Systemic Administration of a Recombinant AAV1 Vector Encoding IGF-1 Improves Disease Manifestations in SMA Mice (original) (raw)
Abstract
Spinal muscular atrophy is a progressive motor neuron disease caused by a deficiency of survival motor neuron. In this study, we evaluated the efficacy of intravenous administration of a recombinant adeno-associated virus (AAV1) vector encoding human insulin-like growth factor-1 (IGF-1) in a severe mouse model of spinal muscular atrophy. Measurable quantities of human IGF-1 transcripts and protein were detected in the liver (up to 3 months postinjection) and in the serum indicating that IGF-1 was secreted from the liver into systemic circulation. Spinal muscular atrophy mice administered AAV1-IGF-1 on postnatal day 1 exhibited a lower extent of motor neuron degeneration, cardiac and muscle atrophy as well as a greater extent of innervation at the neuromuscular junctions compared to untreated controls at day 8 posttreatment. Importantly, treatment with AAV1-IGF-1 prolonged the animals' lifespan, increased their body weights and improved their motor coordination. Quantitative polymerase chain reaction and western blot analyses showed that AAV1-mediated expression of IGF-1 led to an increase in survival motor neuron transcript and protein levels in the spinal cord, brain, muscles, and heart. These data indicate that systemically delivered AAV1-IGF-1 can correct several of the biochemical and behavioral deficits in spinal muscular atrophy mice through increasing tissue levels of survival motor neuron.
Introduction
Spinal muscular atrophy (SMA) is characterized by progressive degeneration of spinal cord motor neurons, resultant muscular atrophy, paralysis, and an attenuated lifespan. Childhood SMA exhibits an autosomal recessive pattern of inheritance with an incidence of ~1 in 6,000–10,000 newborns.1 SMA patients can harbor deletions, mutations, or conversions of the telomeric copy of the survival of motor neuron gene (SMN1). A centromeric SMN gene (SMN2) is present in all SMA patients but is unable to compensate for the gene defect in SMN1 as the primary transcript of the SMN2 gene is defectively spliced.2,3 Currently, there are no therapies for SMA patients that are disease modifying.
Insulin-like growth factor-1 (IGF-1) is a pleiotropic hormone that modulates multiple fundamental cellular processes, such as cellular growth, proliferation, differentiation, and survival.4 IGF-1 is also reportedly necessary for normal brain development and function and enhancing axonal outgrowth of corticospinal motor neurons.5 It therefore represents an attractive therapeutic entity for consideration to treat motor neuron diseases via its neurotrophic effects.6 Indeed, IGF-1 treatment has been shown to improve the disease manifestations in rodent models of amyotrophic lateral sclerosis and spinal and bulbar muscular atrophy;7,8 however, the therapeutic effects of IGF-1 on SMA were modest.9,10,11,12 Plasmid DNA-mediated expression of IGF-1 in skeletal muscle or following intracerebroventricular delivery into SMA mice provided a modest prolongation of median survival but no measurable improvements in motor function.9,10 Similarly, subcutaneous administration of recombinant human IGF-1 into SMA mice reduced the extent of degeneration of motor neurons and improved motor function but did not improve survival or body weight gain.11 In addition, treatment with somatropin, which leads to the release of IGF-1 in the liver and muscles, did not improve muscle strength or function in patients with type II/III SMA.13 Therefore, the utility of IGF-1 as a therapeutic for SMA is equivocal.
As the disease phenotype of SMA patients is diverse (e.g., includes muscular dystrophy and cardiac atrophy in addition to the motor neuron disease), systemic therapy may be necessary to fully alleviate disease progression.14,15,16 An approach to achieve widespread therapy (such as with a secretory factor like IGF-1) is by intravenous injection of a recombinant adeno-associated viral (AAV) vector encoding IGF-1.17 Here, we injected an AAV serotype-1 (AAV1) vector encoding human IGF-1 (AAV1-IGF-1) into the facial vein of a severe mouse model of SMA. Associated with the subsequent hepatic transduction and secretion of IGF-1 into the serum was a significant reduction in motor neuron cell death, muscle degeneration, and cardiac atrophy. Treated SMA mice also exhibited an increased lifespan and improved motor function. We showed that these salutary effects of IGF-1 treatment were likely the result of upregulation of SMN expression.
Results
Intravenous injection of AAV1-IGF-1 into SMA mice resulted in hepatic expression of IGF-1 and secretion into the serum
Intravenous injection of Evans blue dye into the facial vein of wild-type mice (on postnatal day 1 (P1)) resulted in a dose-dependent increase in blue coloration throughout the whole body of the animals after 10 minutes (Figure 1a). Intraperitoneal (i.p.) or subcutaneous injection of Evans blue dye showed a lesser extent and pattern of blue coloration in mice; hence all subsequent studies were by intravenous delivery (Supplementary Figure S1). Following validation of the technique, we next injected AAV1-IGF-1 into the facial vein of P1 heterozygous (Smn +/− SMN2 +/−) and homozygous SMA (Smn −/− SMN2 +/−) mice. Analysis of the injected mice at P7 by reverse transcription polymerase chain reaction (RT-PCR) showed the presence of human IGF-1 transcripts in the liver and heart, but not in the spleen, kidney, brain, spinal cord, and skeletal muscle (Figure 1b). Western blotting and immunostaining confirmed the presence of human IGF-1 protein in tissue lysates and sections from the liver but not the heart (Figure 1c,d). Transcripts of human IGF-1 were detected in the liver as early as 2 days postinjection and that was sustained for 3 months (Figure 1e and Supplementary Figure S2). ELISA analysis of serum from heterozygous mice also showed the presence of human IGF-1 at 3 months posttreatment albeit at lower levels than at 1 month (Figure 1f).
Figure 1.

Expression of human IGF-1 following intravenous administration of adeno-associated virus vector encoding human insulin-like growth factor-1 (AAV1-IGF-1) via the facial vein of mice. (a) Intravenous injection of Evans blue via the facial vein into postnatal day 1 (P1) mice and analyzed at 10 minutes postinjection. (b) RT-PCR analysis of human IGF-1 transcripts in various tissues of heterozygous (Smn _+/−_SMN2 _+/_−) and SMA (Smn_−/−_SMN2 _+/_−) mice at 7 days after intravenous administration of AAV1-IGF-1 (administered on P1). (c) Western blotting for human IGF-1 protein in the liver and heart of heterozygous and SMA mice at 7 days posttreatment. (d) Immunostaining for human IGF-1 (green) in the liver and heart of SMA mice at 7 days after intravenous administration of AAV1-IGF-1. Samples from PBS-treated mice were used as controls. (e) RT-PCR analysis of human IGF-1 transcripts in livers of heterozygous mice at days 7, 30, and 90 after AAV1-IGF-1 treatment. (f) ELISA analysis of serum from heterozygous mice on days 30 and 90 after AAV1-IGF-1 treatment for human IGF-1 protein concentration. B, brain; H, heart; K, kidney ; L, liver; M, muscle; Sp, spleen; Sc, spinal cord; -, no treatment control. Bar = 100 μm; values, mean ± SEM. (*P < 0.05; Wilcoxon ranksum test; n = 6 in each group). PBS, phosphate-buffered saline.
