Dual roles of plcA in Listeria monocytogenes pathogenesis (original) (raw)
. Author manuscript; available in PMC: 2016 Apr 19.
Summary
The plcA gene of Listeria monocytogenes encodes a secreted phosphatidylinositol-specific phospholipase C (PI-PLC). Recent studies have established that transposon mutations within plcA result in avirulence for mice and pleiotropic effects when examined in tissue-culture models of infection. Genetic analysis reveals that many of the effects of the transposon insertions are due to loss of readthrough transcription from plcA into the downstream gene prfA, which encodes an essential transcription factor of numerous L. monocytogenes virulence genes. Construction of an in-frame deletion within plcA had no effect on expression of prfA thus allowing direct assignment of a role of the PI-PLC in pathogenesis. PI-PLC was shown to play a significant role in mediating escape of L. monocytogenes from phagosomes of primary murine macrophages. Interestingly, this defect manifested itself in vivo in the liver but not in the spleen of infected mice.
Introduction
Listeria monocytogenes is a Gram-positive, food-borne human and animal pathogen responsible for serious infections in immunocompromised individuals and pregnant women (Seeliger, 1988). Murine listeriosis has been extensively studied as a model for infections in which antibody plays little or no role (Mackaness, 1962; Hahn and Kaufman, 1981; North, 1970). Immunity to L. monocytogenes requires prior administration of sublethal doses of live bacteria or, alternatively, adoptive transfer of immune CDS+ T-cells, although innate immune mechanisms are also crucial (Portnoy, 1992), Thus, murine listeriosis is an excellent system for the study of host–parasite interactions for infections which require the induction of cell-mediated immunity. During the past few years there has been a surge of interest in the pathogenesis of L. monocytogenes as a model intracellular parasite. Much of this interest reflects the observations that this bacterium enters the host cytoplasm and uses a host system of actin-based motility to mediate movement both within a cell and from cell to cell without leaving the cytoplasm (Tilney and Portnoy, 1989; Dabiri et al., 1990; Mounier et al., 1990; Theriot et al., 1992).
The cell biology of L. monocytogenes intracellular parasitism can be broadly divided into five stages, including (i) internalization, (ii) escape from a vacuole, (iii) intracellular growth, (iv) intracellular movement and pseudopod formation using host actin filaments, and (v) cell-to-cell spread which proceeds through an intermediate double-membraned vacuole (Tilney and Porlnoy. 1989). Several bacterial genes whose products are required for these processes have recently been identified using transposon mutagenesis and characterization of the resulting defects in tissue-culture models of infection (Portnoy et al., 1992a). One region of the L. monocytogenes genome encodes a number of these virulence factors, including a pore-forming haemolysin, Listeriolysin O (LLO), which is largely responsible for lysis of the initial vacuole; two distinct phospholipases C including one. PI-PLC, specific for phosphatidylinositol and the other, PC-PLC, having a broad substrate specificity that includes phosphatidyl-choline (PC); a metalloprotease, Mpl, which apparently can activate PC-PLC; ActA, a surface protein required for actin assembly; and lastly, PrfA, which appears to be a specific transcription factor required for the regulated expression of genes for the above-mentioned proteins (Portnoy et al., 1992a). The precise functions, sites of action, and regulation of these determinants of pathogenicity are current topics of investigation.
During a screen for L. monocytogenes transposon mutants which formed small plaques in tissue-culture monolayers, we identified a mutant which had suffered an insertion within the structural gene encoding PI-PLC (Sun et al., 1990). The mutant was avirulent, was delayed in escape from host vacuoles and, within the cytoplasm, had a severe defect in cell-to-cell spread (Sun et al., 1990; Camilli et al., 1991). The defects of this mutant were due to either loss of PI-PLC activity and/or a polar effect on expression of the downstream prfA gene. Using a genetic approach, we now show that readthrough transcription of prfA from the plcA gene promoter is necessary for expression of genes required for cell-to-cell spread. We also show that PI-PLC is required for efficient escape of L. monocytogenes from host-cell vacuoles and for wild-type levels of growth in the livers of infected mice.
Results
Genetic complementation of _plcA_∷Tn_917_-LTV3 mutation
L. monocytogenes DP-L1054 contains a Tn_917_-LTV3 insertion in plcA (see Fig. 1A, vertical arrow), fails to secrete PI-PLC activity, is defective for cell-to-cell spread and consequently produces small plaques in tissue-culture monolayers, and is avirulent (Camilli et al., 1991; Sun et al., 1990). Transformation of DP-L1054 with a plasmid containing plcA under the expression of its own promoter (Fig. 1B, strain DP-L1341) resulted in a ninefold higher level of secreted PI-PLC activity than that of the wild-type strain (Table 1). The increase in PI-PLC activity over wild-type levels was probably due to an increased gene dosage of plcA on the multicopy plasmid (Wirth et al., 1986), whose replication functions have been reported to maintain a copy number of approximately 10 in Streptococcus sanguis (Behnke and Gilmore, 1981). However, possession of this plasmid (by DP-L1054) failed to complement the defect in cell-to-cell spread as determined by measuring plaque size in L2 fibroblast monolayers (Fig. 2). In contrast, transformation of DP-L1054 with a plasmid containing plcA and the immediate downstream gene prfA (Fig. 1B, strain DP-L1480) resulted in complementation of PI-PLC activity to a level 73 times that of the wild type (Table 1) and partially restored the wild-type plaque phenotype (Fig. 2). Surprisingly, transformation of DP-L1054 with a plasmid containing only prfA (Fig. 1B, strain DP-L1530) complemented plaque formation to nearly wild-type levels (Fig. 2), although it was unable to complement for loss of PI-PLC expression (Table 1). Thus, only plasmids containing prfA could complement the defect in cell-to-cell spread, suggesting that the defective cell-to-cell spread by DP-L1054 was due to loss of prfA expression and not loss of PI-PLC secretion. The most likely explanation for this was that the transposon insertion in DP-L1054 was exerting a polar effect on expression of the downstream prfA gene.
Fig. 1.
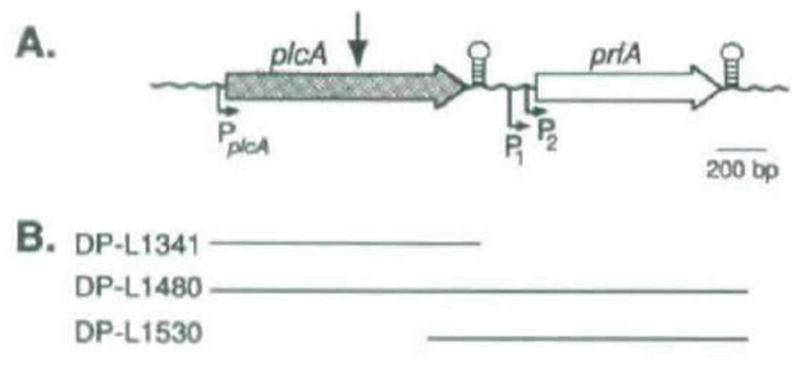
A Physical map of the L. monocytogenes chromosomal region containing plcA and prfA. The site of Tn_917_-LTV3 insertion in DP-L1054 is indicated by the arrow above the plcA gene (Sun et al., 1990). Stem and loop structures represent the locations of rho-independent terminator-like sequences (Mengaud et al., 1989; 1991). Promoters are indicated below the genes by arrows (Mengaud et al., 1989; 1991. N, Freitag and D. A. Portnoy, unpublished).
B. Complementation derivative strains of DP-L1054 and the respective L. monocytogenes DNA sequences (solid lines) contained by their comple-menting plasmids
Table 1.
Bacterial strains and properties.
| Strain | Genotype | Haemolytic activitya | PI-PLC S. A. | LD50 | PC-PLC S.A. | Source/Reference |
|---|---|---|---|---|---|---|
| 10403S | Wild-type Isolate | 220 | 7100 | 9.7 × 103b | ND | Portnoy et al, (1988) |
| 9.6 × 103c | ||||||
| DP-L1054 | 10403S _plcA_∷Tn_917_-LTV3 | 200 | 0 | 1.4 × 107 | ND | Sun et al. (1990) |
| DP-L1341 | DP-L1054 (pAM401∷plcA) | 130 | 62000 | ND | ND | This work |
| DP-L1480 | DP-L1054 (pAM401∷plcA prfA) | 3300 | 520000 | ND | ND | This work |
| DP-11530 | DP-LI054 (pAM40l∷prfA) | 2500 | 0 | ND | ND | This work |
| DP-L1503 | 10403S (pAM401∷plcA promoter) | 170 | 6400 | ND | ND | This work |
| DP-L1483 | 10403S (pAM401) | 200 | 7000 | ND | ND | This work |
| DP-L1510 | 10403S plcA_–_prfA intergenic integration with pKSV7∷prfA promoter | 170 | 7800 | ND | ND | This work |
| DP-L1532 | 10403S random Integration with pKSV7∷random chromosomal DNA fragment | 170 | 9600 | ND | ND | This work |
| DP-L1534 | 10403S random integration with pKSV7∷random chromosomal DNA fragment | 140 | 9600 | ND | ND | This work |
| DP-L1552 | 10403S Δ_plcA_ | 230 | 0 | 2.2 × 104b | ND | This work |
| 36 × 104c | ||||||
| DP-L1044 | 10403S _hly_∷Tn_917_-LTV3 | 0 | 8700 | 6.3 × 108 | ND | Sun et al. (1990) |
| SLCC 5764 | Hypersocreting wild-type isolate | 1250 | 88 000 | 1.3 × 105 | 3600 | Leimeister-Wachter et al. (1989) |
| DP-L1387 | SLCC 5764 _plcA_∷Tn_917_-LTV3d | 770 | 0 | ND | 200 | This work |
| DP-11553 | SLCC 5764 Δ_plcA_ | 1700 | 0 | ND | 3600 | This work |
| DP-L1535 | SLCC 5764 plcA_–_prfA intergenic integration with pKSV7∷prfA promoter | 770 | 5100 | ND | 54 | This work |
| DP-L1536 | SLCC 5764 random integration with pKSV7∷random chromosomal DNA fragment | 1100 | 19000 | ND | 1900 | This work |
Fig. 2.
