CARPEL FACTORY, a Dicer Homolog, and HEN1, a Novel Protein, Act in microRNA Metabolism in Arabidopsis thaliana (original) (raw)
. Author manuscript; available in PMC: 2016 Dec 5.
Summary
Background
In metazoans, microRNAs, or miRNAs, constitute a growing family of small regulatory RNAs that are usually 19–25 nucleotides in length. They are processed from longer precursor RNAs that fold into stem-loop structures by the ribonuclease Dicer and are thought to regulate gene expression by base pairing with RNAs of protein-coding genes. In Arabidopsis thaliana, mutations in CARPEL FACTORY (CAF), a Dicer homolog, and those in a novel gene, HEN1, result in similar, multifaceted developmental defects, suggesting a similar function of the two genes, possibly in miRNA metabolism.
Results
To investigate the potential functions of CAF and HEN1 in miRNA metabolism, we aimed to isolate miRNAs from Arabidopsis and examine their accumulation during plant development in wild-type plants and in hen1-1 and caf-1 mutant plants. We have isolated 11 miRNAs, some of which have potential homologs in tobacco, rice, and maize. The putative precursors of these miRNAs have the capacity to form stable stem-loop structures. The accumulation of these miRNAs appears to be spatially or temporally controlled in plant development, and their abundance is greatly reduced in caf-1 and hen1-1 mutants. HEN1 homologs are found in bacterial, fungal, and metazoan genomes.
Conclusions
miRNAs are present in both plant and animal kingdoms. An evolutionarily conserved mechanism involving a protein, known as Dicer in animals and CAF in Arabidopsis, operates in miRNA metabolism. HEN1 is a new player in miRNA accumulation in Arabidopsis, and HEN1 homologs in metazoans may have a similar function. The developmental defects associated with caf-1 and hen1-1 mutations and the patterns of miRNA accumulation suggest that miRNAs play fundamental roles in plant development.
Introduction
Small double-stranded RNAs of 21–25 nucleotides (nt) in length serve as guide RNAs in related gene silencing phenomena, RNA interference (RNAi) in animals, post-transcriptional gene silencing (PTGS) in plants, and quelling in fungi ([1], reviewed in [2, 3]). These small interfering RNAs (siRNAs) are generated from long double-stranded RNAs by the ribonuclease III enzyme Dicer [4, 5] and are then incorporated into a multiprotein, RNA-induced silencing complex (RISC), which targets homologous RNAs for degradation ([6, 7], reviewed in [2, 8]). One component of the RISC in Drosophila melanogaster is AGO-2, a member of the PPD family of proteins containing PAZ and PIWI domains [9]. PPD proteins are present in metazoans, plants, and fungi, and some have been shown genetically to act in gene silencing in Caenorhabditis elegans, Arabidopsis thaliana, and Neurospora crassa [10–12]. Dicer also contains a PAZ domain, which is postulated to mediate interaction between Dicer and AGO-2 in the RISC [9].
Small single-stranded RNAs of 19–24 nt in length serve as regulators of gene expression during development in invertebrates and vertebrates. Two small temporal RNAs (stRNAs), lin-4 and let-7, regulate developmental timing in C. elegans by inhibiting the translation of target mRNAs through base pairing with the 3′ untranslated regions (UTRs) of these RNAs [13–16]. The stRNAs appear to be members of a growing class of microRNAs (miRNAs) identified from C. elegans, Drosophila, mouse, and human cells [17–20]. miRNAs are associated with an ~550 kDa ribonucleoprotein complex (miRNP), which also contains a PPD protein, eIF2C2 [21]. miRNAs are processed from longer single-stranded precursor RNAs that presumably form stem-loop structures, in which the mature miRNAs reside in the stems. The generation of miRNAs requires Dicer and two members of the PPD family, ALG-1 and ALG-2, in C. elegans [22–24], and this suggests that aspects of siRNA and miRNA metabolism are similar. The presence of siRNAs and core PTGS machinery in plants [1, 12, 25] and the link between miRNA and siRNA metabolism in metazoans (reviewed in [26]) implicate the existence of miRNAs in plants.
CARPEL FACTORY (CAF), the Arabidopsis homolog of Dicer, functions in a variety of developmental pathways in Arabidopsis. This is reflected by the fact that many mutant alleles were isolated in several independent studies and were given different names to describe the phenotypes of the mutants, such as sus1 (abnormal suspensor1; [27, 28]), sin1 (short integument1; [29]), and caf [30]. Several sus1 alleles are defective in embryogenesis such that sus1 embryos do not progress beyond the globular stage [27]. While the sus1 alleles demonstrate the requirement for CAF in embryogenesis, weak sin1/caf alleles show that CAF also acts in a variety of developmental events in the adult plant. The sin1 mutant is defective in ovule development and is female sterile [29, 31]. In addition, the sin1 mutant exhibits altered leaf shape, delayed bolting, and delayed floral transition [31]. The caf-1 mutant exhibits defects in floral meristem determinacy during flower development [30]. However, a role of CAF in PTGS has not been demonstrated yet.