AAV1-IGF-1 treatment attenuated the loss of spinal cord motor neurons in SMA mice
To examine the therapeutic effects of systemically delivered AAV1-IGF-1, spinal cord motor neuron cell counts were performed. SMA mice (n = 5) treated at P1 and analyzed 7 days later showed a significantly higher number of motor neuron cell counts (as visualized by choline acetyltransferase (CHAT) staining) in the lumbar spinal cord than in untreated SMA mice (P = 0.03; Figure 2a). Hematoxylin and eosin staining of spinal cord sections showed untreated SMA mice had significantly lower numbers of motor neurons with large (>600 μm2) and mid-size (400–600 μm2) cross-sectional areas than heterozygous mice (Figure 2b). Treatment of SMA mice with AAV1-IGF-1 increased the number of the large-sized motor neurons over that seen in untreated controls (P = 0.02; Figure 2b).
Figure 2.
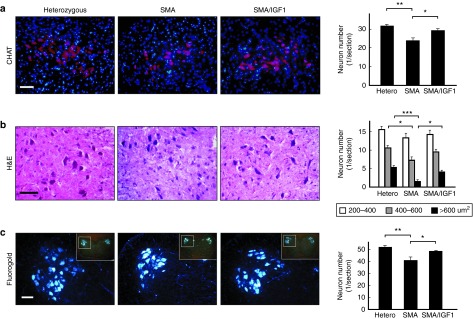
The effects of systemic administration of adeno-associated virus vector encoding human insulin-like growth factor-1 (AAV1-IGF-1) at postnatal day (P1) on the spinal cord motor neurons in spinal muscular atrophy (SMA) mice. (a) Immunostaining with the motor neuron marker CHAT (red), and counter-staining with the nuclear marker 4′,6-diamidino-2-phenylindole (blue), showed the motor neurons in the anterior (ventral) horn of lumbar spinal cord in untreated heterozygous, untreated SMA, and AAV1-IGF-1-treated SMA mice on P8. (b) H&E staining showed the number of neurons according to their cell size: 200–400, 400–600 and >600 μm2, respectively. (c) In retrograde tracing experiments by intragastrocnemius and intraquadriceps injections of a fluorogold neurotracing agent, the number of fluorescent-labeled motor neurons was counted. Bars= 100 μm; values, mean ± SEM; n = 5 in each group. (*P < 0.05; **P ≤ 0.01; ***P ≤ 0.001; one-way analysis of variance with LSD posthoc comparisons). CHAT, choline acetyltransferase; H&E, hematoxylin and eosin; LSD, least significant difference.
Retrograde tracing experiments by gastrocnemius and quadriceps injections of a fluorogold neurotracing agent were used to examine if motor neurons harbored axons that innervated the hindlimb muscles. A positive fluorescent signal in the spinal anterior horn is indicative of motor neurons harboring axons that innervated the limb muscles. The number of motor neurons positive for the fluorogold tracer in the lumbar spinal cord was significantly higher in AAV1-IGF-1-treated than in untreated SMA mice (P = 0.04; Figure 2c), implying that treatment had not only reduced motor neuron degeneration but also rescued neurons harbored axons that innervated the limb muscles.
SMA mice treated with AAV1-IGF-1 prevented muscle atrophy and exhibited greater preservation of the architecture at the neuromuscular junction (NMJ)
The effect of IGF-1 on muscle atrophy and the architecture of the neuromuscular junctions were also examined. Analysis (on day 7) of the cross-sectional area of individual myofibers in the quadriceps of SMA mice (n = 5) treated with AAV1-IGF-1 (at P1) were larger compared to untreated controls (Figure 3a). Histochemical staining of the quadriceps (n = 5) for neurofilament H (axonal marker) and α-bungarotoxin (neuromuscular junction marker) showed significantly less denervated neuromuscular junctions in the AAV1-IGF-1-treated SMA mice compared to control SMA mice (P = 0.02) (Figure 3b). Furthermore, the thickness of the diaphragm of AAV1-IGF-1-treated SMA mice was larger than in untreated SMA mice with or without normalization of the thickness to the body weight (P < 0.01) (Figure 3c).
Figure 3.
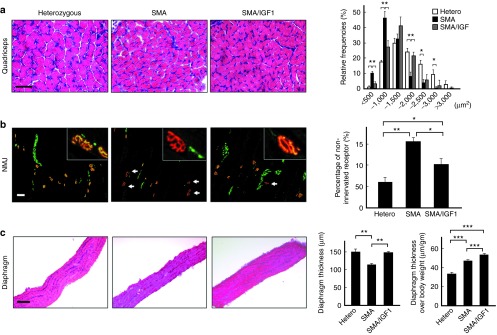
The effects of systemic administration of adeno-associated virus vector encoding human insulin-like growth factor-1 (AAV1-IGF-1) at P1 on the muscle fibers, neuromuscular junction (NMJ), and diaphragm in spinal muscular atrophy (SMA) mice. (a) The frequency distribution of muscle fiber size in quadriceps muscles using H&E stain in untreated heterozygous, untreated SMA, and AAV1-IGF-1-treated SMA mice on postnatal day 8. (b) Immunostaining with the axonal marker neurofilament H (green) and counter-staining with the NMJ marker α-bungarotoxin (red) in the hamstring muscles of SMA mice. The arrow indicates denervated NMJ. (c) H&E staining of the diaphragm for measurement of muscle thickness. Bars= 100 μm; values, mean ± SEM; n = 5 in each group. (*P < 0.05; **P ≤ 0.01; one-way analysis of variance with LSD posthoc comparisons). LSD, least significant difference.