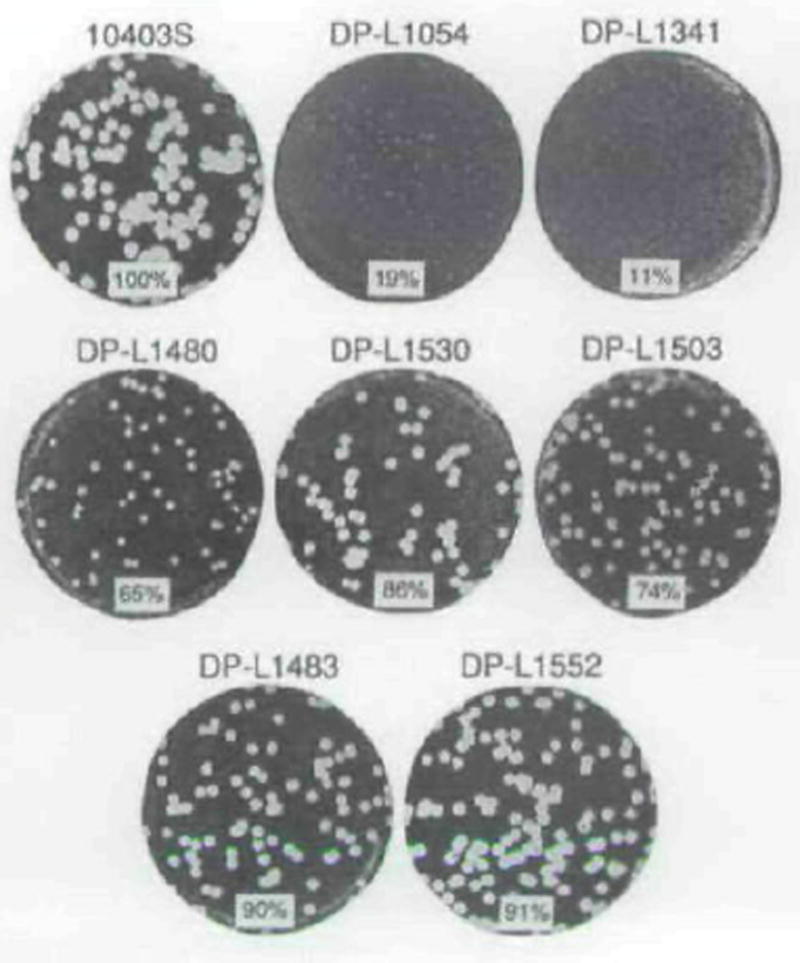
Plaque formation by L. monocytogenes strains in L2 cell monolayers. Plaques were stained with neutral red after 3 d. Strains are indicated above each well. The mean plaque diameter formed by each strain is shown at the bottom of each well as a percentage of the 10403S (wild-type) mean plaque diameter. The standard deviations of ail means was ≤9% of the wild-type mean plaque diameter.
The observation that the complemented strain containing both plcA and prfA (DP-L1480) made smaller plaques than that containing prfA alone (DP-L1530) (see Fig. 2) may have been due to the presence of the plcA promoter in multicopy in the former strain. To test this hypothesis, we transformed the wild-type strain 10403S with a plasmid containing a 616 bp fragment that included the plcA promoter, to generate DP-L1503. DP-L1503 formed smaller plaques than 10403S or 10403S transformed with vector alone (strain DP-L1483) (Fig. 2), suggesting that the presence of the plcA promoter in multicopy was slightly deleterious for cell-to-cell spread of L. monocytogenes. The most likely explanation for this effect was titration of PrfA by the palindromic DNA site recognized by PrfA (Freitag et al. 1992) which was present in the plcA promoter on the multicopy plasmid.
Loss of readthrough transcription in the transposon-containing strain DP-L1054 was difficult to confirm using Northern blot analysis because of low levels of transcription of plcA in the 10403S genetic background. However, we were able to show loss of readthrough transcription from plcA into prfA in another L. monocytogenes genetic background, the hyper-haemolytic isolate SLCC 5764, which exhibited significantly higher levels of plcA and prfA transcription during in vitro growth (compare lanes 1 and 5 in Fig. 3). In wild-type L. monocytogenes plcA and prfA are both transcribed monocistronically as RNA transcripts of approximately 1.1, 0.8 and 0.9 kb, respectively, as well as together in a bicistronic RNA transcript of approximately 2.0 kb (Fig. 3, lane 1) (Leimeister-Wachter et al., 1992; Mengaud et al., 1991). However, the bicistronic RNA transcript was absent in DP-L1387 (Fig. 3, lane 2), which has a Tn_917_-LTV3 insertion in the extreme 3’ end of plcA such that the monocistronic RNA transcript was not noticeably reduced in mobility (a predicted loss of approximately 93 bases from the 3’ end). Note that monocistronic transcription of prfA was unaffected in DP-L1387 (Fig. 3, lane 2). In contrast, monocistronic transcription of plcA was greatly reduced (Fig. 3, lane 2), suggesting that readthrough transcription of prfA contributes significantly to PrfA levels (PrfA activates transcription from the plcA promoter). The loss of secreted PI-PLC activity by DP-11387 (Table 1) was not due to the greatly reduced transcription of plcA (Fig. 3, lane 2) but instead was due to truncation of the _C_-terminal 7 amino acids of the PI-PLC polypeptide caused by the transposon insertion (data not shown). The presence of the bicistronic RNA transcript in 10403S and its corresponding absence in DP-L1054 (_plcA_∷Tn-_917_-LTV3) were confirmed in separate Northern blot experiments after longer autoradiogram exposure times (data not shown).
Fig. 3.

Upper panel. Physical map of the chromosomal region of L. monocytogenes containing the plcA and prfA genes. The directions of transcription and RNA transcripts are indicated schematically below each gene. DNA probes used in the detection of RNA transcripts are lettered A and B. Lower panel. Northern blot analysis autoradiograms of RNA transcripts from strains SLCC 5764 (lane 1), DP-L1387 (lane 2), DP-L1535 (lane 3), DP-L1553 (lane 4), 10403S (lane 5), DP-L1054 (lane 6), DP-L1510 (lane 7), and DP-L1552 (lane 8). After a 24 h exposure to generate the first autoradiogram on the left, the Northern filter membrane was washed to remove probe A and then rehybridized with probe B and exposed for 48 h to generate the second autoradiogram on the right (see the Experimental procedures). Equivalent amounts of RNA (25 μg) were analysed for each strain. The positions and approximate sizes of the 23S, 16S, and 5S ribosomal RNAs served as molecular weight markers and are indicated between the autoradiograms. The slowly migrating bands present for DP- L1054 (lane 6. probe A) represent chimaeric RNA transcripts resulting from a transcriptional fusion with Tn_917_-LTV3 which was inserted into the plcA gene in this strain. Similarly, the taint band of approximately 2.4 kb present for DP-L1510 (lane 7, probe A) resulted from a transcriptional fusion of plcA with one end of the integrated plasmid in this strain. A chimaeric RNA transcript of identical mobility was seen for DP-L1535 (with probe A) upon longer exposures (data not shown).
Loss of readthrough transcription of prfA by intergenic disruption results in small plaque formation
To directly test whether loss of readthrough transcription of prfA affects cell-to-cell spread, a plasmid having temperature-sensitive replication properties was integrated into the plcA_–_prfA intergenic region on the chromosome of wild-type L. monocytogenes to act, in effect, as a transcriptional terminator (Fig. 4). Northern blot analysis confirmed the loss of readthrough transcription in the resulting mutant strain, DP-L1510 (data not shown), and also demonstrated the unaffected monocistronic transcription of both plcA and prfA via their respective promoters (Fig. 3, lane 7). The same plasmid integration mutation made in the SLCC 5764 background more clearly demonstrates loss of the bicistronic RNA transcript (Fig. 3, lane 3). In addition, DP-L1510 secreted wild-type levels of PI-PLC and PrfA-induced haemolytic activity into culture supernatants (Table 1), further suggesting that the plasmid integration did not noticeably affect expression of the adjacent genes in the 10403S genetic background in vitro. However, in vivo, DP-L1510 was defective for cell-to-cell spread forming plaques in L2 cell monolayers that measured less than 25% of wild-type diameter (Fig. 5). As controls, two random integration strains, DP L1532 and DP-L1534, formed medium-sized plaques measuring 62% and 69% of wild-type plaque diameter, respectively (Fig. 5). Thus, readthrough transcription from plcA into prfA was required for large plaque formation in tissue-culture cells. The medium-sized plaques formed after random integration of the plasmids were consistent with our observations that L. monocytogenes strains containing integrated temperature-sensitive plasmids are somewhat defective for cell-to-cell spread (A. Camilli and D. A. Portnoy, unpublished).
Fig. 4.
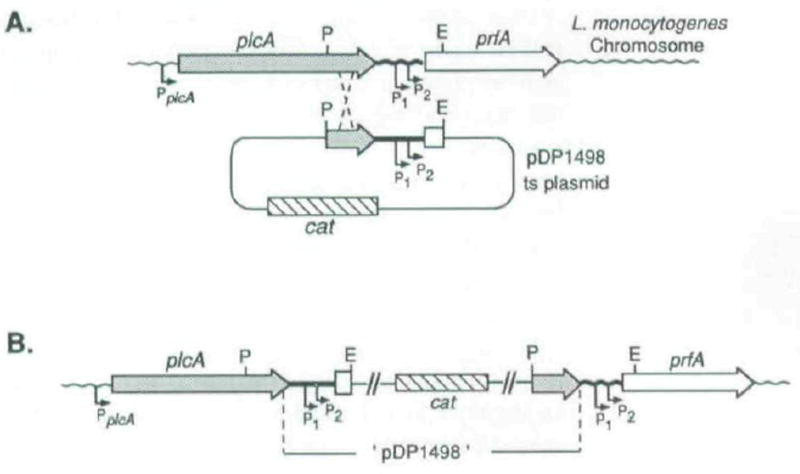
Schematic diagram for construction of the isogenic strains DP-L1510 and DP-L1535, in which readthrough transcription of prfA was blocked.
A. Chromosomal integration of pDP1498 by homologous recombination between the cloned _Pst_l (P)- and _Eco_RI (E)-generated fragment present on the plasmid and the homologous chromosomal sequence. The designated cross-over points are arbitrary. P1 and P2 indicate the two promoter elements immediately upstream of prfA while PplcA indicates the single plcA promoter (Mengaud et al., 1989; 1991; N. Freitag unpublished).
B. The resulting integration structure on the chromosome of both mutant strains. The integration strains were selected for, and maintained by, growth at a non-permissive temperature for plasmid replication in the presence of chloramphenicol. The plasmid was shown by Southern blot analysis to have integrated multiple times in a head-to-head configuration (data not shown).
Fig. 5.
