Increased dietary intake of ω-3-polyunsaturated fatty acids reduces pathological retinal angiogenesis (original) (raw)
. Author manuscript; available in PMC: 2015 Jul 5.
Published in final edited form as: Nat Med. 2007 Jun 24;13(7):868–873. doi: 10.1038/nm1591
Abstract
Many sight-threatening diseases have two critical phases, vessel loss followed by hypoxia-driven destructive neovascularization. These diseases include retinopathy of prematurity and diabetic retinopathy, leading causes of blindness in childhood and middle age affecting over 4 million people in the United States. We studied the influence of ω-3- and ω-6-polyunsaturated fatty acids (PUFAs) on vascular loss, vascular regrowth after injury, and hypoxia-induced pathological neovascularization in a mouse model of oxygen-induced retinopathy1. We show that increasing ω-3-PUFA tissue levels by dietary or genetic means decreased the avascular area of the retina by increasing vessel regrowth after injury, thereby reducing the hypoxic stimulus for neovascularization. The bioactive ω-3-PUFA-derived mediators neuroprotectinD1, resolvinD1 and resolvinE1 also potently protected against neovascularization. The protective effect of ω-3-PUFAs and their bioactive metabolites was mediated, in part, through suppression of tumor necrosis factor-α. This inflammatory cytokine was found in a subset of microglia that was closely associated with retinal vessels. These findings indicate that increasing the sources of ω-3-PUFA or their bioactive products reduces pathological angiogenesis. Western diets are often deficient in ω-3-PUFA, and premature infants lack the important transfer from the mother to the infant of ω-3-PUFA that normally occurs in the third trimester of pregnancy2. Supplementing ω-3-PUFA intake may be of benefit in preventing retinopathy.
Ocular neovascularization is the most common cause of blindness in all age groups: retinopathy of prematurity in children, diabetic retinopathy in working-age adults and age-related macular degeneration in the elderly. In principle, destructive angiogenesis in the eye can be ameliorated by either direct inhibition of neovascularization or by controlling vessel loss in order to reduce the hypoxic stimulus that drives neovascularization. Retinopathy is modeled in the mouse eye with oxygen-induced vessel loss, which precipitates hypoxia-induced retinopathy, allowing for assessment of retinal vessel loss, vessel regrowth after injury and pathological angiogenesis1.
The role of lipids in angiogenesis is just beginning to be defined3,4. The major polyunsaturated fatty acids (PUFA) found in the retina are docosahexaenoic acid (DHA; C22:6ω-3) and arachidonic acid (C20:4ω-6), both found primarily in neural and vascular cell membrane phospholipids5. Eicosapentaenoic acid (EPA; C20:5ω-3), the precursor to DHA, is found in the retinal vascular endothelium6. Dietary sources of ω-3-PUFA and ω-6-PUFA, as well as PUFA released as free fatty acids by phospholipase A2, contribute to a pool of substrates7. These can then be converted to bioactive intermediaries such as eicosanoids from arachidonic acid, neuroprotectins from DHA, D series resolvins from DHA and E series resolvins from EPA8,9. Emerging knowledge of lipid mediators3,6 and epidemiologic data linking PUFA and neovascular age-related macular degeneration indicate that EPA, DHA and arachidonic acid may function in vivo to regulate retinal vaso-obliteration and neovascularization3. We therefore tested whether moderate dietary intake of ω-3-PUFA or ω-6-PUFA can alter retinal angiogenesis. We also tested the role of PUFA using a genetic model in which overexpression of the C. elegans fat-1 gene in transgenic mice converts ω-6-PUFA to ω-3-PUFA, resulting in elevated tissue levels of ω-3-PUFA10.
We subjected mice on a completely defined isocaloric diet enriched with 2% of total fatty acids from either ω-3-PUFA (DHA and EPA) or ω-6-PUFA (arachidonic acid) (Supplementary Table 1 online), modeled after a Japanese or Western diet, respectively, to oxygen-induced retinopathy1. We also examined retinopathy in the Fat-1 mouse on an ω-6-PUFA diet (Supplementary Table 2 online).
Lipid content of milk reflects the lipid profile of the mother's diet9,11. We found that retinal lipid composition in the pups reflected the pups’ (and the mother's) dietary intake of lipids. In the retinas of the milk-fed pups at postnatal day (P) 17, the levels of all of the principal ω-3-PUFA (including EPA, docosapentaenoic acid (DPA)-ω–3 and DHA) were increased with either the ω-3-PUFA-enriched diet or with the expression of the fat-1 gene (P ≤ 0.005). There was also less retinal ω-6-PUFA (including arachidonic acid, docosatetraenoic acid (DTA) and DPA-ω-6) in the ω-3-PUFA group relative to the ω-6-PUFA group, and there was less in Fat-1 mice relative to wild-type mice (P ≤ 0.005). Enriching ω-3-PUFA through diet or through expression of fat-1 resulted in a >50% decrease in the total ω-6/ω-3-PUFA ratio (Table 1).
