, Orcesi S, et al. Recessive mutations in POLR3B, encoding the second largest subunit of pol III, cause a rare hypomyelinating leukodystrophy. Am J Hum Genet 2011;89:652–655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Saitsu H, Osaka H, Sasaki M, et al. Mutations in POLR3A and POLR3B encoding RNA polymerase III subunits cause an autosomal-recessive hypomyelinating leukoencephalopathy. Am J Hum Genet 2011;89:644–651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Daoud H, Tetreault M, Gibson W, et al. Mutations in POLR3A and POLR3B are a major cause of hypomyelinating leukodystrophies with or without dental abnormalities and/or hypogonadotropic hypogonadism. J Med Genet 2013;50:194–197. [DOI] [PubMed] [Google Scholar]
- 9.van der Knaap MS, Breiter SN, Naidu S, Hart AA, Valk J. Defining and categorizing leukoencephalopathies of unknown origin: MR imaging approach. Radiology 1999;213:121–133. [DOI] [PubMed] [Google Scholar]
- 10.Steenweg ME, Vanderver A, Blaser S, et al. Magnetic resonance imaging pattern recognition in hypomyelinating disorders. Brain 2010;133:2971–2982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ozgen HM, Overweg-Plandsoen WC, Blees-Pelk J, Besselaar PP, Hennekam RC. Cerebellar hypoplasia-endosteal sclerosis: a long term follow-up. Am J Med Genet A 2005;134A:215–219. [DOI] [PubMed] [Google Scholar]
- 12.Vanderver A, Tonduti D, Bernard G, et al. More than hypomyelination in Pol-III disorder. J Neuropathol Exp Neurol 2013;72:67–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Billington E, Bernard G, Gibson W, Corenblum B. What is 4H syndrome and how should the endocrinologist manage it? Presented at the Endocrine Society annual meeting; June 15–18, 2013; San Francisco, CA. 2013499.
- 14.Piana RL, Tonduti D, Dressman HG, et al. Brain magnetic resonance imaging (MRI) pattern recognition in pol III-related leukodystrophies. J Child Neurol 2014;29:214–220. [DOI] [PubMed] [Google Scholar]
- 15.Takanashi J, Osaka H, Saitsu H, et al. Different patterns of cerebellar abnormality and hypomyelination between POLR3A and POLR3B mutations. Brain Dev 2014;36:259–263. [DOI] [PubMed] [Google Scholar]
- 16.Simons C, Wolf NI, McNeil N, et al. A de novo mutation in the beta-tubulin gene TUBB4A results in the leukoencephalopathy hypomyelination with atrophy of the basal ganglia and cerebellum. Am J Hum Genet 2013;92:767–773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Garbern JY, Yool DA, Moore GJ, et al. Patients lacking the major CNS myelin protein, proteolipid protein 1, develop length-dependent axonal degeneration in the absence of demyelination and inflammation. Brain 2002;125:551–561. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Supplement
Accompanying Editorial