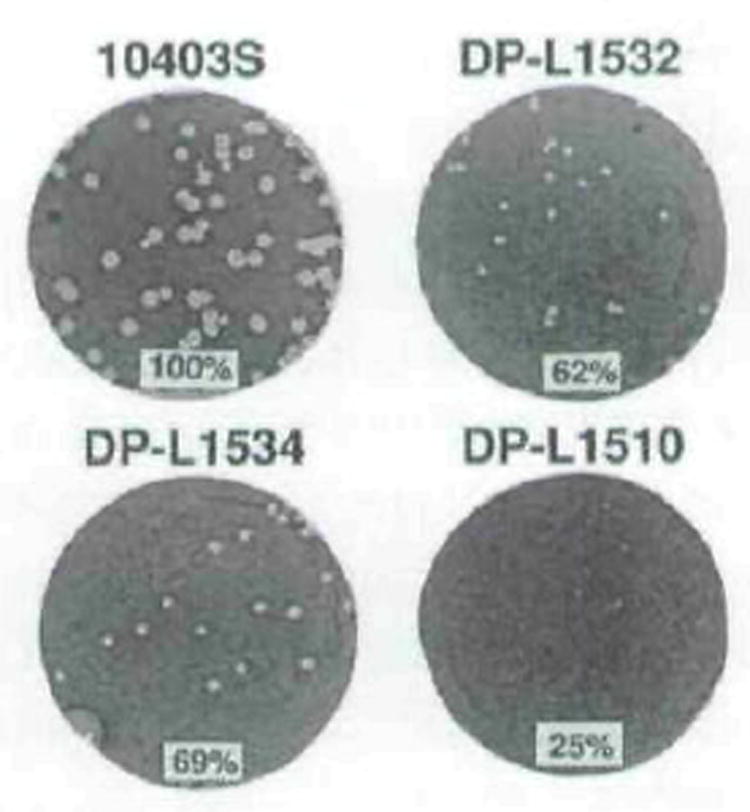
Plaque formation by wild-type L. manocytogenes (10403S), a mutant strain having readthrough transcription of prfA blocked by an integrated plasmid (DP-Ll510), and two control random integration strains (DP-L1532 and DP-L1534). Plaques were stained with neutral red after 3 d. Strains are indicated above each well. The mean plaque diameter formed by each strain is shown at the bottom of each well as a percentage of the 10403S (wild-type) mean plaque diameter. The standard deviations of all means was ≤9% of the wild-type mean plaque diameter.
Disruption of plcA by internal deletion does not affect plaque size
To independently confirm that PI-PLC was not required for cell-to-cell spread, we developed a method using allelic exchange to generate a strain containing an internal, in-frame, deletion in plcA (Fig. 6) to abolish PI-PLC expression but maintain readthrough transcription of prfA. The resulting strain, DP-L1552, had no detectable PI-PLC activity in culture supernatants (Table 1), secreted wild-type levels of haemolytic activity (Table 1) and, transcribed wild-type levels of the now truncated monocistronic and bicistronic RNA transcripts (Fig. 3, lanes 4 and 8). i.e., due to the internal Δ_plcA_. As shown in Fig. 2, DP-L1552 made large plaques indistinguishable from those of the wild-type strain thus confirming that (i) PI-PLC plays no apparent role in cell-to-cell spread, and (ii) that readthrough transcription of prfA from the upstream plcA gene promoter plays a major quantitative role in plaque formation. Hence, loss of readthrough transcription of prfA in the original transposon mutant strain, DP-L1054, appears to have been entirely responsible for its defective cell-to-cell spread.
Fig. 6.
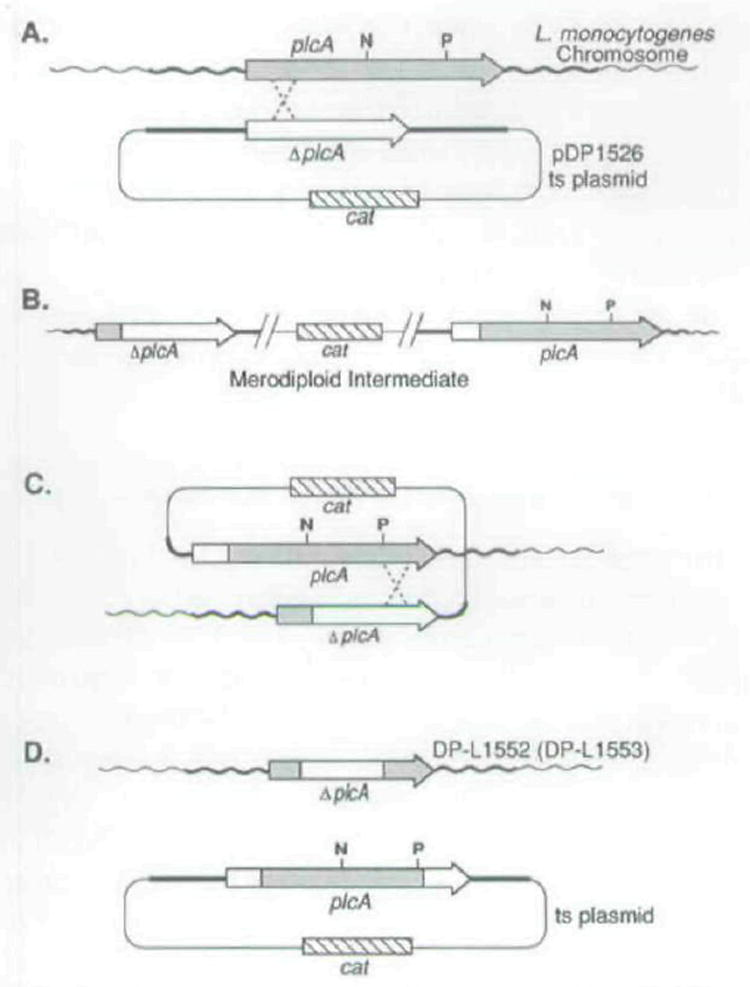
Schematic diagram for construction of the isogenic strains DP-L1552 and DP-L1553 each containing an in-frame Internal deletion in plcA.
A. Chromosomal integration of pDP1526 by homologous recombination between the Δ_plcA_ allele on the plasmid, which had the internal _Nsi_l (N) to _Pst_l (P) fragment deleted, and the wild-type plcA chromosomal allele. The designated cross-over points are arbitrary; however, recombination occurred on either side of the internal deletion.
B. The resulting integration structure on the chromosome which was selected for by growth at a non-permissive temperature for plasmid replication in the presence of chloramphenicol. Upon passage of the merodiploid intermediate strains for several generations without selective pressure, spontaneous excision of the integrated plasmid from the chromosome occurred (C). After curing of the excised plasmids at a non-permissive temperature for plasmid replication (D), L. monocylogenes Cms revertants were recovered at a frequency of = 1 %. Approximately 50% of the Cms revertants retained the Δ_plcA_ allele on the chromosome. These resulted from excision of the integrated plasmid via homologous recombination on the opposite side of the deletion allele as shown in (C).
Loss of readthrough transcription of prfA in L. monocytogenes SLCC 5764 results in reduced secretion of several polypeptides in vitro
In addition to plcA, at least four other linked L. monocytogenes genes encode secreted proteins including the structural genes for LLO, PC-PLC, Mpl, and ActA (Mengaud et al., 1989, Vazquez-Boland et al., 1992; Domann et al., 1992). The prfA gene product has recently been shown to be necessary for transcriptional activation of all five of these linked genes, and perhaps others (Freitag et al., 1992; Chakraborty et al., 1992; Mengaud et al., 1991). The experiments described in the sections above demonstrated that readthrough transcription of prfA was required for cell-to-cell spread. Accordingly, the defect in cell-to-cell spread may have resulted from reduced expression of _prfA_-activated genes. However, strains DP-L1054 and DP-L1510, in which readthrough transcription was blocked, still expressed wild-type levels of the _prfA_-activated gene product LLO in vitro (Table 1 and Sun et al., 1990) and had secreted protein profiles indistinguishable from that of the wild-type strain (data not shown). Thus, the polar effect was manifested while inside host cells but had no measurable effect on secreted levels of _prfA_-induced polypeptides in vitro.
The majority of L. monocytogenes clinical isolates resemble 10403S in the levels of secreted proteins in vitro. However, a minority of isolates such as SLCC 5764 exhibit a hypersecreting phenotype that includes high levels of secretion of the PrfA-induced gene products LLO, PC-PLC, PI-PLC, and ActA (A. Camilli and D. A. Portnoy, unpublished). Although not proven, the hypersecretory phenotype of SLCC 5764 in vitro may approximate the in vivo behaviour of 10403S at an intracellular stage of infection with respect to secreted levels of PrfA-induced proteins. Given the technical difficulties of assessing the levels of protein secretion of 10403S and derivative strains when intracellular, we therefore examined whether blocking readthrough transcription of prfA in SLCC 5764 would result in reduced secretion of _prfA_-regulated proteins in vitro. A SLCC 5764 derivative strain, DP-L1387, containing a Tn_917_-LTV3 insertion in plcA, had readthrough transcription blocked (Fig. 3, lane 2). This mutant strain secreted into culture supernatants only 6% and 62% of the wild-type levels of PC-PLC and haemolysin activities, respectively (Table 1), and accordingly showed reduced amounts of PC-PLC and LLO polypeptides on an SDS-PAGE gel of secreted proteins (compare lanes B and D in Fig. 7 and see legend). In addition to LLO and PC-PLC, at least six other polypeptide species were present in reduced amounts compared to the parental strain or to DP-L1553 which had an inframe internal deletion in plcA (Fig. 7, lane C). Furthermore. DP-L1535, which had readthrough transcription blocked by integration of a temperature-sensitive plasmid into the _plcA_-prfA intergenic region, had a secreted protein profile similar to that of DP-L1387 and, in addition, showed reduced secretion of PI-PLC (Table 1; Fig. 7, lane E and legend), while a random integration control strain, DP-L1536, had essentially a wild-type profile (Fig. 7, lane F). Thus, readthrough transcription of prfA does occur in vitro in the hypersecreting strain SLCC 5764 and was necessary for full expression of several secreted polypeptides, some of which represent known virulence factors.
Fig. 7.
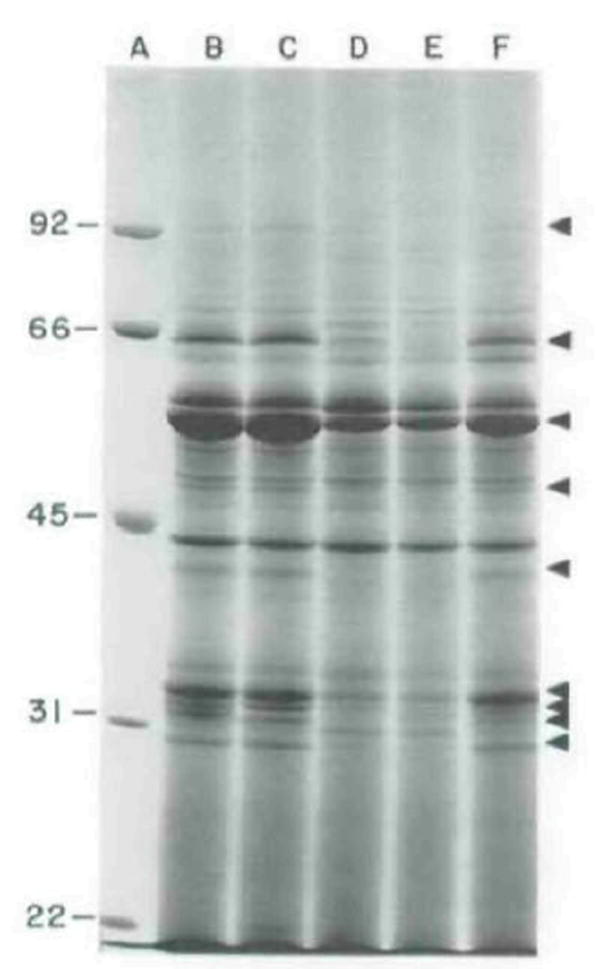
Coomassie-brilliant-blue stained SDS–PAGE of secreted polypeplides of mutant and wild-type L. monocytogenes SLCC 5764. Proteins were precipitated from culture supernatant fluids with 10% trichloroacetic acid and separated on a 7% SDS-polyacrylamide gel.