Table 1.
Fatty acyl composition (%) of retinas from P17 pups
| Fatty acid | ω-6 diet (n = 6) | ω-3 diet (n = 6) | Wild type (n = 6) | Fat-1 (n = 4) |
|---|---|---|---|---|
| Saturates | ||||
| PA (16:0) | 22.53 (0.14) | 22.81 (0.24) | 21.40 (0.05) | 21.83 (0.88) |
| SA (18:0) | 20.30 (0.12) | 20.51 (0.26) | 19.01 (0.06) | 19.26 (0.14) |
| Total SFA | 44.85 (0.21) | 45.39 (0.43) | 41.84 (0.43) | 42.31 (1.14) |
| Monounsaturates | ||||
| OA (18:1ω9) | 8.43 (0.03) | 8.79 (0.09)a | 6.61 (0.13) | 6.91 (0.62) |
| VA (18:1ω7) | 2.40 (0.03) | 2.21 (0.04)a | 2.01 (0.05) | 1.82 (0.13) |
| Total MUFA | 11.98 (0.10) | 12.15 (0.12) | 9.45 (0.21) | 9.61 (0.99) |
| ω-6-Polyunsaturates | ||||
| LA (18:2ω6) | 0.74 (0.01) | 0.90 (0.04)a | 1.65 (0.04) | 1.76 (0.09) |
| AA (20:4ω6) | 8.87 (0.34) | 7.11(0.35)a | 11.40 (0.21) | 8.41 (0.00)a |
| DTA (22:4ω6) | 1.25(0.15) | 0.57(0.09)a | 2.25 (0.03) | 0.85 (0.05)a |
| DPA (22:5ω6) | 4.29 (0.29) | 0.96 (0.08)a | 4.93 (0.12) | 0.29 (0.01)a |
| Total ω-6-PUFA | 15.82 (0.39) | 10.50 (0.30)a | 21.66 (0.28) | 12.69 (0.10)a |
| ω-3-Polyunsaturates | ||||
| ALA (18:3ω3) | 0.03 (0.003) | 0.03 (0.01) | 0.01 (0.00) | 0.03 (0.01)a |
| EPA (20:5ω3) | 0.02 (0.0002) | 0.25 (0.02)a | 0.00 (0.00) | 0.52 (0.01)a |
| DPA (22:5ω3) | 0.17 (0.01) | 0.47 (0.03)a | 0.15 (0.01) | 0.76 (0.01)a |
| DHA (22:6ω3) | 12.65 (0.93) | 17.92 (1.07)a | 17.58 (0.22) | 26.58 (0.39)a |
| Total ω-3-PUFA | 12.87 (0.93) | 18.68 (1.07)a | 17.74 (0.22) | 27.93 (0.37)a |
| DHA/DPAω6 | 3.08 (0.40) | 19.63 (2.32)a | 3.57 (0.13) | 92.36 (5.14)a |
| ω-6/ω-3 ratio | 1.23 | 0.56 | 1.22 | 0.45 |
We found that a lower retinal ω-6/ω3-PUFA ratio had a protective effect against pathological angiogenesis. At P17, mice on the ω-6-PUFA versus the ω-3-PUFA diet had a vaso-obliterated/total retinal area of 21.5 ± 0.4% versus 13.7 ± 0.5%, P ≤ 0.0001 (Fig. 1a,b). Moreover, P17 pups fed an ω-3-PUFA versus an ω-6-PUFA diet were significantly protected from pathologic neovascularization (5.7 ± 0.4% versus 9.0 ± 0.6% neovascularization, P ≤ 0.0001 (Fig. 1a,c).
Figure 1.
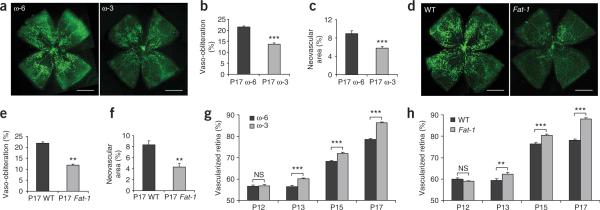
Elevated levels of ω-3-PUFAs result in decreased vaso-obliteration and retinal neovascularization in mice. Retinas from C57BL/6 mice fed an ω-3- or ω-6-PUFA enhanced diet were examined by whole-mount microscopy after oxygen-induced retinopathy. (a) P17 retinal vasculature stained with isolectin B4-FITC showing vaso-obliteration and neovascularization in the ω-6- and ω-3-PUFA fed mice (ω-6, n = 14, and ω-3, n = 27) (scale bar, 1 mm). (b) Vaso-obliteration and (c) neovascularization in the ω-3- and ω-6-PUFA-fed mice at P17. (d) Vaso-obliteration and neovascularization in wild-type (WT) versus Fat-1 retinas at P17 (scale bar, 1 mm). (e) Vaso-obliteration and (f) neovascularization in the _fat-1_-expressing mice compared with that in controls (wild-type, n = 20; Fat-1, n = 16). (g,h) Percentage vascularized area in retinal whole-mounts analyzed at P12, P13, P15 and P17 after exposure to 75% oxygen from P7–P12 from mice on a ω-6-PUFA or a ω-3-PUFA diet (g) or in Fat-1 mice or their wild-type controls (h), n = 8–20 per group. **P ≤ 0.001, ***P ≤ 0.0001; NS, not significant.