AAV1-IGF-1 treatment abated the development of cardiac atrophy in SMA mice
As SMA mice exhibit cardiac atrophy, we examined if IGF-1 treatment also addressed this abnormality.15 As expected, the hearts of untreated SMA mice were smaller than their heterozygous counterparts. Treatment of SMA mice with AAV1-IGF-1 partially reduced the extent of cardiac atrophy (Figure 4a,b). This was illustrated by the finding that the weights of hearts from SMA mice treated with AAV1-IGF-1 were significantly higher than from untreated controls after normalization of the heart weight to the body weight (P = 0.01) (Figure 4c). However, they were still smaller than those from heterozygous mice (P < 0.001). Hematoxylin and eosin staining of heart tissues from untreated SMA mice showed significantly lesser thickness of the left ventricular wall and interventricular septum compared to the heterozygous mice (P < 0.001) (Figure 4d). AAV1-IGF-1 treatment significantly increased the thickness of the left ventricular wall (P = 0.004) and interventricular septum with or without normalization of the thickness to the body weight (P < 0.001) (Figure 4d).
Figure 4.
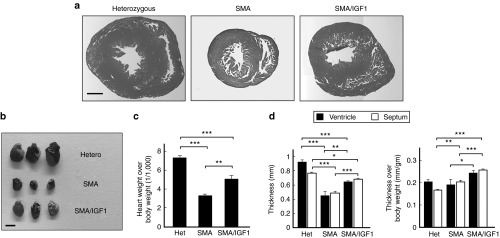
The effects of systemic administration of adeno-associated virus vector encoding human insulin-like growth factor-1 (AAV1-IGF-1) at postnatal day 1 (P1) on the heart in spinal muscular atrophy (SMA) mice. (a) Cross-section of the heart following H&E staining, (b) pictures of the heart, (c) the heart weight, and (d) the thickness of the left ventricular wall and interventricular septum in untreated heterozygous, untreated SMA, and AAV1-IGF-1-treated SMA mice on P8. Bars = 500 μm as in a and 2 mm as in b; values, mean ± SEM; at least n = 6 as in c and n = 5 as in d in each group. (*P < 0.05; **P ≤ 0.01; ***P ≤ 0.001; one-way analysis of variance with LSD posthoc comparisons). LSD, least significant difference.
AAV1-IGF-1 treatment improved motor function and prolonged the lifespan of SMA mice
We next determined if AAV1-IGF-1 treatment impacted the body weight, motor function, and lifespan of the SMA mice. Both untreated (n = 14) and phosphate-buffered saline (PBS)-treated SMA mice (n = 16) exhibited a median lifespan of 9 days. Interestingly, SMA mice administered AAV1-IGF-1 exhibited a median lifespan of 12 days (n = 24) which represents a 33% increase relative to controls (P < 0.001) (Figure 5a). Similarly, treatment of SMA mice with AAV1-IGF-1 provided a modest increase in weight compared to untreated and PBS-treated controls at P8 and P11 (P < 0.05) (Figure 5b,c). There was no significant difference in body weight gain between heterozygous mice with and without AAV1-IGF-1 treatment.
Figure 5.
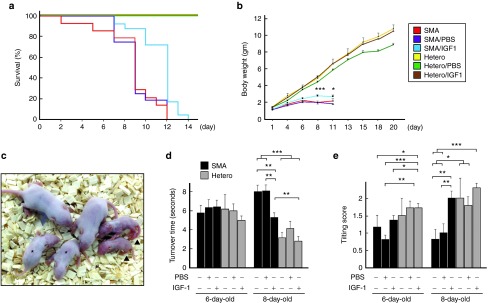
The effects of systemic administration of adeno-associated virus vector encoding human insulin-like growth factor-1 (AAV1-IGF-1) at postnatal day 1 (P1) on the survival, fitness, and motor function in spinal muscular atrophy (SMA) mice. (a) lifespan, (b) body weight, (c) size of mice on P8, (d) turnover time, and (e) the tilting score in untreated SMA (n = 14), phosphate-buffered saline (PBS)-treated SMA (SMA/PBS, n = 16), AAV1-IGF-1-treated SMA (SMA/IGF, n = 24), untreated heterozygous (n = 6), PBS-treated heterozygous (Hetero/PBS, n = 16), and AAV1-IGF-1-treated heterozygous (Hetero/IGF, n = 35) mice. Asterisk and arrowhead in c indicate AAV1-IGF-1-treated SMA mice and untreated SMA mice, respectively. (a) P < 0.001; log-rank test. (b) *P < 0.05 and ***P ≤ 0.001, AAV1-IGF-1-treated SMA mice versus untreated and PBS-treated SMA mice; one-way analysis of variance (ANOVA) with LSD posthoc comparisons. (d,e) *P < 0.05; **P ≤ 0.01; ***P ≤ 0.001; one-way ANOVA with LSD posthoc comparisons; LSD, least significant difference.
Assessment of motor function using the turnover test to investigate the righting reflex showed no difference in performance between groups on P6 (Figure 5d). However, on P8, untreated and PBS-treated SMA mice exhibited a longer latency in their turnover time than heterozygous mice (P < 0.001). Treatment with AAV1-IGF-1 corrected this aberration (P < 0.01). Measurement of the tilting scores of untreated and PBS-treated SMA mice on P6 and P8 indicated they were lower than their age-matched heterozygous counterparts (Figure 5e). Again, treatment of SMA mice with AAV1-IGF-1 increased their tilting scores on P8 (P < 0.01) to those noted in heterozygous mice.
Intravenous administration of AAV1-IGF-1 increased tissue levels of SMN protein
Measurement of tissue levels of SMN protein in SMA mice (n = 4 in each group) administered AAV1-IGF-1 (at P1) on day 7 showed they were ~twofold higher in the spinal cord, brain, muscle, and heart (Figure 6a–d) than in untreated controls (P < 0.05). However, the absolute levels were still significantly lower than those noted in heterozygous mice (P = 0.003).
Figure 6.

Levels of survival motor neuron (SMN) in untreated heterozygous (H), untreated spinal muscular atrophy (S−), and AAV1-IGF-1-treated SMA (S+) mice on postnatal day 8 (P8). AAV1-IGF-1 was injected via the facial vein into SMA mice at P1. (a,b) Western blots of homogenized spinal cords, brain, heart, and muscles stained for SMN and β-actin. (c,d) Analysis of the ratio of SMN levels to β-actin. (e) Immunostaining for SMN (green) and choline acetyltransferase (CHAT) (red), and counter-stained with 4′,6-diamidino-2-phenylindole (blue), in the lumbar spinal cord sections. Cells that were immunopositive for both SMN and CHAT-stained yellow, while cells that were immunonegative for SMN and immunopositive for CHAT-stained red (arrow). Gems bodies (arrowhead) located in nucleus are shown in panel. (f) The number of cells per tissue section that were double-labeled for SMN and CHAT. (g) The number of gems per CHAT immunopositive cell. Bars = 20 μm; n = 4 in each group. (*P < 0.05; **P ≤ 0.01; ***P ≤ 0.001; one-way analysis of variance with LSD posthoc comparisons). LSD, least significant difference.