A. Molecular mass markers of the sizes indicated (in kDa) on the left; B, parental strain SLCC 5764; C, DP-L1553; D, DP-L1387; E, DP-L1535; F, DP-L1536. Arrows indicate polypeptide species of reduced expression in DP-L1387 and DP-L1535. LLO is the major band migrating at =58 kDa indicated by the third arrow from the top (Kathariou et al., 1987). The two secreted forms of PC-PLC are indicated by the bottom and fourth from bottom arrows (Raveneau et al., 1992; Kathariou et al., 1990; D. A. Portnoy, unpublished). The second arrow from the bottom indicates a doublet consisting of PI-PLC (top band), which accordingly is absent from DP-L1553 (compare lanes B and C), and an unknown polypeptide (bottom band).
PI-PLC is necessary for full virulence in mice
The small plaque mutant DP-L1054 (plcA_∷Tn_917_-LTV3) was previously reported to have a 50% lethal dose (LD50) in mice three logs higher than that of the wild-type strain (Camilli et al., 1991. and Table 1). Given the large plaque phenotype of the Δ_plcA strain, DP-L1552 (Fig. 2), we predicted that its LD50 would be close to that of the wild-type strain. Indeed, the LD50, for DP-L1552 was only two- to fourfold higher than that of 10403S (Table 1). Recall that DP-L1552 had no detectable secretion of PI-PLC but was competent for readthrough transcription from plcA into prfA.
To determine whether or not the reduced virulence of DP-L1552 in mice was due to a decreased ability to multiply within the murine host, we determined the ratios of 10403S to DP-L1552 in the livers and spleens of mice that had been simultaneously co-infected with equal numbers of the two strains 48 h previously (see Experimental procedures). It is known that ≈90% of intravenously administered L. monocytogenes are immediately sequestered in the livers and ≈10% in the spleens of mice and that these organs are the primary sites of subsequent bacterial multiplication during murine listeriosis (Mandel and Cheers, 1980). The ratios of 10403S to DP-L1552 in the spleens were not significantly different from a value of 1 (_P_>0.5) (see Fig. 8, closed circles). However, in the livers the ratios of 10403S to DP-L1552 were greater than 1 (P < 0.001) with a mean ratio of 33 (Fig. 8, open circles). Thus, although PI-PLC was not required for growth in vitro (DP-L1552 exhibited growth kinetics identical to wild-type in rich medium; data not shown) or in the spleens of mice, this secreted factor was required for wild-type levels of net bacterial growth in the murine liver following intravenous infection.
Fig. 8.
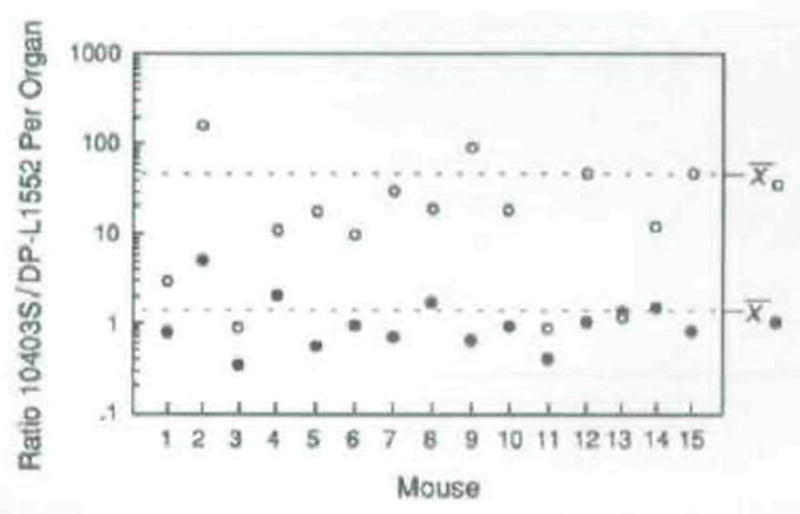
Ratio of wild-type L. monocytogenes to DP-L1552 in livers (open circles) and spleens (closed circles) of mice 48 h after infection. Note that the ratio ordinate is a log scale. Fifteen mice were injected intravenously with a sublethal dose of log-phase bacteria, sacrificed after 48 h, and the number of colony-forming units per organ was determined by plating serial dilutions of organ homogenates on agar media. The ratios of wild-type (PI-PLC+) to DP-L1552 (PI-PLC−) were determined by patching 200 colonies (derived from each organ homogenate) onto BHI/egg yolk agar and scoring for an opaque zone surrounding each PI-PLC+ isolate, after 24 h of bacteria growth (see the Experimental procedures). The mean ratios in liver and spleen are indicated on the right side of the figure. The ratio values for liver and spleen were statistically analysed for deviation from a value of 1 by the Wilcoxon paired sample test (Zar, 1974).
PI-PLC is necessary for wild-type growth in primary cultures of murine macrophages
DP-L1552 (Δ_plcA_) formed plaques whose sizes were almost indistinguishable from those of the wild-type strain in L2 cell monolayers (Fig. 2), suggesting that PI-PLC was not required for intracellular growth or cell-to-cell spread in this fibroblast cell line. However, it remained possible that PI-PLC would play a role in survival and/or growth within macrophages, which are the first host cells to encounter L. monocytogenes after i.v. infection. Therefore, we compared the growth of the wild-type strain 10403S with that of its isogenic derivative DP-L1552 in both established and primary cultures of murine macrophages. As previously reported (Portnoy et al., 1988; Sun et al., 1990) 10403S and DP-L1054 (_plcA_∷Tn_917_-LTV3) grew logarithmically in the J774 macrophage-like cell line while a non-haemolytic derivative, DP-L1044, incapable of lysing the phagosomal membrane (Sun et al., 1990), did not grow (Fig. 9A). The growth of DP-L1552 was similar to that of 10403S in this cell line, confirming that PI-PLC was not necessary for growth in J774 cells.
Fig. 9.

Intracellular growth of L. monocytogenes strains in primary and established murine macrophages cells. A, J774 cell-line; B, peritoneal-derived macrophages; C, bone marrow-derived macrophages, ○, wild-type strain 10403S; ●, DP-L1552; □, DP-L1054: ■, DPL1044. Data points and error bars represent the mean and standard deviations of the number of viable bacteria recovered from three coverslips.
We next tested bacterial growth in the more stringent cell types: peritoneal- and bone marrow-derived macrophages. In contrast to the J774 cell-line, these primary cell populations are capable of killing L. monocytogenes in tissue culture (Fig. 9B and C; Portnoy et al., 1988; 1989) and thus are thought to have more physiological relevance to in vivo macrophage populations. DP-L1054, DP-L1044, and DP-L1552 were continually killed in peritoneal macrophages while 10403S showed limited growth after 3 h (Fig. 9B). Light-microscopic examination of the 10403S-infected peritoneal macrophage monolayers 8h after infection (data not shown) revealed occasional foci of infection resulting from bacterial multiplication and cell-to-cell spread. In contrast, no foci of infection were found for the negative control strain DP-L1044 which is non-haemolytic and thus incapable of escaping from host vacuoles (Gaillard et al., 1987; Tilney and Portnoy, 1989). Strains DP-L1054 and DP-L1552, despite net killing over the course of the experiment, made rare foci of infection. Although in DP-L1054 and DP-L1552 foci occurred less frequently than in 10403S-infected monolayers, they were, nonetheless, identical in appearance to wild-type foci with respect to the number of bacteria and extent of their cell-to-cell spread which, for DP-L1552, suggested that PI-PLC was not important for events subsequent to escape from the initial host cell phagosome. In bone marrow-derived macrophage cultures all strains tested except for the non-haemolytic strain DP-L1044 showed net growth (Fig. 9C). Again, microscopic examination of infected monolayers revealed less frequent foci of infection for both DP-L1054 and DP-L1552 compared to 10403S infected monolayers, i.e. 9.5% and 38% of the foci per unit area for 10403S-infected monolayers, respectively (data not shown). The reduced net growth and less frequent foci of infection formed by DP-L1552 compared to 10403S in peritoneal and bone marrow-derived macrophages was consistently seen in multiple experiments (data not shown).
The possibility existed that an unlinked spontaneous mutation had occurred during the construction of DP-L1552 which was responsible for defective intracellular growth in macrophages. To test for this possibility a revertant of DP-L1552 was constructed using allelic exchange to replace the Δ_plcA_ allele with the wild-type plcA allele (essentially the reverse of the method shown schematically in Fig. 6). The revertant strain secreted wild-type levels of PI-PLC into culture supernatants and exhibited wild-type intracellular growth in peritoneal-derived macrophages (data not shown), thus confirming that the Δ_plcA_ was responsible for loss of PI-PLC expression and defective intracellular growth of DP-L1552.
PI-PLC is required for efficient lysis of host vacuoles in bone marrow-derived macrophages
The reduced frequency of infectious foci for DP-L1552 compared to 10403S-infected peritoneal- and bone marrow-derived macrophages was not due to reduced invasiveness, as the number of intracellular bacteria at early time points was essentially identical for all strains tested (Fig. 9, A, B and C). It was possible that inefficient lysis of phagosome membranes by DP-L1552 led to increased residence times within the potentially harsh phagosomal environment thus resulting in increased killing of the bacteria. Accordingly, inefficient escape from phagosomes may have been responsible for the reduced number of infectious foci seen for DP-L1552 versus wild-type- infected monolayers. We tested this hypothesis by direct quantification of membrane-bound versus cytoplasmic bacteria after infecting bone marrow-derived macrophages with 10403S or DP-L1552. Peritoneal-derived macrophages were not used for this experiment because the number of cytoplasmic bacteria was too low (because of the extensive killing of L. monocytogenes by these cells; Fig. 9B) to allow electron-microscopic quantification of significant numbers. Both stationary and log-phase cultures of L. monocytogenes, representing two different bacterial physiological states which may occur in vivo during infection, were used to infect bone marrow-derived macrophages in separate experiments. Electron-microscopic examination of infected monolayers fixed in situ clearly revealed the presence or absence of a vacuolar membrane surrounding the intracellular bacteria (Fig. 10). At 1.5 h after identical infections with stationary-phase bacteria, 7.5% of wild-type bacteria had lysed the phagosomal membrane and were found free in the cytoplasm while only one DP-L1552 bacterium (0.4%) was found free in the cytoplasm (Table 2). At 1.5h after identical infections with log-phase bacteria, 44% of wild-type bacteria had lysed the phagosomal membrane and were found free in the cytoplasm while only 15% of DP-L1552 bacteria were found free in the cytoplasm (Table 2). Thus, PI-PLC was necessary for efficient lysis of the phagosomal membrane in primary cultures of bone marrow-derived macrophages. In addition, log-phase L. monocytogenes appears to escape the phagosomal compartment of bone marrow-derived macrophages more efficiently than stationary-phase bacteria.