We confirmed in Fat-1 mice10 that increased retinal ω-3-PUFA levels (Table 1) inhibited neovascularization. At P17, after oxygen-induced retinopathy1 wild-type mice lacking the fat-1 transgene had more extensive vaso-obliteration (21.9 ± 0.7% versus 11.9 ± 0.5%) of total retinal area (P ≤ 0.001) (Fig. 1d,e) and more severe retinal neovascularization (8.3 ± 0.8% versus 4.3 ± 0.7%) of total retinal area (P ≤ 0.001) (Fig. 1d,f) than Fat-1 homozygotes that had a decreased ω-6/ω-3-PUFA ratio (Table 1).
The ω-3-PUFA-induced protection against retinopathy at P17 was due to increased regrowth of vessels and not to decreased oxygen-induced vessel loss during hyperoxia. No difference in vessel loss was observed following 24 h of 75% oxygen exposure between the different experimental groups (data not shown). After 5 d exposure to 75% oxygen, retinas from mice (P12) on a ω-3-PUFA or ω-6-PUFA diet, and from Fat-1 mice or controls, showed no difference in vaso-obliteration (Fig. 1g,h). However, the ω-3-PUFA-fed mice showed less avascular retinal area than did ω-6-PUFA-fed mice starting between P12 and P13. This reduction was maintained through P17 (Fig. 1g). Similar results were obtained in Fat-1 transgenic mice and controls (Fig. 1h).
Resolvins (resolution phase interaction products) and neuroprotectins are ω-3-PUFA bioactive products derived from EPA and DHA, respectively (Fig. 2a). They were first identified in resolving inflammatory exudates in DHA-enriched tissues12. Their contribution to the regulation of angiogenesis has not been investigated12.
Figure 2.

ResolvinD1, resolvinE1 and neuroprotectinD1, derived from ω-3-PUFAs, protect against retinopathy with reductions in vaso-obliteration and neovascularization. (a) Biosynthetic pathway of ω-3-PUFA metabolite formation from EPA and DHA. ω-22-Hydroxy-PD1 is a biosynthetic marker of NPD1. RvE2 is a biosynthetic marker of the E series EPA resolvins. (b,c) Liquid chromatography MS-MS spectrum of RvE2 (b) and of ω-22-hydroxy-PD1 (c) from retinal extracts of ω-3-PUFA-fed mice. (d) Vaso-obliteration at P17 in RvD1, RvE1 or NPD1-treated mice compared with vehicle-treated control. (e) Neovascularization at P17 in mice injected i.p. from P5–P8 with RvD1 (n = 14), RvE1 (n = 10) or NPD1 (n = 14) compared with that in saline-treated mice (n = 14). (f) Vaso-obliteration at P8 with RvD1 (n = 7), RvE1 (n = 9), NPD1 (n = 7) or saline (n = 6) treatment. (g) Left column, retinal whole-mounts from mice with oxygen-induced retinopathy at P14 on an ω-6-PUFA diet stained for endothelial cells (red), Csf1r (green) and ChemR23 (magenta). Central panel, merged image (scale bar, 50 μm); white indicates the colocalization of all three stains. Right column, four images of one Csf1r+ cell at indicated focal planes (z = 1.1 μm, 2.1 μm, 3.0 μm and 4.0 μm). The red lines on the cross-section (far right column) indicate the relative z depth that corresponds to the section shown directly to the left. #P ≤ 0.05; **P ≤ 0.0001.
Neither resolvins nor neuroprotectins were detected by liquid chromatography followed by tandem mass spectroscopy (MS-MS) in the ω-6-PUFA-diet retinas. However, in the ω-3-PUFA-diet retinas, we identified ω-22-hydroxy-protectinD1 (PD1) and resolvinE2 (RvE2), which are markers of the biosynthesis of neuroprotectinD1 (NPD1) and resolvinE1 (RvE1), respectively (Fig. 2b,c, Supplementary Table 3 online)13,14.
In mice without ω-3-PUFA diet supplementation, a very low intraperitoneal (i.p.) dose of resolvinD1 (RvD1), RvE1 or NPD1 (10 ng/d, an amount comparable to the total found in the retinas of mice on the ω-3-PUFA diet, Supplementary Table 3) conferred significant protection from vaso-obliteration (P ≤ 0.0001) (Fig. 2d) and neovascularization (P ≤ 0.03) (Fig. 2e) at P17 compared with the control.