HEN1 was identified in our previous studies as a gene that plays a role in the specification of stamen and carpel identities during flower development [32]. Recessive mutations in HEN1, hen1-1 and hen1–2, cause stamen-to-petal and carpel-to-sepal transformation in the hua1-1 hua2-1 background, which is weakly compromised in stamen and carpel identities [32, 33]. Although the floral homeotic phenotypes are only present in the hua1-1 hua2-1 hen1 triple mutants, the hen1 single mutants exhibit a wide array of developmental defects, including reduced leaf size, altered leaf shape, reduced plant height, reduced carpel fusion, and reduced female fertileity [32]. Intriguingly, many aspects of the mutant phenotypes exhibited by the stronger hen1-1 allele are almost identical to those in the sin1/caf-1 mutants, and this leads us to hypothesize that HEN1, like CAF, acts in miRNA metabolism in Arabidopsis. Here, we report the isolation of 11 miRNAs from Arabidopsis and show that HEN1 and CAF are both required for miRNA accumulation.
Results
Isolation of miRNAs from Arabidopsis
To begin to test our hypothesis that HEN1 plays a role in miRNA metabolism, we aimed to clone small RNAs, 18–28 nt in size, from Arabidopsis using a protocol previously employed to isolate siRNAs and later used to isolate miRNAs from animals ([17, 34]; see the Experimental Procedures). We obtained 230 unique, putative small RNA sequences, which were subjected to BLAST [35] analyses against the Arabidopsis genome (http://www.Arabidopsis.org/Blast/). A total of 176 sequences corresponded to known noncoding RNAs, such as rRNAs, tRNAs, and snRNAs, and were not further analyzed. A total of 15 had 2-nt or more mismatches with the Arabidopsis genome and were not further analyzed. Five were found to be in exons or introns of protein-coding genes. The remaining 34 sequences were in intergenic regions. The 39 sequences from the last two categories were compared with the Arabidopsis genome to identify the genomic sequences surrounding the putative miRNAs. Using the m-fold program, longer genomic sequences (60–110 nt) containing the small RNAs were used to predict secondary structures ([36]; see Experimental Procedures). A total of 29 were capable of forming stem-loop structures (Figure 1A and data not shown), 23 of which were tested by expression studies. We were able to detect small transcripts of the expected size (20–31 nt) for 11 of the 23 tested sequences. Interestingly, the 11 miRNAs appeared to belong to 2 groups. For seven miRNAs, which we named group I (Table 1), we were able to detect a single 21–24-nt transcript by RNA filter hybridization (Figures 2 and 3; data not shown). For the other four miRNAs, which we named group II (Table 1), we were able to detect longer 65–100-nt transcripts as well as small microRNA-sized transcripts (Figures 2 and 3; data not shown). In fact, all four group II miRNAs, or their putative precursors, exhibited sequence similarity to known noncoding RNAs from other plant species (Table 1) and were not eliminated from our first screen because the corresponding Arabidopsis genes were not annotated as noncoding RNAs.
Figure 1. miRNA Precursors and Complementarity of miRNAs to mRNAs.

(A) Stem-loop structures of putative miRNA precursors. Genomic sequences (62–102 nt) containing the miRNAs were used for RNA secondary structure prediction with the m-fold program (see the Experimental Procedures). The structures shown are the outputs from m-fold without manual modification. The miRNA sequences are underlined. Two miRNA sequences in the same precursors are differentiated by colors (one in black and the other in gray). A hyphen indicates the absence of nucleotides. For MIR167, MIR172, MIR175, MIR176, MIR177, and MIR179, identical miRNA sequences were found at multiple genomic loci (as indicated by numbers after dashes; MIR167a-2 not shown). For MIR167, MIR172, MIR175, and MIR179, the putative precursors of the different identical miRNA copies were related but were not identical in sequence, but they were able to form stem-loop structures. For MIR176 and MIR177, which were found in the same precursor, the precursor sequences of the identical miRNA copies at different genomic loci were identical with one exception (MIR176-4 and MIR177-4).
(B) Complementarity of selected miRNAs to their putative target mRNAs.
Table 1.