Costaining of tissue sections from spinal cords of AAV1-IGF-1-treated SMA mice for choline acetyltransferase and SMN (n = 4 in each group) showed they harbored a significantly greater number of cells that stained positively for both as compared to untreated SMA mice (P = 0.02). However, these were still significantly lower than noted in heterozygous mice (P = 0.03) (Figure 6e,f). SMN is typically concentrated within discrete nuclear compartments called gems, where it is thought to play an essential role in the assembly of the spliceosomal small ribonucleoproteins.18 The number of gems were determined to be lower in untreated SMA mice as compared to heterozygous mice (P < 0.01) (Figure 6e,g). AAV1-IGF-1 treatment significantly increased the number of gems in motor neurons of SMA mice (P = 0.03) suggesting the augmented SMN levels were functional.
AAV1-IGF-1 treatment increased transcription of SMN2
To investigate the basis for the increase in SMN protein levels following AAV1-IGF-1 treatment, both RT-PCR and quantitative real-time PCR (qRT-PCR) were performed on tissue lysates. RT-PCR analysis of lysates from the spinal cord, brain, muscles, and heart of AAV1-IGF-1-treated SMA mice revealed a higher level of SMN expression (both full-length and truncated transcripts; P = 0.04) than in untreated animals (Figure 7a,c). As these animals lack SMN1, this increase in SMN transcripts was likely from activation of the SMN2 promoter. Moreover, AAV1-IGF-1 treatment increased the ratio of full-length to truncated SMN transcripts (P = 0.04) in the spinal cord, brain, muscles, and heart (Figure 7b,d), suggesting that it also partially corrected the aberrant splicing of the SMN2 pre-mRNA. To confirm these results, we reanalyzed SMN transcript levels using different primer pairs by qRT-PCR. Similar findings were noted in mice treated with AAV1-IGF-1 that showed higher levels of SMN transcripts and an increased ratio of full-length to truncated SMN transcripts (P = 0.04) in the spinal cord, brain, muscles, and heart (Figure 7e,f). To rule out the possibility that the cryoanethesia procedure influences SMN expression, we compared the SMN levels in the spinal cords of SMA mice with and without cryoanesthesia. There was no significant difference in levels of SMN transcripts or protein between the two groups (Supplementary Figure S3).
Figure 7.

Levels of survival motor neuron (SMN) transcript in SMA mice on postnatal day 8 with or without systemic administration of AAV1-IGF-1 at P1. RT-PCR of homogenized spinal cord, brain, muscles, and heart with primer pairs for (a) total SMN and β-actin and (b) full-length SMN, truncated SMN, and β-actin. (c,d) Quantified PCR of homogenized spinal cord, brain, muscles, and heart with primer pairs for full-length SMN, truncated SMN, and glyceraldehyde 3-phosphate dehydrogenase (GAPDH). Analysis of the ratio of (c) total SMN levels to GAPDH and (d) full-length SMN to truncated SMN levels. n = 4 in each group. (*P < 0.05; Wilcoxon ranksum test).
Intravenous administration of AAV1-IGF-1 did not alter the blood chemistry of SMA mice
To evaluate the safety of AAV1-IGF-1 for treating SMA, blood cell counts and serum chemistries were performed on heterozygous mice that received a single intravenous injection of the viral vector (on P1) on day 30 posttreatment. The results revealed no significant differences in blood cell count (e.g., white blood cells, lymphocytes, or granulocytes) between AAV1-IGF-1-treated (n = 7) and untreated mice (n = 6) (Supplementary Table S1). The levels of glutamate oxaloacetate transaminase, glutamate pyruvate transaminase, and total bilirubin were also similar between untreated mice and animals treated with AAV1-IGF-1. This suggests that hepatic transduction with AAV1-IGF-1 was without overt hepatic toxicity. Since previous studies suggested that IGF-1 may induce hypoglycemia,19 we also analyzed serum glucose levels but saw no effect of AAV1-IGF-1 treatment on this parameter. The levels of blood urea nitrogen (23 ± 1 versus 19 ± 1 mg/dl) and creatinine (0.58 ± 0.03 versus 0.4 ± 0.03 mg/dl), which reflect renal function, were higher in AAV1-IGF-1-treated mice but the levels were within normal limits (normal = 8–33 mg/dl for blood urea nitrogen and 0.2−0.9 mg/dl for creatinine).20
Discussion
IGF-1, a pleiotropic hormone, has been reported to be meritorious for treating motor neuron diseases, such as amyotrophic lateral sclerosis and spinal and bulbar muscular atrophy.7,8,21,22 However, the benefits of IGF-1 in mouse models of SMA were modest.9,10,11,12 Here, we report our findings of administering a recombinant AAV1 vector encoding human IGF-1 systemically into a severe mouse model of SMA. We showed treatment with AAV1-IGF-1 abated the degeneration of spinal motor neurons, increased the size of myofibers in the hindlimbs and muscle thickness in the diaphragm. Expression of IGF-1 also improved the extent of innervation at the neuromuscular junctions and lessened the atrophy in the cardiac muscles of SMA mice. Importantly, these improvements translated to an increase in the overall health of the mice as illustrated by greater body weight gain, improved motor function, and a longer lifespan. In addition to these neurotrophic, myotrophic, and cardiotrophic effects of IGF-1,6,23,24 this report also showed for the first time that systemically delivered AAV1-IGF-1 increased the levels of SMN transcripts and protein in the spinal cord, brain, skeletal muscles, and heart of treated SMA mice. Indeed, it is possible that the noted benefits of IGF-1 were primarily through its activity at augmenting tissue levels of SMN, albeit through mechanisms that are as yet unclear.