Fig. 10.
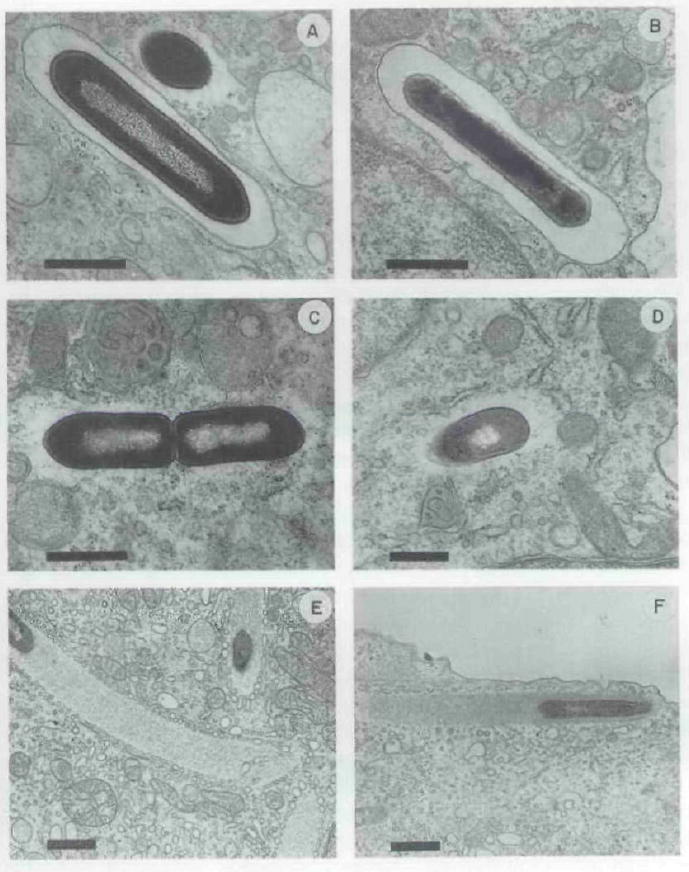
Representative electron micrographs of thin sections of portions of bone marrow-derived macrophages infected with stationary-phase L-monocytogenes and fixed in situ. A, C. E, wild-type strain 10403S after 1, 1.5, and 4 h of infection, respectively; B, D, F, DP-L1552 alter 1, 1.5, and 4 h of infection, respectively. The presence of host membrane surrounding bacteria is shown in (A) and (B). while bacteria free in the cytoplasm are shown in the remaining panels. Actin tails were seen trailing both the wild-type and mutant L. monocytogenes bacteria after 4 h of infection (panels E and F). It is worth noting the presence in panel E, and to a lesser extent in panel F, of many small vesicles ‘coating’ the periphery of the actin tails. This was observed infrequently in electron micrographs from this experiment as well as from other experiments using different host cell types (data not shown), and thus may be highly dependent upon variable fixation conditions. The function and/or significance of these small vesicles is not known. Bars = 0.1 μm.
Table 2.
Electron-microscopic quantification of vacuolar versus cytoplasmic bacteria in bone marrow-derived macrophages after infection with stationary- or log-phase L. monocytogenes.
| Strain | Bacterial phase of growtha | Total number of bacteria examined | Number of bacteria in vacuoles | Number (% of total) of bacteria in cytoplasm |
|---|---|---|---|---|
| 10403S | Stationary | 322 | 298 | 24 (7) |
| DP-L1552 | Stationary | 262 | 261 | 1 (0.4) |
| 10403S | Logarithmic | 186 | 105 | 81 (44) |
| DP-L1552 | Logarithmic | 170 | 144 | 26 (15) |
Electron micrographs of cytoplasmic 10403S or DP-L1552, 4 h after infection, clearly revealed the presence of electron-dense actin tails trailing the bacteria (Fig. 10, E and F) (Tilney and Portnoy, 1989), suggesting that loss of PI-PLC did not affect actin assembly. In addition, video microscopy of infected PTK2 cells revealed no measurable difference in intracellular movement of DP-L1552 versus wild type (J. Theriot and D. A. Portnoy, unpublished).
Discussion
The data presented here show that the L. monocytogenes plcA gene plays a dual role in pathogenesis. Perhaps its most critical role is in regulation, as transcripts which initiate at the plcA promoter and read through a putative terminator (Mengaud et al., 1989) in vivo are required for full expression of the positive regulatory factor PrfA which, in turn, regulates genes necessary for efficient cell-to-cell spread. A second role of plcA is to encode PI-PLC which is required for bacterial growth in the murine liver, which may reflect its role in lysis of macrophage phagosomes. Thus, L. monocytogenes has adopted a strategy for coupling the expression of one virulence factor to that of a positive regulatory factor which is necessary for expression of other genes.
plcA provides readthrough transcription of prfA which is required for cell-to-cell spread
In a previous study, we found that a _L. monocytogenes plcA_∷Tn_917_-LTV3 mutant (DP-L1054) was defective in the nucleation of host actin filaments and showed impaired cell-to-cell spread (Sun et al., 1990). Since the results of the present study show that PI-PLC has no apparent role in cell-to-cell spread, we suggest that readthrough transcription of prfA occurs in the host cell cytoplasm and is required for actin-based motility and cell-to-cell spread. In this respect, an intact actA gene has recently been shown to be required for bacterial-mediated nucleation of host actin (Kocks et al., 1992; Domann et al., 1992) and requires prfA for its transcription activation (Mengaud et al., 1991; Chakraborty et al., 1992). Thus, it is likely that actA is one of the downstream genes not properly expressed in loss of prfA readthrough transcription mutant strains. Indeed, DP-L1054 makes greatly reduced levels of ActA in infected J774 cells (R. A. Brundage and D. A. Portnoy, unpublished). Our data showing reduced secretion of at least eight polypeptides after blocking of readthrough transcription in a hypersecreting L. monocytogenes strain suggests that PrfA also regulates expression of other bacterial factors which may interact with host molecules at other stages of intracellular parasitism. Indeed, four of the eight polypeptides of reduced expression are known virulence factors. These include LLO, which is required tor lysis of the host phagosome, two forms of PC-PLC, which are required for efficient escape from the double-membraned vacuole formed after spreading cell-to-cell, and PI-PLC (Vazquez-Boland et al., 1992), which we now show plays a role in lysis of the host phagosome formed after the initial invasion event. Our data showing reduced expression of the above-mentioned virulence factors are consistent with previous reports indicating that PrfA activates transcription of plcA, hly, actA, and plcB (Chakraborty et al., 1992; Mengaud et al., 1991; Freitag et al., 1992). Interestingly, the differing levels of reduction of PC-PLC versus LLO expression after blocking of readthrough transcription of prfA (94% and 38% reduction compared with control strain activities, respectively) suggest a model of differential regulation for these two determinants in vitro and perhaps in vivo.
Readthrough transcription of a gene that encodes a positive transcription factor of the upstream gene as well as other genes involved in virulence was, to our knowledge, unprecedented in bacteria. In addition to the promoter provided by the plcA gene, prfA has its own two promoters immediately upstream of the PrfA coding sequence, providing other possible levels of PrfA expression in vivo (Mengaud et al., 1991; N. E. Freitag and D. A. Portnoy, unpublished). The relative contributions of these three promoters in pathogenesis are currently under investigation. It is likely that the expression of prfA is highly regulated via all three promoters to allow precise control over the levels of PrfA in the bacterial cell during various stages of intracellular parasitism. Readthrough transcription may be one mechanism that L. monocytogenes uses to temporally regulate PrfA levels during infection.
Role of PI-PLC in pathogenesis
Our studies show that PI-PLC is required for full virulence of L. monocytogenes in a murine model of infection. Interestingly, PI-PLC was necessary for growth in the liver but not the spleen. In the liver, resident macrophages (Kupffer cells) ingest the majority of intravenously injected L. monocytogenes (Lepay et al., 1985). The surviving bacteria spread into hepatocytes and grow logarithmically until host defence mechanisms are sufficiently mobilized to limit bacterial growth (Rosen et al., 1989). The precise role of PI-PLC in vivo is not known, but our results using bone marrow-derived macrophages suggest that PI-PLC plays a role in mediating efficient lysis of host phagosomes. Why PI-PLC has no apparent role in the spleen remains a mystery. In contrast to the liver, innate immunity to L. monocytogenes in the spleen is rather ineffective, as shown by a lack of bacterial killing in the spleen during the first hours of infection (Havell, 1989). Thus, the lack of a demonstrable role for PI-PLC in growth within the spleen may simply reflect a lack of efficient bactericidal properties of spleen cells harbouring L. monocytogenes. Specifically, delayed escape from phagosomes by PI-PLC-deficient L. monocytogenes may be tolerated in spleen cells perhaps because of inefficient listericidal activity of the phagosome compartments. This may reflect a different population of cells infected by L. monocytogenes in the spleen from that in the liver, and/or a different cellular environment.
What are the host target molecules of L. monocytogenes PI-PLC? Our results showing that PI-PLC was required for efficient bacterial escape from phagosomes in primary cultures of bone marrow-derived macrophages strongly suggests that PI-PLC may hydrolyse host PI and/or glycosyl-phophatidylinositol (GPI)-anchored proteins within the extracytoplasmic leaflet of the phagosomal membrane to facilitate escape into the cytoplasm. Goldfine and Knob (1992) have purified the L. monocytogenes PI-PLC to homogeneity and found that it specifically hydrolyses PI with high activity levels extending into the acidic pH range. In addition, LLO haemolytic activity has been found to have a low pH optimum (Geoffroy et al., 1987; Portnoy et al., 1992b). Thus, it is likely that PI-PLC may act in concert with LLO, possibly within acidified vacuoles of the host cell to mediate lysis of the vacuolar membrane.