We saw no difference in normal vascular development at P6 between ω-3- and ω-6-PUFA-fed mice nor between controls and RvD1, RvE1 or NPD1-treated (P4–P5) mice (Supplementary Fig. 1 online). No differences in vessel loss were observed between controls and mice treated from P5–P8 with either NPD1, RvD1or RvE1 (Fig. 2f). At P17 there was considerably less vaso-obliteration in all treatment groups compared with controls (Fig. 2d), indicating that these compounds also confer their protective actions against retinopathy by means of enhanced vessel regrowth, not protection from vessel loss. These findings coincide with our dietary PUFA results and demonstrate that the protective effect of ω-3-PUFAs against retinal neovascularization is mediated in part through the potent bioactive mediators RvD1, RvE1 and NPD1.
We next addressed the mechanism by which RvD1, RvE1 and NPD1 might mediate the ω-3-PUFA protection against retinal neovascularization. RvE1 can block the production of proinflammatory cytokines when it binds to the receptor ChemR23 (refs. 15,16). In retinal whole-mounts we localized ChemR23 to a subset of colony-stimulating factor-1-receptor–positive (Csf1r+) microglia that were morphologically distinct from resident microglia. These cells were closely associated with retinal blood vessels (Fig. 2g). ChemR23 was not detected elsewhere in the retina under these conditions. These findings are consistent with the idea that ω-3-PUFA-derived biomediators interact with receptors on microglial cells to decrease proinflammatory cytokine production.
We next examined the role of the inflammatory cytokine tumor necrosis factor (TNF)-α. Modulation of TNF-α production influences vascular growth in other systems through endothelial cell cycle regulation17,18. Moreover, mice lacking TNF-α have less oxygen-induced retinopathy19. Therefore, we hypothesized that ω-3-and ω-6-PUFAs and their bioactive metabolites act in part through the regulation of TNF-α expression.
Consistent with our hypothesis, we found that an ω-3-PUFA diet potently suppressed retinal expression of Tnf mRNA, encoding TNF-α, by ~90% (P ≤ 0.0001) during hyperoxia (P8) and hypoxia (P14) compared with expression in mice on an ω-6-PUFA diet (Fig. 3a). Retinal TNF-α protein levels were also suppressed by ~30% (P ≤ 0.001) compared with those in mice on an ω-6-PUFA diet (Fig. 3a). Suppression of TNF-α in the ω-6-PUFA diet group through i.p. injection of a TNF-α receptor fusion protein (etanercept), which sequesters TNF-α, resulted in a reduction in avascular area at P17 (P ≤ 0.001) (Fig. 3b), as well as a reduction in pathologic neovascularization (P ≤ 0.05) (Fig. 3c), to levels seen in ω-3-PUFA fed mice. Mice on an ω-3-PUFA diet with suppressed but not ablated levels of TNF-α (Fig. 3b) had further suppression of both vaso-obliteration (P ≤ 0.001) and neovascularization (P ≤ 0.02) with i.p. etanercept treatment (Fig. 3b,c). Similar results were observed with intraocular injections of etanercept (Supplementary Fig. 2 online). These data indicate that partial reduction of TNF-α levels with an ω-3-PUFA diet seems to be sufficient to help mediate a protective effect against retinopathy.
Figure 3.

Dietary PUFA regulation of TNF-α in retinopathy. (a) Left panel, mean total retinal Tnf mRNA expression in ω-6-PUFA and ω-3-PUFA-fed mice (n = 6) normalized to that of cyclophilin. Right panel, retinal TNF-α protein levels, normalized to total protein, at P14 in ω-6-PUFA and ω-3-PUFA-fed mice (n = 4). (b,c) Inhibition of ω-6-PUFA-induced retinopathy with suppression of TNF-α using etanercept. (b) Vaso-obliteration in etanercept treatment versus saline (i.p.) in ω-6-PUFA-fed (n = 8) and ω-3-PUFA-fed mice (n = 10) at P17. (c) Neovascularization in etanercept-treated ω-6-PUFA-fed mice versus saline-treated controls (n = 8) and in ω-3-PUFA-fed mice (n = 10) at P17. (d) Vaso-obliteration at P17 in Tnf+/+ and Tnf−/− mice on either an ω-3-PUFA diet (n = 15 and n = 11, respectively) or an ω-6-PUFA diet (n = 22 and n = 10, respectively). (e) Neovascularization at P17 in Tnf+/+ and _Tnf_−/− mice on either an ω-3-PUFA diet (n = 15 and n = 11, respectively) or an ω-6-PUFA diet (n = 22 and n = 10, respectively). (f) Retinal whole-mount from a P17 mouse on an ω-6-PUFA diet with oxygen-induced retinopathy stained (left column) for endothelial cells (red), Csf1r (green) and Tnf-α (magenta). Central panel, merged image of the three stains (scale bar, 50 μm). Right column, one Csf1r+ cell at focal planes z = 1.1 μm, 1.9 μm, 2.8 μm and 3.6 μm. Red lines (far right column) indicate relative z depth that corresponds to the section immediately to the left. (g) NF-κB (luciferase) activity (relative luminescence units) in P14 oxygen-treated retinas from NF-κB-Luc reporter mice on ω-6-PUFA (n = 10) or ω-3-PUFA (n = 11) diets. (h) Left column, retina with oxygen-induced retinopathy from a P14 mouse on an ω-6-PUFA diet stained for endothelial cells (red), CD11b (green) and luciferase (magenta). Right panel, merged image. Scale bar, 20 μm; #P ≤ 0.05, **P ≤ 0.001, ***P ≤ 0.0001.