Intravenous administration of AAV1-IGF-1 led to the transduction of primarily the liver and heart; however, expression of IGF-1 was only manifested in hepatocytes and not cardiomyocytes. Levels of human IGF-1 protein were also detected in the serum, presumably secreted from the transduced hepatic cells. Circulating IGF-1 is reportedly able to penetrate the blood–brain barrier.25 It is anticipated that it was this human IGF-1 in circulation that was responsible for the salutary effects noted in the other tissues (spinal motor neurons, skeletal muscles, and heart). In contrast to the positive results noted here with intravenous administration of AAV1-IGF-1, local delivery of either a plasmid or viral vector encoding IGF-1 to the muscle or central nervous system (CNS) reportedly only partially alleviated disease progression in SMA mice.9,10,12 This discrepancy in the robustness of therapeutic responses implies that rescue of cells not only in the spinal cord, but also in the muscle and heart, may be necessary for an optimal outcome. The notion that organs other than the CNS are also affected in SMA is supported by the demonstration that deletion of exon 7 of the murine SMN gene solely in skeletal muscle of mice led to presentation of a severe muscular dystrophy.14 Congenital heart defects have also been recognized as additional important phenotypes in SMA patients.15 Notably, systemic delivery of antisense oligonucleotide to correct the splicing aberration in SMN2 translated to an extension in the lifespan of SMA mice that was greater than that realized with intracerebroventricular delivery.16 Together, these finding suggest that reconstituting SMN protein levels in affected cells in the CNS and visceral organs may be necessary for an optimal therapeutic outcome.
Subcutaneous administration of IPLEX (recombinant human IGF-1 complexed with recombinant human IGF-1 binding protein 3) into SMA mice resulted in a modest reduction in the extent of degeneration of spinal motor neurons and improved motor function. However, this approach did not influence the body weight of the mice or their survival.11 Although coadministering IGF-1 binding protein-3 with IGF-1 prolonged the circulating half life from minutes to hours,26 therapy required daily subcutaneous injection for a therapeutic effect. Gene therapy with AAV1-IGF-1 offers the benefit of sustained production of the neurotrophic agent and may account for the more robust outcome than noted with periodic administrations of IPLEX. Indeed, in our studies, hepatic-derived IGF-1 was detected for at least 3 months after a single treatment with AAV1-IGF-1. Heterozygous mice treated with AAV1-IGF-1 did not show abnormal body weight gain or serum chemistry. Hence, intravenous delivery of AAV1-IGF-1 may represent an alternative therapeutic strategy for SMA as well as other motor neuron and metabolic diseases, such as IGF-1 deficiency. The SMA mouse model used in these studies had a severe disease phenotype and a median lifespan of 9 days. Although IGF-1 expression was detected in the liver at day 2 posttreatment, it was evident that this did not confer a robust effect on their lifespan and motor function. Additional studies in SMA mice with milder disease severity are necessary to fully vet the benefits of AAV1-IGF-1 treatment, such as using the SMNDelta7 SMA mouse model that have a median lifespan of 15 days.27
The mechanistic basis for some of the pathophysiology in SMA remains poorly understood. Serum levels of IGF-1 level are decreased in SMA mice11,16 and increasing SMN levels such as with antisense oligonucleotide restores the IGF-1 levels to normal.16 IGF-1-null mice also exhibit a phenotype that is similar to that noted in SMA mice; e.g., animals have low body weights and present with generalized muscle dystrophy.28 Hence, IGF-1 may be a potential downstream effector of SMN and supplementation with exogenous IGF-1 can partially overcome the deficiency in SMN in SMA.
We were surprised by the finding that systemically delivered AAV1-IGF-1 increased SMN protein levels in both CNS and extra-CNS tissues. In the spinal cord motor neuron, the increase in SMN protein levels was associated with greater numbers of gems bodies suggesting that the SMN produced was able to form a functional complex. While the basis for the increase in SMN levels following expression of IGF-1 is unclear, we presume it was likely from activation of the SMN2 gene. We showed that expression of IGF-1 also led to partial correction of the splicing defect as illustrated by a change in the ratio of full-length and truncated SMN proteins. Shababi et al. 10 had also previously shown that plasmid DNA-mediated expression of IGF-1 in the CNS of SMA mice modestly increased SMN protein levels in the spinal cord. Murdocca et al. 11 also demonstrated that subcutaneous administration of IPLEX into SMA mice resulted in a significant increase in the amount of truncated SMN (SMNd7) transcript levels in muscle.11 However, intracerebellar delivery of AAV1-IGF-1 attenuated motor neuron cell death without increasing SMN expression in a mouse model of SMA with lesser disease severity.12 Hence, the noted discrepancies on the effect of SMN expression following IGF-1-related treatment may be due to differences in treatment methods and the disease severity of the SMA mouse models. The relationship between IGF-1 and SMN is complex and may include roles that extend beyond the modulation of SMN levels.
Materials and Methods
Recombinant vectors. Recombinant AAV vectors that contained serotype-2 inverted terminal repeats and the human IGF-1 cDNA under the control of the cytomegalovirus enhancer and chicken β-actin promoter were packaged into serotype-1 capsids as described.21,22 Titers of AAV1-IGF-1 used in the studies were 2.8 × 1012 DRP/ml, as determined by quantitative PCR.
Mice. SMA model mice were generated by deletion of exon 7 of the Smn gene and knock-in of the human SMN2 (Smn −/− SMN2 +/−).29 We have since generated a variant that presents with more severe disease through backcrosses to obtain a more homogenous genetic background. This more severe mouse model of SMA harbor two copies of SMN2 transgene (Smn_−/−_SMN2+/−) and was generated through intercrossing the heterozygous knock-out mice (Smn+/−) and homozygous knock-out mice with two alleles of the SMN2 transgene (Smn_−/−_SMN2 +/+). Mouse genotypes were confirmed by PCR analysis as previously described.30 All procedures were approved by the Institutional Animal Care and Use Committee (Protocol # 20110468). Mice were under the care of the laboratory animal center of the National Taiwan University College of Medicine, and were supplied with sterile water and rodent pellets ad libitum.
Intravenous injection. On P1, neonatal SMA mice were cryoanesthetized as described.31 Using a beveled syringe with a 30-gauge needle, 3 μl of AAV1-IGF-1 or PBS was injected into the blood stream via the facial vein. The maximum volume for intravenous injection in adult mice is suggested to be 200 μl.32 Considering the small body size of neonatal mice (1 g), we only injected 3 μl of volume to avoid intravascular fluid overload of mice. Animals were injected with a dose of 8.4 × 109 genome copies AAV1-IGF-1/mouse.