The precise molecular events mediated by LLO and PI-PLC to cause lysis of host vacuoles are not known. LLO is a pore-forming protein which requires cholesterol as a membrane receptor (Smyth and Duncan, 1978; Geoffroy et al., 1987). Cholesterol is usually equally distributed between the cytoplasmic and extracytoplamic leaflets of eukaryotic plasma cell membranes (Fisher, 1976). In contrast, PI is predominantly found on the cytoplasmic leaflet but can be found in the extracytoplamic leaflet in large quantities in some cell types such as hepatocytes (Higgins et al., 1989). In addition, PI can be found in the extra-cytoplasmic leaflet in small quantities as lipid anchors for GPI-linked proteins (Cross, 1990). Interestingly, Ikezawa and Taguchi (Ikezawa and Taguchi, 1981) have reported that erythrocytes treated with purified Bacillus cereus PI-PLC are more sensitive to osmotic lysis. It is conceivable that L. monocytogenes PI-PLC may function similarly on phagosome membranes within other cell types of haematopoeitic origin in order to increase their sensitivity to lysis by LLO. In addition, PI-PLC may at some stage of lysis gain access to and hydrolyse PI within the cytoplasmic leaflet to facilitate dissolution of the phagosomal membrane. Whatever its role(s) may be, PI-PLC is not absolutely required for lysis of the phagosomal membrane, as PI-PLC-deficient L. monocytogenes can escape to the cytoplasm in all tissue-culture cells examined thus far (D. A. Portnoy, unpublished). In addition to PI-PLC, L. monocytogenes secretes a broad-substrate PLC (PC-PLC) which may function redundantly with PI-PLC to mediate lysis of phagosomal membranes in concert with LLO (see below).
The importance of PI-PLC, as well as LLO, in escape from the double-membraned vacuoles formed during cell-to-cell spread is not known. The presence of two host membranes (inverted with respect to each other) surrounding the bacteria may necessitate two distinct mechanisms for sequential membrane lysis. A L. monocytogenes PC-PLC mutant strain has been shown to be partially defective in escape from these double-membraned vacuoles, suggesting that PC-PLC plays a role in lysis of the inner membrane (Vazquez-Boland et al., 1992). PI-PLC-deficient L. monocytogenes appears to have little or no defect in cell-to-cell spread in tissue-culture. However, recent studies using Δ_plcA_ Δ_plcB_ single and double mutant strains have shown a major role for these two PLCs in production of wild-type size plaques, i.e. a synergistic reduction of plaque size was noted for the double mutant strain versus either single mutant strain alone (G. A. Smith and D. A. Portnoy, unpublished). Thus, PI-PLC and PC-PLC appear to have redundant functions.
Given that L. monocytogenes is an oral pathogen that must cross through the intestinal epithelial barrier, it is possible that PI-PLC may act at other stages of infection that were bypassed in the present studies using intravenous infections. In this respect, it is known that GPI-anchored proteins are found exclusively on the apical surface of intestinal epithelial cells (Lisanti et al., 1990). Therefore, L. monocytogenes PI-PLC may act on intestinal epithelia to somehow promote survival and/or invasion. However, it has recently been found that the purified L. monocytogenes PI-PLC has relatively little activity on eukaryotic GPI-anchored proteins relative to other bacterial PI-PLCs (H. Goldfine, personal communication). In addition, in preliminary experiments we were unable to detect a defect in the ability of a PI-PLC-deficient strain (DP-L1552) to gain access to and multiply within the livers and spleens of mice after oral challenge (data not shown).
The use of PI-PLC by L. monocytogenes to directly facilitate lysis of host phagosomes appears to be unique among intracytoplasmic pathogens. Although other bacterial species, including B. cereus. Bacillus thuringiensis, Clostridium novyi, and Staphylococcus aureus, secrete related PI-PLC enzymes, none of these is known to be an intracellular pathogen. However, this does not preclude action of these enzymes on host membranes extracellularly, particularly in the case of S. aureus for which there is evidence that PI-PLC contributes to the pathogenesis of infections (Marques et al., 1989). Interestingly, several protozoan intracellular parasites express developmentally regulated PI-PLCs hypothesized to mediate invasion of host cells indirectly. The most studied systems are the merozoite stage of malarial parasites (Breton et al., 1990) and Trypanosoma cruzi (Fenton Hall et al., 1992), which apparently use PI-PLCs to release endogenous GPI-anchored substrates which, in turn, act on host cell membrane to mediate invasion of erythrocytes (malarial parasites) and on host glycoproteins to mediate invasion of the cytoplasm of macrophages (T. cruzi).
Although PI-PLC is not an essential determinant of pathogenicity in the mouse model (in contrast to LLO) it nevertheless clearly plays a significant role in the growth of L. monocytogenes in some host tissues. It is reasonable that many virulence determinants will play subtle roles in pathogenesis when assayed in the laboratory, but in fact may contribute significantly to the overall fitness of a pathogenic species in nature. Molecular dissection of virulence determinants of this class will be necessary for a detailed understanding of host–parasite interactions.
Experimental procedures
Plasmid constructions
The Escherichia coli host for all plasmid constructions was DH5αMCR (F− mcrA, mcrB, mrr, φ80d_lacZ_ΔM15, Δ(lacZYA_–_argF)U169, recA1. endA1, hsdR, hsdM, supE44, λ− thi-1, gyrA, relA1) (Bethesda Research Laboratories Life Technologies, Inc.). Derivatives of pAM401 (Wirth et al., 1986) were maintained in E. coli by growth in the presence of 10 μ9 of chloramphenicol per ml of media. Plasmid pKSV7 is a shuttle vector capable of replication in E. coli and temperature-sensitive replication in B. subtilis and other Gram-positive bacteria such as L. monocytogenes (Smith and Youngman, 1992) and was maintained in E. coli in the presence of 50 μg of ampicillin per ml of media.
The L. monocytogenes plcA gene, bases 279 to 1349 (Mengaud et al., 1989), was amplified from 10403S chromosomal DNA using the polymerase chain reaction (PCR), and ligated into pAM401 using _Sal_I- and _Xba_I-generated DNA ends to generate pDP1339. The _Sal_I and _Xba_I restriction sites of the amplified product were created as non-complementary ends of the amplification primers. Primers used were 5′-GGGTCGACTAGGAATAATATATGTTGTTG-3′ and 5′-GGTCTAGATTCTAGTCCTGCTGTCC-3′. The plcA and prfA genes, bases −810 to 1349 (Mengaud et al., 1989), were amplified together from 10403S chromosomal DNA using the second primer above and 5′-GGGTCGACCAGCTCTTCTTGGTGAAG-3′. The amplified product was then ligated into pAM401 using _Sal_I- and _Xba_I-generated DNA ends to generate pDP1462. Plasmid PDP1500, containing prfA alone, was constructed by deleting the plcA gene, bases 429 to 1349 (Mengaud et al., 1989), from pDP1462 after restriction with _Xba_I and _Pst_l, treatment of the DNA ends with T4 DNA polymerase to make them blunt, and intramolecular ligation. Plasmid pDP1499, containing the plcA promoter and a portion of the 3′ end of plcA, was constructed by deleting a plcA internal fragment, bases 428 to 882 (Mengaud et al., 1989). from pDP1339 after restriction with _Pst_l and Nsi_l and intramolecular ligation. pDP1526 (pKSV7∷Δ_plcA) was constructed by a single three-part ligation of pKSV7 restricted with _Bam_HI and _Xba_I, the 468 bp _Xba_I and Nsi_l-generated fragment from pAM401∷_plcA containing the 5′ end of plcA (bases 882 to 1351; Mengaud et al., 1989) and the 501 bp _Pst_l- and Bam_HI -generated fragment from pAM401∷_plcA prfA containing the 3′ end of plcA (bases −77 to 429; Mengaud et al., 1989). The prfA promoter, bases 1–429 (Mengaud et al.,1989), was isolated by _Eco_RI and _Pst_l double digestion of pDP1462 and the fragment was subsequently ligated into _Eco_RI- and _Pst_l-restricted pKSV7 to generate pDP1498. Two random _Hind_III-generated 10403S chromosomal DNA fragments, approximately 3kb in length, were ligated into _Hind_III-restricted pKSV7, to generate the random integration control plasmids pDP1519 and pDP1521.
Construction of L. monocytogenes mutant strains
L. monocytogenes strain DP-L1387 was isolated as a mutant with reduced lecithinase (PC-PLC) from a Tn_917_-LTV3 bank of SLCC 5764, constructed as previously described (Camilli et al., 1990). The site of Tn_917_-LTV3 insertion was determined by sequencing one transposon-chromosomal DNA junction as previously described (Sun et al., 1990). L. monocytogenes was transformed with plasmid DNA as previously described (Camilli et al., 1990). Selective pressure for maintenance of pAM401, pKSV7, and their derivatives in L. monocytogenes was exerted in the presence of 10 μg of chloramphenicol per ml of media. In addition, maintenance of pKSV7 derivatives required growth at 30°C, a permissive temperature for plasmid replication in Gram-positive bacteria.
Integration of pKSV7 derivatives into the L. monocytogenes chromosome occurred by homologous recombination between L. monocytogenes DNA sequences on the plasmids and their corresponding chromosomal alleles (see Figs 4 and 6). Integration mutants were enriched by growth for approximately 30 generations at 40°C, a non-permissive temperature for pKSV7 replication, in Brain Heart Infusion (BHI) broth containing 10 μg chloramphenicol per ml of media. Each integration strain was subsequently colony purified on BHI agar containing 10 μg chloramphenicol per ml of media and incubated at 40°C. Southern blot analyses of chromosomal DNA isolated from each integration strain confirmed the presence of the integrated plasmid.
Construction of DP-L1552 containing a Δ_plcA_ allele on the chromosome is shown schematically in Fig. 6. Integration of the pKSV7 derivative, pDPl526, to generate a merodiploid intermediate was done as described above. Spontaneous excision of the integrated plasmid, through intramolecular homologous recombination, occurred at a low frequency. Bacteria in which the plasmid had excised from the chromosome were enriched by growth at 30°C in BHI broth for approximately 50 generations. The nature of the selective pressure during this step was not known but may be due to a slight growth defect of strains containing integrated temperature-sensitive plasmids. Approximately 50% of excision events, i.e. those resulting from homologous recombination between sequences 3′ of the deletion, resulted in allelic exchange of Δ_plcA_ for the wild-type allele on the chromosome. The excised plasmids were cured by growing the bacteria at 40°C in BHI for approximately 30 generations. Cured bacteria retaining the Δ_plcA_ allele on the chromosome were identified by their failure to produce a zone of turbidity surrounding colonies after growth on BHI agar plates containing a 5 ml overlay of BHI agar/2.5% egg yolk/2.5% phosphate-buffered saline (PBS) (BHI/egg yolk agar). The turbid zones resulted from PI-PLC hydrolysis of PI in the egg yolk, giving an insoluble diacylglycerol precipitate. The correct plcA deletion on the L. monocytogenes chromosome was confirmed by amplifying the deleted allele using PCR and sequencing across the deletion.