The complete loss of TNF-α had a more significant effect on retinal neovascularization and vessel loss compared with the partial suppression with etanercept. As expected, Tnf+/+ wild type mice fed an ω-3-PUFA diet were protected from vaso-obliteration and neovascularization compared with their ω-6-PUFA-fed counterparts (P ≤ 0.00001) (Fig. 3d,e). However, in _Tnf_−/− mice at P17 there was a much larger but equal reduction in both diet groups in both vaso-obliteration and neovascularization. Under the condition of complete loss of TNF-α there was no further protection against retinopathy (vaso-obliteration or neovascularization) in ω-3-PUFA fed mice and no increase in retinopathy in ω-6-PUFA fed mice (Fig. 3d,e) indicating that TNF-α has a crucial role in the disease process.
We then sought the cellular source of TNF-α in the retina. Although several cell types can produce TNF-α, monocyte-derived cells are a major source20. Microglia, macrophages and dendritic cells are Csf1r+ inflammatory cells that are critical to the development and repair of the retinal vasculature21,22. To determine whether these cells or vascular endothelial cells produce TNF-α in retinopathy, we immunohistochemically labeled and localized TNF-α, Csf1r and vascular endothelial cells with confocal microscopy in retinal whole-mounts from mice on an ω-6-PUFA diet (Fig. 3f). TNF-α was found only in the subset of Csf1r+ cells that were associated with the retinal vasculature. This implies that under these experimental conditions, the primary source of TNF-α is a subpopulation of Csf1r+ macrophages or microglial cells that are in close proximity to blood vessels.
Monocyte or macrophage induction of Tnf gene expression is predominately dependent upon an upstream promoter region containing NF-κB DNA-binding motifs23. Using NF-κB–luciferase (NF-κB-Luc) reporter mice24, we found that NF-κB activity was increased ~100% (P ≤ 0.00001) in the retinas of ω-6-PUFA versus ω-3-PUFA-fed mice (Fig. 3g). In retinal whole-mounts, NF-κB transcriptional activity was found in a subset of microglia closely associated with the retinal vasculature (Fig. 3h), as were ChemR23 (Fig. 2g) and TNF-α (Fig. 3f). These results indicate that a subset of retinal macrophages or microglia may be involved in retinal angiogenesis, acting in part through the regulation of NF-κB-driven TNF-α production.
DHA and EPA acted on Csf1r+ microglia or macrophages in culture to inhibit Tnf mRNA production (Supplementary Fig. 3 online). Furthermore, the metabolites NPD1, RvD1 and RvE1 dose-dependently reduce Tnf mRNA expression in macrophages in culture and in other inflammatory models15,25–27.
In summary, ω-3- and ω-6-PUFAs significantly influence vascular growth and pathology. EPA, DHA and their potent bioactive products (NPD1, RvD1 and RvE1) at physiological levels reduce pathologic neovascularization through enhanced vessel regrowth after vascular loss and injury. Macrophages or microglia are a critical component of retinal vascular growth and repair21,22. In the retina, increased dietary ω-6-PUFA (arachidonic acid) increases activated microglial production of TNF-α, which is suppressed with elevated ω-3-PUFA levels (DHA, EPA). These effects on angiogenesis are likely to be important for retinopathy as well as other pathologies where vascular loss precipitates disease. The ω-3-PUFA suppressive effect on retinopathy in the mouse eye is comparable in magnitude to that of treatment with a vascular endothelial growth factor (VEGF) inhibitor28 and it is likely to be additive to anti-VEGF therapy, as VEGF was not significantly suppressed with the ω-3-PUFA diet (data not shown).
These results indicate that enriching the sources of ω-3-PUFA may be an effective therapeutic approach to help prevent proliferative retinopathy. The resolvins RvD1 and RvE1 and the neuroprotectin NPD1 are potent anti-inflammatory and pro-resolving mediators29. The present study establishes the first results to our knowledge indicating that these recently discovered mediators are also potent regulators of angiogenesis. If supplementation with DHA, EPA or their bioactive mediators is found to be as effective in ameliorating retinal vascular disease in humans as was the case in mice in the present study, this cost-effective intervention could benefit millions of people.