RT-PCR and quantified real-time PCR. Total RNA from mouse tissues were extracted with TRIZOL (Invitrogen, Carlsbad, CA) and subjected to reverse transcription using SuperScript III (Invitrogen) according to the manufacturer's instructions. For RT-PCR, the cDNAs of human IGF-1, human total SMN (identifying exon 2a), human full-length and exon 7 truncated SMN (identifying exon 7), and mouse β-actin were amplified using the specific primers: human IGF-1, 5′-CAGCAGTCTTCCAACCCAAT-3′ (forward) and 5′-CTGCACTCCCTCTACTTGC-3′ (reverse); human total SMN, 5′-CTGACATTTGGGATGATACAGCAC-3′ (forward) and 5′-TGGTGGAGGGAGAAAAGAGTTCC-3′ (reverse); human full-length and exon 7 truncated SMN, 5′-CTCCCATATGTCCAGATTCTCTTGATGATGC-3′ (forward) and 5′-ACTGCCTCACCACCGTGCTGG-3′ (reverse); mouse β-actin, 5′-CCGTAAAGACCTCTATGCCAACAC-3′ (forward) and 5′-AAAGAAAGGGTGTAAAACGCAGC-3′ (reverse). PCR products were visualized on 1.5% agarose gels and the intensities of the signals were determined with ImageJ (National institutes of Health, Bethesda, MD) (Free download at http://rsbweb.nih.gov/ij/). Quantitative real-time PCR was undertaken as previously described, using different primer pairs from those in RT-PCR studies.33 Quantitative PCR reactions were run in triplicate for each sample on a IQ5 Multicolor Real-Time PCR Detection System (Bio-Rad Laboratories, Hercules, CA). The SMN primer sets (full-length SMN and exon 7 truncated SMN) and an endogenous control (glyceraldehyde 3-phosphate dehydrogenase; GAPDH) primer set were used on each sample. Mouse “calibrator” samples were run on every plate, and the values for these samples were set to 1.
Western blot. Western blot analysis was conducted as previously reported.34 Briefly, mouse tissues were homogenized in ice-cold modified RIPA buffer (50 mmol/l Tris–HCl, pH 7.4, 1% NP-40, 0.25% deoxycholic acid, 0.15 mol/l NaCl, 1 mmol/l EDTA, 1 mmol/l PMSF/NaF/sodium orthovanadate, and protease inhibitors cocktail). The supernatants were collected after centrifugation, separated on a 12% sodium dodecyl sulphate–polyacrylamide gel, and electro-transferred to a polyvinylidene fluoride membrane. The membranes were incubated in blocking solution for 1 hour at room temperature, and then overnight at 4 °C with the following primary antibodies: anti-human-IGF-1 (1:1,000, Abcam, Cambridge, UK), anti-SMN (1:500, BD Biosciences, San Diego, CA), and anti-β-actin (1:5,000, Sigma Aldrich, St Louis, MO). The membranes were washed and incubated in blocking solution with the appropriate HRP-conjugated secondary antibody for 2 hours at room temperature (1:5,000, GeneTex, Irvine, CA). The signals were visualized directly using a UV transilluminator (BioDoc-It Imaging System, UVP, Upland, CA). Semi-quantitative evaluation of the bands was performed by densitometric analysis using ImageJ and the protein expression levels were normalized to β-actin.
Immunohistochemistry. The liver, spinal cord, and muscle were removed, placed in 10% formaldehyde at room temperature for 3 hours, followed by 15% sucrose for 1 day and 30% sucrose at 4 °C overnight, and then rapidly frozen in liquid nitrogen-cooled isopentane. Frozen serial sections of spinal cord and muscle were cut to 7- and 15-μm thickness, respectively. Tissue sections were blocked with 5% serum and incubated overnight at 4 °C with the following primary antibodies: anti-human IGF-1 (1:100; Abcam), anticholine acetyltransferase (1:100; Millipore, Billerica, MA), anti-SMN (1:50; BD Biosciences), anti-neurofilament 200 kDa (1:200; Millipore) or α-bungarotoxin Alexa Fluor 555 conjugate (1:1,000; Molecular Probes, Eugene, OR). Samples were washed with Tris buffered saline with Tween 20 (10 mmol/l Tris–HCl, pH 8.0, 150 mmol/l NaCl, and 0.1% Tween 20) and incubated for 2 hours at room temperature with the appropriate fluorescence-conjugated secondary antibodies (1:200; Molecular Probes). 4′,6-diamidino-2-phenylindole (Sigma) was used for counter-staining the cell nucleus. Tissues were examined on a fluorescence microscope (Zeiss AXIO Imager A1, Carl Zeiss, Göttingen, Germany) or laser-scanning confocal microscope (Leica TCS SP2 Spectral Confocal System, Leica Microsystems, Mannheim, Germany) .
ELISA. Blood samples were collected from the heart of heterozygous mice at 1 or 3 months posttreatment with AAV1-IGF-1. Following centrifugation, mouse sera were collected and stored at −20 °C. Human IGF-1 levels were measured (n = 6 in each group) using a human IGF-1 ELISA kit (Life Sciences, Oakland, CA) according to the manufacturer's instructions.
Motor function tests. Two behavioral tests were used to evaluate the motor function of mice.35 In the righting reflex test, each pup was turned onto its back and the time it took to stably place all its four paws on the ground was recorded (cutoff time of 10 seconds). For the negative geotaxis study, each pup was placed on a 45° incline with its head pointing down the incline. The responses of the mice were scored as follows: 0, slipped down immediately; 1, held onto the incline with its head still pointing down; 2, turned around on the incline by 90°; 3, turned around by 180° with its head pointing up the incline; and 4, turned around and climbed up the incline.
Hematoxylin and eosin staining. A subset of frozen tissue sections (7 μm thickness), from spinal cord, muscles, diaphragm, and heart, was stained for hematoxylin and eosin as reported.30 Neurons with a size greater than 200 µm2 located in the anterior (ventral) horn of the spinal cord were analyzed. We counted the number of neurons (only neurons showing nuclei) according to their cell size: 200–400, 400–600, and >600 μm2, respectively. The size of myocytes from quadriceps was also analyzed and we calculated the number of myocytes according to their cross-sectional size.
Retrograde tracing. For retrograde tracing in motor neurons, 4 μl of fluorogold (Fluorochrome, Denver, CO) was injected into bilateral gastrocnemius and quadriceps muscles of mice on P4 using a 30-gauge needle at a rate of 1 μl/minute. Mice were killed 3 days postinjection and their spinal cord was then removed. After formaldehyde fixation, frozen serial sections of whole lumbar spinal cord were cut at 12-μm thickness and processed for histological analysis.