SDS–PAGE and activity assays
Each L. monocytogenes strain was grown overnight, from a single fresh colony, in a 2 ml BHI stationary culture containing 10 μg of chloramphenicol per ml of media either at 30°C for plasmid containing strains or at 40°C for integration strains; 1.5 ml of each overnight culture was subsequently used to inoculate 13.5 ml of BHI and the inoculated cultures were incubated with aeration either at (i) 37°C for 6h for 10403S integration derivative strains, SLCC 5764, and SLCC 5764 plasmid-containing derivatives, (ii) 37°C for 7h for SLCC 5764 integration derivative strains or, (iii) 37°C for 5h for all other strains. These variable times of incubation were empirically determined, by optical density measurements of growing cultures of all strains used, prior to producing final cultures of approximately the same stage of growth (early stationary phase). The resulting cultures were immediately chilled on ice for 5 min, and the bacteria sedimented at 4300 × g for 10 min at 4°C; 1 ml of each clear supernatant was transferred to a sterile polypropylene tube and kept on ice to be used immediately for haemolytic. PI-PLC, and PC-PLC activity assays. Haemolytic assays were done as previously described (Portnoy et al., 1989), PI hydrolysis assays were done essentially as described (Low, 1981) except that [3H-inositol]-PI, 0,045 μCi, 17.4 Ci mmol−1 (Amersham Corp.), and 13.5μg of PI (Sigma Chemical Co.) were sonicated in 100μ1 of 40 mM Tris-HCI, pH 7.2, and 0.2% deoxycholate (per assay), and the Pl-sonicate was then incubated with culture supernatant in a final volume of 200 μl at 37°C. PC hydrolysis assays were done exactly as above for PI hydrolysis except that [3H]-PC, 0.045μCi, 17.4 Ci mmol−1 (Amersham Corp.), and 13.5μg of PC (Sigma Chemical Co.) were used. When necessary, BHI broth was used as a diluent for high-activity supernatants; 9 ml of each clear supernatant was used for trichloroacetic acid precipitation and SDS–PAGE analysis of secreted polypeptides as previously described (Portnoy et al., 1988). Multiple activity and SDS–PAGE experiments were performed using separately grown cultures of the strains listed in Table 1 to confirm the relative activity levels and secreted protein profiles.
Northern blot analysis
L. monocytogenes wild-type and mutant strains were grown to early stationary phase as described in the section above (SDS–PAGE and activity assays). RNA was prepared from each strain according to the following method (E. Domann and T. Chakraborty, personal communication). Bacteria were sedimented from 10 ml of culture fluid at 4300 × g for 5 min at 20°C and the cell pellet resuspended in 1 ml of 25% sucrose, 25 mM Tris-HCI (pH 8), and 10 mg ml−1 lysozyme. The bacteria were incubated at 37°C with vigorous shaking (220 r.p.m.) for 1h, at which time 1 ml of 2% SDS and 100μl of 5 mg ml−1 proteinase K was added, and the bacteria were incubated at 37°C with vigorous shaking for an additional 30 min; 800μ1 of the viscous suspension (due to lysis of bacteria) was aliquoted to a micro-centrifuge tube and bacterial lysis was then brought to completion by freeze-thawing three times by incubation for 4 min each time in a dry ice-ethanol bath and then a 37°C water bath. The lysate was incubated at 65°C for 4min, at which time 400μl of 65°C phenol (previously equilibrated with 50 mM sodium acetate, pH 5.5) was added. The tube was mixed briefly by inversion and incubated for 5 min at 65°C. The aqueous and phenol phases were separated by centrifugation at 12000 × g for 5 min at room temperature. The aqueous phase was subsequently transferred to a clean microcentrifuge tube and extracted two more times with 400 μl of room-temperature phenol equilibrated as above. A final extraction of the aqueous phase was performed with an equal volume of water-saturated ether. RNA was precipitated with 0.1 volume of 3M sodium acetate (pH 5.2) and 2vols 100% ethanol at−70°C.
Twenty-five micrograms of RNA from each L. monocytogenes strain was separated on 1.1% agarose formaldehyde gels, blotted to nylon, and hybridized with random-primed plcA and _prfA_-specific DNA probes according to standard methods (Sambrook et al., 1989), The _plcA_-specific DNA probe extended from base number 882 lo 1350 (Mengaud et al., 1989), which includes the plcA promoter and the 5′ end but does not include the site of Tn_917_-LTV3 insertion in DP-L1054. The _prfA_-specific DNA probe extended from base number −396 to 1 (Mengaud et al., 1989), which includes the first two-thirds of the prfA coding sequence.
Virulence and plaquing assays
LD50 experiments were performed to assess the precise virulence of L. monocytogenes. LD50 values were determined after intravenous injection of BALB/c mice (Charles River) with bacteria grown overnight in Trypticase soy broth (TSB) as described (Portnoy et al., 1988), with the following modification; DP-L1341 was grown overnight in TSB containing 10 μg of chloramphenicol per ml of media.
Plaquing of L. monocytogenes strains was done as described previously (Sun et al., 1990), with the following modifications; all plasmid-containing strains were grown overnight at 30°C in BHI containing 10 μg chloramphenicol per ml of media, and all integration strains were grown overnight at 40°C in BHI containing 10 μg of chloramphenicol per ml of media. Plaques of plasmid-containing and integration strains were grown in the presence of 10 μg of chloramphenicol per ml of media at 37°C in a 5% CO2 atmosphere, All plaques were grown for 3d before staining with neutral red (Sigma).
Tissue-culture growth assays
Intracellular growth assays in J774 cells were done as previously described (Sun et al., 1990). Growth in resident peritoneal macrophages and bone marrow-derived macrophages was performed as described previously (Portnoy et al., 1988; 1989).
Growth in liver and spleen
10403S and DP-L1552 were grown overnight, from a single fresh colony, in 2 ml of BHI stationary cultures at 30°C; 1 ml of each overnight culture was used to inoculate 19 ml of BHI and the cultures were incubated at 37°C with aeration until an optical density of 0.8 was attained, representing =109 cfu per ml of log-phase bacteria. The cultures were immediately chilled on ice for 5 min, and the bacteria kept at 4°C in all subsequent steps. An equal volume of each of the 10403S and DP-L1552 cultures were combined and the bacteria washed twice in lipopolysaccharide-free PBS in a microcentrifuge. The bacteria were diluted to a final concentration of 1 × 109cfu ml−1 in lipopolysaccharide-free PBS and the bacterial titre measured by plating serial dilutions on BHI agar, A 1:1 ratio of 10403S to DP-L1552 in the final suspension was confirmed by testing 200 colonies from the BHI titre plates for their PI-PLC reaction on BHI/egg yolk agar. Approximately 1 × 103 cfu were given intravenously by tail-vein injection into BALB/c female mice 7–10 weeks old. The mice were given food and water ad libitum, and were sacrificed after 48 h by CO2-suffocation. The liver and spleen from each mouse were immediately removed and each homogenized in 12 ml of 4°C sterile water. Serial dilutions of each homogenate were plated onto Luria-Bertani agar containing 150 μg of streptomycin per ml of media, 10403S (and derivatives) is a spontaneous streptomycin-resistant derivative of 10403. The plates were incubated at 37°C for approximately 30 h, then 200 single colonies from each organ homogenate were patched using sterile toothpicks onto BHI/egg yolk agar to score for their PI-PLC-reaction. The ratio of PI-PLC-positive to PI-PLC negative bacteria represented the ratio of 10403S to DP-L1552 in the organs.
Electron microscopy
Bone marrow-derived macrophages were infected with stationary-phase bacteria as previously described (Portnoy et al., 1988) such that every cell contained approximately 10–20 bacteria after 30 min of incubation. Alternatively, log-phase bacteria, OD (600 nm) = 0.8 in BHI broth at 37°C with aeration, were used for infection such that every cell contained approximately 10–20 bacteria after 30 min of incubation. After 1.5 h of incubation, the infected monolayers were fixed in situ, and subsequently examined by electron microscopy as previously described (Tilney et al., 1990).
Acknowledgments
We thank E. Domann and T. Chakraborty for their advice on RNA preparation, R Barry and D, Hinrichs for the LD50 determinations, P. Connelly for doing the electron microscopy, R. Shaffer for training and advice on the mouse experiments. P. Youngman for his advice and enthusiasm, H. Goldfine for advice on the biochemistry of phospholipases, and J. Theriot, V Dirita. S. Moseley for (heir comments on the manuscript. This research was supported by grants HD-14474 (L.G.T.) AI-26919 and AI-27655 (D.A.P.), and AI-31537 (to H. Goldfine), from the National Institutes of Health.