METHODS
Mice
These studies adhered to the Association for Research in Vision and Ophthalmology Statement for the Use of Animals in Ophthalmic and Vision Research and were approved by Children's Hospital Animal Care and Use Committee. For diet studies, C57BL/6 mothers at delivery were fed a defined rodent diet with 10% (w/w) safflower oil containing either 2% ω-6-PUFAs (arachidonic acid) and no ω-3-PUFAs (DHA and EPA), or 2% ω-3-PUFAs and no ω-6-PUFAs (Supplementary Table 1). Lipids in the diet were stable over time with respect to oxygen exposure, as there was no detectable lipid oxidation nor lipid decomposition over the course of the experiments nor with cold storage of feed for a year (Supplementary Table 4 online). Dietary PUFAs were produced by DSM Nutritional Products under the trade name ROPUFA. The arachidonic acid and DHA supplements, under the trade names ARASCO and DHASCO respectively, were obtained from Martek Biosciences Corporation. The feed was produced at Research Diets Incorporated. Fat-1 transgenic mice contain a humanized fat-1 cDNA driven by the cytomegalovirus enhancer and a chicken β-actin promoter10. Fat-1 and control dams were fed a defined diet with elevated ω-6-PUFAs (Supplementary Table 2). TNF-α-deficient (_Tnf_−/−, Jackson Laboratories stock no. 003008) and Tnf+/+ wild type (stock no. 101045) mice were administered the same diets as the C57BL/6 mice above. The NF-κB Luc reporter mice (stock no. 006100) were made using the pBIIX-luciferase targeting construct with two copies of the NF-κB binding sites from the immunoglobulin κ light chain intronic enhancer in front of a minimal Fos promoter as described previously24.
O2-induced retinopathy (vessel degeneration, regrowth and pathological neovascularization)
To induce vessel loss, mice were exposed to 75% oxygen from P7 to P12 (ref. 1). Retinal vessel loss was assessed at P12 and vessel regrowth at P13, P15 and P17. Retinal neovascularization was evaluated at P17, 5 d after return to room air, when the neovascular response is greatest.
Quantification of vaso-obliteration and retinal neovascularization
See Supplementary Methods online.
Resolvins and neuroprotectins
Lipid mediator profiles were analyzed from the retinas of P17 pups fed either ω-3- or ω-6-PUFA enriched diets and exposed to oxygen from P7–P12. PUFA-derived products were extracted, identified and quantified using a deuterium-labeled internal standard and MS-MS based mediator informatics program. Results were obtained from retinas of six mice, each from a separate litter. See Supplementary Methods for further details.
Lipid extraction and fatty acid analysis
Retinal samples containing one retina each were analyzed and results were averaged to derive a mean score. Extracted methyl esters were quantified on a model 6890 series gas chromatograph (Agilent Technologies) using a fast gas chromatography method using a 1-μl injection at a 25:1 split ratio. See Supplementary Methods for further details.
TNF-α receptor (etanercept) treatment
Intraperitoneal injections of a soluble TNF-α receptor (etanercept, 500 μg per mouse) were given at P7, P12, P14 and P16 to mice raised on an ω-3-or ω-6-PUFA-rich diet as previously described30. Retinas were isolated, fixed, stained and whole-mounted at P17 to evaluate vaso-obliteration and retinal neovascularization, as previously described1,21.
Immunohistochemistry
Eyes were enucleated from mice fed an elevated ω-6-PUFA diet. Following a 1-h fixation in 4% paraformaldehyde at 25 °C, the retinas were dissected, permeabilized overnight at 4 °C with 1% Triton X-100 (T-8787, Sigma) in PBS, and stained with isolectin B4, as described in Supplementary Methods. Retinal microglia were visualized in retinas from mice with green fluorescent protein expression under the control of Csf1r regulatory elements (Jackson Laboratories stock no. 005070). Fluorescence was enhanced with a fluorescein isothiocyanate (FITC)-labeled secondary antibody to green fluorescent protein (F6005, Sigma). Antibodies to TNF-α (ab39542, Abcam) or ChemR23 (sc-32652, Santa Cruz) were used for colocalization studies according to manufacturers’ recommendations. Retinal whole-mounts were prepared as described in Supplementary Methods and visualized with a Leica SP2 confocal microscope using a ×40 objective with taken ×2 zoom. A stack of optical sections was at intervals of 0.16 μm and compiled to form a three-dimensional image in the yz plane. See Supplementary Methods.
Western Blotting
Quantitative analysis of gene expression (quantitative real-time PCR)
NF-κB activity analysis
Female NF-κB-Luc reporter mice were placed on experimental diets following birth and subjected to oxygen-induced retinopathy as described. At P14 we collected retinas from the pups and homogenized them in Glow Lysis Buffer (E2661, Promega). We analyzed 50 μg of retinal homogenate for luciferase activity using Promega's Bright-Glo Luciferase Assay system (E2610). Retinal whole-mounts were also taken at P14 and stained for luciferase protein with rabbit antibody to luciferase (Abcam, ab21176) and to CD11b (Serotec, MCA74G).