Statistical analysis. Values are expressed as mean ± SEM from at least four independent experiments. Statistical significance was analyzed by one-way analysis of variance followed by least significant difference posthoc comparisons or a Wilcoxon-ranksum test. A Kaplan–Meier analysis was used for estimation of survival curves and a log-rank test was used to determine differences of two survival curves. Two-tailed P values of less than 0.05 were considered statistically significant. The SPSS software was used for statistical analyses.
SUPPLEMENTARY MATERIAL Figure S1. Intraperitoneal (i.p.), subcutaneous (SC), and intravenous (IV, via the facial vein) injection of Evans blue into postnatal day 1 wild-type mice and analyzed at 10 minutes postinjection.Figure S2. RT-PCR analysis of human IGF-1 transcripts in livers of heterozygous mice at postnatal days 2, 4, and 6 after injection of AAV1-IGF-1 at postnatal day 1.Figure S3. Levels of SMN transcripts and protein in the spinal cord of SMA mice on postnatal day 8 with or without cryoanesthesia on postnatal day 1.Table S1. Blood cell count and serum biochemistry studies in mice with or without AAV1-IGF-1 treatment.
Acknowledgments
We thank the Department of Medical Research, National Taiwan University Hospital, Taiwan for excellent technical assistance and equipment support. This work was supported by grants from the National Science Council (100-2314-B-002-050-MY2 and 102-2314-B002-065-MY3). J.C.D., S.H.C. and M.A.P. are paid employees of Genzyme, a Sanofi Company. The other authors declared no conflict of interest.
Supplementary Material
Supplementary Figure S1
Intraperitoneal (i.p.), subcutaneous (SC), and intravenous (IV, via the facial vein) injection of Evans blue into postnatal day 1 wild-type mice and analyzed at 10 minutes postinjection.
Supplementary Figure S2
RT-PCR analysis of human IGF-1 transcripts in livers of heterozygous mice at postnatal days 2, 4, and 6 after injection of AAV1-IGF-1 at postnatal day 1.
Supplementary Figure S3
Levels of SMN transcripts and protein in the spinal cord of SMA mice on postnatal day 8 with or without cryoanesthesia on postnatal day 1.
Supplementary Table S1
Blood cell count and serum biochemistry studies in mice with or without AAV1-IGF-1 treatment.
References
- Czeizel A, Hamula J. A hungarian study on Werdnig-Hoffmann disease. J Med Genet. 1989;26:761–763. doi: 10.1136/jmg.26.12.761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lefebvre S, Bürglen L, Reboullet S, Clermont O, Burlet P, Viollet L, et al. Identification and characterization of a spinal muscular atrophy-determining gene. Cell. 1995;80:155–165. doi: 10.1016/0092-8674(95)90460-3. [DOI] [PubMed] [Google Scholar]
- Wirth B. An update of the mutation spectrum of the survival motor neuron gene (SMN1) in autosomal recessive spinal muscular atrophy (SMA) Hum Mutat. 2000;15:228–237. doi: 10.1002/(SICI)1098-1004(200003)15:3<228::AID-HUMU3>3.0.CO;2-9. [DOI] [PubMed] [Google Scholar]
- Vardatsikos G, Sahu A, Srivastava AK. The insulin-like growth factor family: molecular mechanisms, redox regulation, and clinical implications. Antioxid Redox Sign. 2009;11:1165–1190. doi: 10.1089/ars.2008.2161. [DOI] [PubMed] [Google Scholar]
- Ozdinler PH, Macklis JD. IGF-I specifically enhances axon outgrowth of corticospinal motor neurons. Nat Neurosci. 2006;9:1371–1381. doi: 10.1038/nn1789. [DOI] [PubMed] [Google Scholar]
- Sakowski SA, Schuyler AD, Feldman EL. Insulin-like growth factor-I for the treatment of amyotrophic lateral sclerosis. Amyotroph Lateral Scler. 2009;10:63–73. doi: 10.1080/17482960802160370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaspar BK, Lladó J, Sherkat N, Rothstein JD, Gage FH. Retrograde viral delivery of IGF-1 prolongs survival in a mouse ALS model. Science. 2003;301:839–842. doi: 10.1126/science.1086137. [DOI] [PubMed] [Google Scholar]
- Palazzolo I, Stack C, Kong L, Musaro A, Adachi H, Katsuno M, et al. Overexpression of IGF-1 in muscle attenuates disease in a mouse model of spinal and bulbar muscular atrophy. Neuron. 2009;63:316–328. doi: 10.1016/j.neuron.2009.07.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bosch-Marcé M, Wee CD, Martinez TL, Lipkes CE, Choe DW, Kong L, et al. Increased IGF-1 in muscle modulates the phenotype of severe SMA mice. Hum Mol Genet. 2011;20:1844–1853. doi: 10.1093/hmg/ddr067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shababi M, Glascock J, Lorson CL. Combination of SMN trans-splicing and a neurotrophic factor increases the life span and body mass in a severe model of spinal muscular atrophy. Hum Gene Ther. 2011;22:135–144. doi: 10.1089/hum.2010.114. [DOI] [PubMed] [Google Scholar]
- Murdocca M, Malgieri A, Luchetti A, Saieva L, Dobrowolny G, de Leonibus E, et al. IPLEX administration improves motor neuron survival and ameliorates motor functions in a severe mouse model of spinal muscular atrophy. Mol Med. 2012;18:1076–1085. doi: 10.2119/molmed.2012.00056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsai LK, Chen YC, Cheng WC, Ting CH, Dodge JC, Hwu WL, et al. IGF-1 delivery to CNS attenuates motor neuron cell death but does not improve motor function in type III SMA mice. Neurobiol Dis. 2012;45:272–279. doi: 10.1016/j.nbd.2011.06.021. [DOI] [PubMed] [Google Scholar]
- Kirschner J, Schorling D, Hauschke D, Rensing-Zimmermann C, Wein U, et al. Somatropin treatment of spinal muscular atrophy: a placebo-controlled, double-blind crossover pilto study. Neuromuscular Dis. 2013;24:134–142. doi: 10.1016/j.nmd.2013.10.011. [DOI] [PubMed] [Google Scholar]