References
- Behnke D, Gilmore MS. Location of antibiotic resistance determinants, copy control, and replication functions on the double-selective streptococcal cloning vector pGB301. Mot Gen Genet. 1981;184:115–120. doi: 10.1007/BF00271206. [DOI] [PubMed] [Google Scholar]
- Breton CB, Langsley G, Barale J, Pereira da Silva LH. A malaria phosphatidylinositol-specific phospholipase C: a possible role in merozoite maturation and erythrocyte invasion. In: Cohen CM, Palek J, editors. Cellular and Molecular Biology of Normal and Abnormal Erythroid Membranes. New York: Wiley-Liss; 1990. pp. 315–332. [Google Scholar]
- Camilli A, Portnoy DA, Youngman P. Insertional mutagenesis of Listeria monocytogenes with a novel Tn917 derivative that allows direct cloning of DNA flanking transposon insertions. J Bacteriol. 1990;172:3738–3744. doi: 10.1128/jb.172.7.3738-3744.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Camilli A, Goldfine H, Portnoy DA. Listeria monocytogenes mutants lacking phosphatidylinositol-specific phospholipase C are avirulent. J Exp Med. 1991;173:751–754. doi: 10.1084/jem.173.3.751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chakraborty T, Leimeister-Wachter M, Domann E, Hartl M, Goebel W, Nichterlein T, Notermans S. Coordinate regulation of virulence genes in Listeria monocytogenes requires the product of the prfA gene. J Bacteriol. 1992;174:568–574. doi: 10.1128/jb.174.2.568-574.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cross GAM. Glycolipid anchoring of plasma membrane proteins. Annu Rev Cell Biol. 1990;6:1–39. doi: 10.1146/annurev.cb.06.110190.000245. [DOI] [PubMed] [Google Scholar]
- Dabiri GA, Sanger JM, Portnoy DA, Southwick FS. Listeria monocytogenes moves rapidly through the host cytoplasm by inducing directional actin assembly. Proc Natl Acad Sci USA. 1990;87:6068–6072. doi: 10.1073/pnas.87.16.6068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Domann E, Wehland J, Rohde M, Pistor S, Hartl M, Goebel W, Leimeister-Wachter M, Wuenscher M, Chakraborty T. A novel bacterial virulence gene in Listeria monocytogenes required for host cell microfilament interaction with homology to the proline-rich region of vinculin. EMBO J. 1992;11:1981–1990. doi: 10.1002/j.1460-2075.1992.tb05252.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fisher KA. Analysis of membrane halves: cholesterol. Proc Nal Acad Sci USA. 1976;73:173–177. doi: 10.1073/pnas.73.1.173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freitag NE, Youngman P, Portnoy DA. Transcriptional activation of the Listeria monocytogenes hemolysin gene in Bacillus subtilis. J Bacteriol. 1992;174:1293–1298. doi: 10.1128/jb.174.4.1293-1298.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaillard JL, Berche P, Mounier J, Richard S, Sansonetti P. In vitro model of penetration and intracellular growth of Listeria monocytogenes in the human enterocyte-tike cell line Caco-2. Infect Immun. 1987;55:2822–2829. doi: 10.1128/iai.55.11.2822-2829.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geoffroy C, Gaillard JL, Alouf JE, Berche P. Purification, characterization, and toxicity of the sulfhydryl- activated hemolysin listeriolysin O from Listeria monocytogenes. Infect Immun. 1987;55:1641–1646. doi: 10.1128/iai.55.7.1641-1646.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldfine H, Knob C. Purification and characterization of Listeria monocytogenes phosphatidylinositol-specific phospholipase C. Infect Immun. 1992;60:4059–4067. doi: 10.1128/iai.60.10.4059-4067.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hahn H, Kaufmann SHE. The role of cell-mediated immunity in bacterial infections. Rev Inf Dis. 1981;3:1221–1250. doi: 10.1093/clinids/3.6.1221. [DOI] [PubMed] [Google Scholar]
- Fenton Hall BF, Webster P, Ma AK, Joiner KA, Andrews NW. Desialylation of lysosomal membrane glycoproteins by Trypanosoma cruzi: a proposed role for the surface neuraminidase in facilitating parasite entry into the host cell cytoplasm. J Exp Med. 1992;176:313–325. doi: 10.1084/jem.176.2.313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Havell EA. Evidence that tumor necrosis factor has an important role in antibacterial resistance. J Immunol. 1989;143:2894–2899. [PubMed] [Google Scholar]
- Higgins JA, Hitchin BW, Low MG. Phosphatidylinositol-specific phospholipase C of Bacillus thuringiensis as a probe for the distribution of phosphatidylinositol in hepatocyte membranes. Biochem J. 1989;259:913–916. doi: 10.1042/bj2590913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ikezawa H, Taguchi R. Phosphatidylinositol-specific phospholipase C from Bacillus cereus and Bacillus thuringiensis. Meth Enzymol. 1981;71:731–741. [Google Scholar]
- Kathariou S, Metz P, Hof H, Goebel W. Tn916-induced mutations in the hemolysin determinant affecting virulence of Listeria monocytogenes. J Bacteriol. 1987;169:1291–1297. doi: 10.1128/jb.169.3.1291-1297.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kathariou S, Pine L, George V, Carlone GM, Holloway BP. Nonhaemolytic Listeria monocytogenes mutants that are also noninvasive for mammalian cells in culture: evidence for coordinate regulation of virulence. Infect Immun. 1990;58:3988–3995. doi: 10.1128/iai.58.12.3988-3995.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kocks C, Gouin E, Tabouret M, Berche P, Ohayon H, Cossart P. L. monocytogenes-induced actin assembly requires the actA gene product, a surface protein. Cell. 1992;68:521–531. doi: 10.1016/0092-8674(92)90188-i. [DOI] [PubMed] [Google Scholar]
- Leimeister-Wachter M, Goebel W, Chakraborty T. Mutations offering hemolysin production In Listeria monocytogenes located outside the listeriolysin gene. FEMS Microbiol Lett. 1989;65:23–30. doi: 10.1016/0378-1097(89)90360-1. [DOI] [PubMed] [Google Scholar]
- Leimeister-Wachter M, Domann E, Chakraborty T. The expression of virulence genes in Listeria monocytogenes is thermoregulated. J Bacteriol. 1992;174:947–952. doi: 10.1128/jb.174.3.947-952.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lepay DA, Steinman RM, Nathan CF, Murray HW, Cohn ZA. Liver macrophages in murine listeriosis. J Exp Med. 1985;161:1503–1512. doi: 10.1084/jem.161.6.1503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lisanti MP, Rodriguez-Boulan E. Glycophospholipid membrane anchoring provides clues to the mechanism of protein sorting in polarized epithelial ceils. Trends Biochem Sci. 1990;15:113–118. doi: 10.1016/0968-0004(90)90195-h. [DOI] [PubMed] [Google Scholar]
- Low MG. Phosphatidylinositol-specific Phospholipase C from Staphylococcus aureus. Meth Enzymol. 1981;71:741–746. doi: 10.1016/0076-6879(81)71087-5. [DOI] [PubMed] [Google Scholar]
- Mackaness GB. Cellular resistance to infection. J Exp Med. 1962;116:381–406. doi: 10.1084/jem.116.3.381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mandel TE, Cheers C. Resistance and susceptibility of mice to bacterial infection: histopathology of Listeriosis in resistant and susceptible strains. Infect Immun. 1980;30:851–861. doi: 10.1128/iai.30.3.851-861.1980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marques MB, Weller PF, Parsonnet J, Ransil BJ, Nicholson-Weller A. Phosphatidylinositol-specific phospholipase C, a possible virulence actor of Staphylococcus aureus. J Clin Microbiol. 1989;27:2451–2454. doi: 10.1128/jcm.27.11.2451-2454.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mengaud J, Vicente MF, Cossart P. Transcriptional mapping and nucleotide sequence of the Listeria monocytogenes hlyA region reveal structural features that may be involved in regulation. Infect Immun. 1989;57:3695–3701. doi: 10.1128/iai.57.12.3695-3701.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mengaud J, Dramsi S, Gouin E, Vazquez-Boland JA, Milon G, Cossart P. Pleiotropic control of Listeria monocytogenes virulence factors by a gene that is autoregulated. Mol Microbiol. 1991;5:2273–2283. doi: 10.1111/j.1365-2958.1991.tb02158.x. [DOI] [PubMed] [Google Scholar]
- Mounier J, Ryter A, Coquis-Rondon M, Sansonetti PJ. Intracellular and cell-to-cell spread of Listeria monocytogenes involves interaction with F-actin in the enterocytelike cell line Caco-2. Infect Immun. 1990;58:1048–1058. doi: 10.1128/iai.58.4.1048-1058.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- North RJ. The relative importance of blood monocytes and fixed macrophages to the expression of cell-mediated immunity to infection. J Exp Med. 1970;132:521–534. doi: 10.1084/jem.132.3.521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Portnoy DA. Innate immunity to a facultative intracellular bacterial pathogen. Curr Opin Immunol. 1992;4:20–24. doi: 10.1016/0952-7915(92)90118-x. [DOI] [PubMed] [Google Scholar]
- Portnoy DA, Jacks PA, Hinrichs DJ. Role of hemolysin for the intracellular growth of Listeria monocytogenes. J Exp Med. 1988;167:1459–1471. doi: 10.1084/jem.167.4.1459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Portnoy DA, Schreiber RD, Connelly P, Tilney LG. Gamma interferon limits access of Listeria monocytogenes to the macrophage cytoplasm. J Exp Med. 1989;170:2141–2146. doi: 10.1084/jem.170.6.2141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Portnoy DA, Chakraborty T, Goebel W, Cossart P. Molecular determinants of Listeria monocytogenes pathogenesis. Intect Immun. 1992a;60:1263–1267. doi: 10.1128/iai.60.4.1263-1267.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Portnoy DA, Tweten RK, Kehoe M, Bielecki J. Capacity of Listeriolysin O, Streptolysin O. and Perfringolysin O to mediate growth of Bacillus subtilis within mammalian cells. Infect Immun. 1992b;60:2710–2717. doi: 10.1128/iai.60.7.2710-2717.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raveneau J, Geoffroy C, Beretti J, Gaillard J, Alouf JE, Berche P. Reduced virulence of a Listeria monocytogenes phospholipase-deficient mutant obtained by transposon insertion into the zinc metalloprotease gene. Infect Immun. 1992;60:916–921. doi: 10.1128/iai.60.3.916-921.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosen H, Gordon S, North RJ. Exacerbalion of murine listeriosis by a monoclonal antibody specific for the type 3 complement receptor of myelomonocytic cells. Absence of monocytes at infective foci allows Listeria to multiply in nonphagocytic cells. J Exp Med. 1989;170:27–37. doi: 10.1084/jem.170.1.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sambrook J, Fritsch EF, Maniatis T. Molecular Cloning: A Laboratory Manual. 2. Cold Spring Harbor, New York: Cold Spring Harbor Laboratory Press; 1989. [Google Scholar]
- Seeliger HPR. Listeriosis-history and actual developments. Infection. 1986;16:81–85. doi: 10.1007/BF01639726. [DOI] [PubMed] [Google Scholar]
- Smith K, Youngman P. Use of a new integrational vector to investigate compartment-specific expression of the Bacillus subtilis spollM gene. Biochimie. 1992;74:705–711. doi: 10.1016/0300-9084(92)90143-3. [DOI] [PubMed] [Google Scholar]
- Smyth CJ, Duncan JL. Thiol-activated (oxygenlabile) cytolysins. In: Jeljaszewicz J, Wadström T, editors. Bacterial Toxins and Cell Membranes. New York: Academic Press Inc; 1978. pp. 129–183. [Google Scholar]
- Sun A, Camilli A, Portnoy DA. Isolation of Listeria monocytogenes small-plaque mutants defective for intracellular growth and cell-to-cell spread. Infect Immun. 1990;58:3770–3778. doi: 10.1128/iai.58.11.3770-3778.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Theriot JA, Mitchison TJ, Tilney LG, Portnoy DA. The rate of actin-based motility of intracellular Listeria monocytogenes equals the rate of actin polymerization. Nature. 1992;357:257–260. doi: 10.1038/357257a0. [DOI] [PubMed] [Google Scholar]
- Tilney LG, Portnoy DA. Actin filaments and the growth, movement, and spread of the intracellular bacterial parasite, Listeria monocytogenes. J Cell Biol. 1989;109:1597–1608. doi: 10.1083/jcb.109.4.1597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tilney LG, Connelly PS, Portnoy DA. The nucleation of actin filaments by the bacterial intracellular pathogen, Listeria monocytogenes. J Cell Biol. 1990;111:2979–2988. doi: 10.1083/jcb.111.6.2979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vazquez-Botand J, Kocks C, Dramsi C, Ohayon H, Geoffrey C, Mengaud J, Cossart P. Nucleotide sequence of the lecithinase operon of Listeria monocytogenes and possible role of lecithinase in cell-to-cell spread. Infect Immun. 1992;60:219–230. doi: 10.1128/iai.60.1.219-230.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wirth R, An FY, Clewell DB. Highly efficient protoplast transformation syslem for Streptococcus faecalis and a new Escherichia coli-S. faecalis shuttle vector. J Bacteriol. 1986;165:831–836. doi: 10.1128/jb.165.3.831-836.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zar JH. Biostatistical Analysis. 2. Chapter 10. Englewood, NJ: Prentice-Hall, Inc; 1974. p. 124. [Google Scholar]