Statistical analysis
Results are presented as mean ± s.e.m. for mouse studies and mean ± s.d. for other studies. Analysis of variance with significance α = 0.05 was used for processing the data. A two-sample _t_-test was used as a post-test unless otherwise indicated.
Supplementary Material
S1
S2
S3
S4
S5
S6
S7
S8
ACKNOWLEDGMENTS
We thank C. DiMartino, N. Liu, J.-R. Mo and K. Percarpio for technical help and J.-Y. Tsai for discussions. We thank the US National Institutes of Health Office of Dietary Supplements. This research was generously supported by the V. Kann Rasmussen Foundation and the US National Institutes of Health (EY008670, EY017017, EY14811 (L.E.H.S.); 1F32 EY017789, 5 T32 EY07145 (K.M.C.); P50-DE016191, GM38765 (C.N.S.); and Children's Hospital Boston Mental Retardation and Developmental Disabilities Research Center, P01 HD18655). We thank the Juvenile Diabetes Research Foundation for fellowship support (J.C.). This work was also supported by the Research to Prevent Blindness Lew Wasserman Merit Award (L.E.H.S.). The sponsors had no role in the design or conduct of the study, in the collection, analysis and interpretation of data or in the preparation, review or approval of the manuscript.
Footnotes
Supplementary information is available on the Nature Medicine website.
References
- 1.Smith LE, et al. Oxygen-induced retinopathy in the mouse. Invest. Ophthalmol. Vis. Sci. 1994;35:101–111. [PubMed] [Google Scholar]
- 2.Crawford MA, et al. Are deficits of arachidonic and docosahexaenoic acids responsible for the neural and vascular complications of preterm babies? Am. J. Clin. Nutr. 1997;66:1032S–1041S. doi: 10.1093/ajcn/66.4.1032S. [DOI] [PubMed] [Google Scholar]
- 3.Kermorvant-Duchemin E, et al. Trans-arachidonic acids generated during nitrative stress induce a thrombospondin-1-dependent microvascular degeneration. Nat. Med. 2005;11:1339–1345. doi: 10.1038/nm1336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Fierro IM, Kutok JL, Serhan CN. Novel lipid mediator regulators of endothelial cell proliferation and migration: aspirin-triggered-15R-lipoxin A(4) and lipoxin A(4). J. Pharmacol. Exp. Ther. 2002;300:385–392. doi: 10.1124/jpet.300.2.385. [DOI] [PubMed] [Google Scholar]
- 5.Fliesler SJ, Anderson RE. Chemistry and metabolism of lipids in the vertebrate retina. Prog. Lipid Res. 1983;22:79–131. doi: 10.1016/0163-7827(83)90004-8. [DOI] [PubMed] [Google Scholar]
- 6.SanGiovanni JP, Chew EY. The role of omega-3 long-chain polyunsaturated fatty acids in health and disease of the retina. Prog. Retin. Eye Res. 2005;24:87–138. doi: 10.1016/j.preteyeres.2004.06.002. [DOI] [PubMed] [Google Scholar]
- 7.Calder PC. Polyunsaturated fatty acids and inflammation. Prostaglandins Leukot. Essent. Fatty Acids. 2006;75:197–202. doi: 10.1016/j.plefa.2006.05.012. [DOI] [PubMed] [Google Scholar]
- 8.Salem N, Jr., Litman B, Kim HY, Gawrisch K. Mechanisms of action of docosahexaenoic acid in the nervous system. Lipids. 2001;36:945–959. doi: 10.1007/s11745-001-0805-6. [DOI] [PubMed] [Google Scholar]
- 9.Serhan CN, Savill J. Resolution of inflammation: the beginning programs the end. Nat. Immunol. 2005;6:1191–1197. doi: 10.1038/ni1276. [DOI] [PubMed] [Google Scholar]
- 10.Kang JX, Wang J, Wu L, Kang ZB. Transgenic mice: fat-1 mice convert n-6 to n-3 fatty acids. Nature. 2004;427:504. doi: 10.1038/427504a. [DOI] [PubMed] [Google Scholar]
- 11.Moriguchi T, et al. Effects of an n-3-deficient diet on brain, retina, and liver fatty acyl composition in artificially reared rats. J. Lipid Res. 2004;45:1437–1445. doi: 10.1194/jlr.M400087-JLR200. [DOI] [PubMed] [Google Scholar]
- 12.Serhan CN, Arita M, Hong S, Gotlinger K. Resolvins, docosatrienes, and neuroprotectins, novel omega-3-derived mediators, and their endogenous aspirin-triggered epimers. Lipids. 2004;39:1125–1132. doi: 10.1007/s11745-004-1339-7. [DOI] [PubMed] [Google Scholar]
- 13.Hong S, Gronert K, Devchand PR, Moussignac RL, Serhan CN. Novel docosatrienes and 17S-resolvins generated from docosahexaenoic acid in murine brain, human blood, and glial cells. Autacoids in anti-inflammation. J. Biol. Chem. 2003;278:14677–14687. doi: 10.1074/jbc.M300218200. [DOI] [PubMed] [Google Scholar]