- Cifuentes-Diaz C, Frugier T, Tiziano FD, Lacène E, Roblot N, Joshi V, et al. Deletion of murine SMN exon 7 directed to skeletal muscle leads to severe muscular dystrophy. J Cell Biol. 2001;152:1107–1114. doi: 10.1083/jcb.152.5.1107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rudnik-Schöneborn S, Heller R, Berg C, Betzler C, Grimm T, Eggermann T, et al. Congenital heart disease is a feature of severe infantile spinal muscular atrophy. J Med Genet. 2008;45:635–638. doi: 10.1136/jmg.2008.057950. [DOI] [PubMed] [Google Scholar]
- Hua Y, Sahashi K, Rigo F, Hung G, Horev G, Bennett CF, et al. Peripheral SMN restoration is essential for long-term rescue of a severe spinal muscular atrophy mouse model. Nature. 2011;478:123–126. doi: 10.1038/nature10485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gray SJ. Gene therapy and neurodevelopmental disorders. Neuropharmacology. 2013;68:136–142. doi: 10.1016/j.neuropharm.2012.06.024. [DOI] [PubMed] [Google Scholar]
- Gubitz AK, Feng W, Dreyfuss G. The SMN complex. Exp Cell Res. 2004;296:51–56. doi: 10.1016/j.yexcr.2004.03.022. [DOI] [PubMed] [Google Scholar]
- Roith DL. Insulin-like growth factors. N Eng J Med. 1997;336:633–640. doi: 10.1056/NEJM199702273360907. [DOI] [PubMed] [Google Scholar]
- Liu HC, Ting CH, Wen HL, Tsai LK, Hsieh-Li HM, Li H, et al. Sodium vanadate combined with L-ascorbic acid delays disease progression, enhances motor performance, and ameliorates muscle atrophy and weakness in mice with spinal muscular atrophy. BMC Med. 2013;11:38. doi: 10.1186/1741-7015-11-38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dodge JC, Haidet AM, Yang W, Passini MA, Hester M, Clarke J, et al. Delivery of AAV-IGF-1 to the CNS extends survival in ALS mice through modification of aberrant glial cell activity. Mol Ther. 2008;16:1056–1064. doi: 10.1038/mt.2008.60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dodge JC, Treleaven CM, Fidler JA, Hester M, Haidet A, Handy C, et al. AAV4-mediated expression of IGF-1 and VEGF within cellular components of the ventricular system improves survival outcome in familial ALS mice. Mol Ther. 2010;18:2075–2084. doi: 10.1038/mt.2010.206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar A, Yamauchi J, Girgenrath T, Girgenrath M. Muscle-specific expression of insulin-like growth factor 1 improves outcome in Lama2Dy-w mice, a model for congenital muscular dystrophy type 1A. Hum Mol Genet. 2011;20:2333–2343. doi: 10.1093/hmg/ddr126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L, Ma W, Markovich R, Chen JW, Wang PH. Regulation of cardiomyocyte apoptotic signaling by insulin-like growth factor I. Circ Res. 1998;83:516–522. doi: 10.1161/01.res.83.5.516. [DOI] [PubMed] [Google Scholar]
- Carro E, Nuñez A, Busiguina S, Torres-Aleman I. Circulating insulin-like growth factor I mediates effects of exercise on the brain. J Neurosci. 2000;20:2926–2933. doi: 10.1523/JNEUROSCI.20-08-02926.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adams S, Moore J, Chu S, Bagi C, DeLeon L, Liu C, et al. Pharmacokinetics and bioavailability of rhIGF-I/IGFBP-3 in the rat and monkey. Prog Growth Factor Res. 1995;6:347–356. doi: 10.1016/0955-2235(95)00003-8. [DOI] [PubMed] [Google Scholar]
- Porensky PN, Mitrpant C, McGovern VL, Bevan AK, Foust KD, Kaspar BK, et al. A single administration of morpholino antisense oligomer rescues spinal muscular atrophy in mouse. Hum Mol Genet. 2012;21:1625–1638. doi: 10.1093/hmg/ddr600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Powell-Braxton L, Hollingshead P, Warburton C, Dowd M, Pitts-Meek S, Dalton D, et al. IGF-I is required for normal embryonic growth in mice. Genes Dev. 1993;7:2609–2617. doi: 10.1101/gad.7.12b.2609. [DOI] [PubMed] [Google Scholar]
- Hsieh-Li HM, Chang JG, Jong YJ, Wu MH, Wang NM, Tsai CH, et al. A mouse model for spinal muscular atrophy. Nat Genet. 2000;24:66–70. doi: 10.1038/71709. [DOI] [PubMed] [Google Scholar]
- Tsai LK, Tsai MS, Lin TB, Hwu WL, Li H. Establishing a standardized therapeutic testing protocol for spinal muscular atrophy. Neurobiol Dis. 2006;24:286–295. doi: 10.1016/j.nbd.2006.07.004. [DOI] [PubMed] [Google Scholar]
- Phifer CB, Terry LM. Use of hypothermia for general anesthesia in preweanling rodents. Physiol Behav. 1986;38:887–890. doi: 10.1016/0031-9384(86)90058-2. [DOI] [PubMed] [Google Scholar]
- Donovan J, Brown P. Use of mouse, rat, hamster, and rabbit. Curr Protocol Immunol. 2006. pp. 1.6.1–1.6.10.
- Tsai LK, Yang CC, Ting CH, Su YN, Hwu WL, Li H. Correlation of survival motor neuron expression in leukocytes and spinal cord in spinal muscular atrophy. J Pediatr. 2009;154:303–305. doi: 10.1016/j.jpeds.2008.08.050. [DOI] [PubMed] [Google Scholar]
- Tsai LK, Tsai MS, Tin JH, Li H. Multiple therapeutic effects of valproic acid in spinal muscular atrophy model mice. J Mol Med. 2008;86:1243–1254. doi: 10.1007/s00109-008-0388-1. [DOI] [PubMed] [Google Scholar]
- Butchbach MER, Edwards, Burghes AHM. Abnormal motor phenotype in the SMNΔ7 mouse model of spinal muscular atrophy. Neurobiol Dis. 2007;27:207–219. doi: 10.1016/j.nbd.2007.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure S1
Intraperitoneal (i.p.), subcutaneous (SC), and intravenous (IV, via the facial vein) injection of Evans blue into postnatal day 1 wild-type mice and analyzed at 10 minutes postinjection.
Supplementary Figure S2
RT-PCR analysis of human IGF-1 transcripts in livers of heterozygous mice at postnatal days 2, 4, and 6 after injection of AAV1-IGF-1 at postnatal day 1.
Supplementary Figure S3
Levels of SMN transcripts and protein in the spinal cord of SMA mice on postnatal day 8 with or without cryoanesthesia on postnatal day 1.
Supplementary Table S1
Blood cell count and serum biochemistry studies in mice with or without AAV1-IGF-1 treatment.