- 14.Tjonahen E, et al. Resolvin E2: identification and anti-inflammatory actions: pivotal role of human 5-lipoxygenase in resolvin E series biosynthesis. Chem. Biol. 2006;13:1193–1202. doi: 10.1016/j.chembiol.2006.09.011. [DOI] [PubMed] [Google Scholar]
- 15.Arita M, et al. Stereochemical assignment, antiinflammatory properties, and receptor for the omega-3 lipid mediator resolvin E1. J. Exp. Med. 2005;201:713–722. doi: 10.1084/jem.20042031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Meder W, et al. Characterization of human circulating TIG2 as a ligand for the orphan receptor ChemR23. FEBS Lett. 2003;555:495–499. doi: 10.1016/s0014-5793(03)01312-7. [DOI] [PubMed] [Google Scholar]
- 17.Goukassian DA, et al. Tumor necrosis factor-α receptor p75 is required in ischemia-induced neovascularization. Circulation. 2007;115:752–762. doi: 10.1161/CIRCULATIONAHA.106.647255. [DOI] [PubMed] [Google Scholar]
- 18.Kishore R, et al. The cytoskeletal protein ezrin regulates EC proliferation and angiogenesis via TNF-α–induced transcriptional repression of cyclin A. J. Clin. Invest. 2005;115:1785–1796. doi: 10.1172/JCI22849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gardiner TA, et al. Inhibition of tumor necrosis factor-α improves physiological angiogenesis and reduces pathological neovascularization in ischemic retinopathy. Am. J. Pathol. 2005;166:637–644. doi: 10.1016/s0002-9440(10)62284-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Vassalli P. The pathophysiology of tumor necrosis factors. Annu. Rev. Immunol. 1992;10:411–452. doi: 10.1146/annurev.iy.10.040192.002211. [DOI] [PubMed] [Google Scholar]
- 21.Ritter MR, et al. Myeloid progenitors differentiate into microglia and promote vascular repair in a model of ischemic retinopathy. J. Clin. Invest. 2006;116:3266–3276. doi: 10.1172/JCI29683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Checchin D, Sennlaub F, Levavasseur E, Leduc M, Chemtob S. Potential role of microglia in retinal blood vessel formation. Invest. Ophthalmol. Vis. Sci. 2006;47:3595–3602. doi: 10.1167/iovs.05-1522. [DOI] [PubMed] [Google Scholar]
- 23.Collart MA, Baeuerle P, Vassalli P. Regulation of tumor necrosis factor alpha transcription in macrophages: involvement of four κB-like motifs and of constitutive and inducible forms of NF-κB. Mol. Cell. Biol. 1990;10:1498–1506. doi: 10.1128/mcb.10.4.1498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Millet I, et al. Inhibition of NF-κB activity and enhancement of apoptosis by the neuropeptide calcitonin gene-related peptide. J. Biol. Chem. 2000;275:15114–15121. doi: 10.1074/jbc.275.20.15114. [DOI] [PubMed] [Google Scholar]
- 25.Duffield JS, et al. Resolvin D series and protectin D1 mitigate acute kidney injury. J. Immunol. 2006;177:5902–5911. doi: 10.4049/jimmunol.177.9.5902. [DOI] [PubMed] [Google Scholar]
- 26.Mukherjee PK, Marcheselli VL, Serhan CN, Bazan NG. Neuroprotectin D1: a docosahexaenoic acid-derived docosatriene protects human retinal pigment epithelial cells from oxidative stress. Proc. Natl. Acad. Sci. USA. 2004;101:8491–8496. doi: 10.1073/pnas.0402531101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Arita M, et al. Resolvin E1, an endogenous lipid mediator derived from omega-3 eicosapentaenoic acid, protects against 2,4,6-trinitrobenzene sulfonic acid-induced colitis. Proc. Natl. Acad. Sci. USA. 2005;102:7671–7676. doi: 10.1073/pnas.0409271102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Aiello LP, et al. Suppression of retinal neovascularization in vivo by inhibition of vascular endothelial growth factor (VEGF) using soluble VEGF-receptor chimeric proteins. Proc. Natl. Acad. Sci. USA. 1995;92:10457–10461. doi: 10.1073/pnas.92.23.10457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bannenberg GL, et al. Molecular circuits of resolution: formation and actions of resolvins and protectins. J. Immunol. 2005;174:4345–4355. doi: 10.4049/jimmunol.174.7.4345. [DOI] [PubMed] [Google Scholar]
- 30.Grounds MD, et al. Silencing TNFα activity by using Remicade or Enbrel blocks inflammation in whole muscle grafts: an in vivo bioassay to assess the efficacy of anti-cytokine drugs in mice. Cell Tissue Res. 2005;320:509–515. doi: 10.1007/s00441-005-1102-z. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
S1
S2
S3
S4
S5
S6
S7
S8