Sequences and Genomic Locations of miRNA Genes
| miRNA Genes | Sequence Length | Chromosome Locationa | Homologsb | Expression in Maize and Tobacco/Other Remarksc |
|---|---|---|---|---|
| Group I | ||||
| MIR159 | TTTGGATTGAAGGGAGCTCTA-21 | 159a: 1; F25P22 (41198–41178) | 159b: 1–20; 1; T10F20 (26891-26910)159c: 1–19; 2; T3F17 (28742–28724) | 18/21 nt complementary to myb-related transcription factors At5g55020, At2g26950, At3g11440, and At5g06100Tobacco flower, maizeRice (20/21) |
| MIR163 | TTGAAGAGGACTTGGAAC TTCGAT-24 | 163a: 1; F4N21 (72531–72508) | 163b-1: 1–24 (no 20th T); 1; F4N21 (82583–82561)163b-2: 1–24 (no 20th T); 1; F4N21 (868488–86466 163c: 1–24 (T12C, no 20th T); 1; F4N21 (75264–75286) | 21/24 nt complementary to unknown proteinsAt1g66700 and At1g66690 |
| MIR167 | TGAAGCTGCCAGCATGAT CTA-21 | 167a-1: 3; F5N5 (20031–20051)167a-2: 3; F16M2 (73860–73880) | 167b: 1–20; 1; F28K20 (48695–48714) | 17–18/21 nt complementary to auxin-responsive factor8 (ARF8) and _ARF6_Tobacco flower, maizeRice (21/21) |
| MIR172 | AGAATCTTGATGATGCTG CAT-21 | 172a-1: 2; F24D13 (18082–18102)172a-2: 5; T19N18 (91778–91758) | 172b: 1–20; 3; F24K9 (42346–42327)172c: 2–21; 5; F20I5 (52036–52055) | 18–20/21 nt complementary to APETALA2 (AP2) and _AP2_-like genes At5g60120, At5g67180, and At2g28550Tobacco flowerRice (20/21) |
| MIR173d | aTTCGCTTGCAGAGAGAAAT CAC-22/23 | 3; MXC7 (76775–76796) | 17/22 nt complementary to unknown protein At3g28460 | |
| MIR174 | TTGAAAGCTGAGGTGGAG GGTGTT-24 | 4; T27D20 (72299–72322) | 15/24 nt complementary to putative disease resistance gene At2g17050 | |
| MIR175 | TTCTAAGTCCAACATAGCG TA-21 | 175a-1: 1; F2J10 (26516–26536)175a-2: 2; F12K2 (4441–4461)175a-3: 2; F17A14 (11326–13306) | 175b: 1–21 (G19A); 1; F2J10 (26495–26515)175c: 3–20; 2; F12K2 (4359–4376)175d: 4–20; 2; F12K2 (4402–4418) | 17–18/21 nt complementary to hypothetical proteins At5g18040, At3g43200, and At1g51670 |
| Group II | ||||
| MIR176 | TGTCGTCCAGCGGTTAGG ATATCTGGC-27 | 176a-1: 1; F7G19 (3766–3740)176a-2: 1; T20H2 (16588–16614)176a-3: 3; T3A5 (37876–37902) 176a-4: 4; AP22 (66811–66837) 176a-5: 5; T1G16 (35722–35696) 176a-6: 5; MRA19 (47092–47066) 176a-7: 5; MSN2 (58741–58715) | 176b: 1–27 (C23T); 1; F25P22 (16112–16138)176c: 1–27 (G2A); 3; T20E23 (34829–34805) | 6 nt ahead of _MIR177_Present in the same stem-loop structure with _MIR177_Rice (27/27) |
| MIR177d | CCAGGAGACCCGGGTTCG aTTCCCGGC-27 | 177a-1: 1; F7G19 (3733–3707)177a-2: 1; T20H2 (16621–16647) 177a-3: 3; T3A5 (37909–37935) 177a-4: 4; AP22 (66844–66870) 177a-5: 5; T1G16 (35689–35663) 177a-6: 5; MRA19 (47059–47033) 177a-7: 5; MSN2 (58708–58682) | 177b: 1–26 (C23T, C24T); 1; F25P22 (16145–16166)177c: 1–26; 3; T20E23 (34798–34773) | 6 nt behind _MIR176_Present in the same stem-loop structure with _MIR176_Rice (27/27)22/27(6–27), Triticum aestivum tRNA-Gly21/27 nt complementary to hypothetical protein At1g29200 |
| MIR178 | AGCGGGGTAGAGGAATT GGT-20 | 2; T5E7 (61199–61180) | 1–20, Brassica oleracea tRNA-Met, mt genomeRice (20/20) | |
| MIR179d | CAGAGGGGGTTTGCGTTCG CGCAGCCCCTACt-31/32 | 179a-1: 1; F12F1 (107356–107386)179a-2: 4; F16G20 (37795–37825179a-3: 5; F18A17 (5745–5715)179a-4: 5; T1G16 (53959–52989)179a-5: 5; MI024 (795–825) | 1–31 (T10C, T11C), tomato U1 snRNARice (29/31) |
Figure 2. Spatial and Temporal Patterns of miRNA Accumulation as Revealed by RNA-Filter Hybridization with Antisense Oligonucleotide Probes Complementary to MIR Sequences.

MIR163 and MIR167 showed distinct temporal patterns of expression during development (RNAs from aerial portions of plants were used). The numbers of days were calculated from the time stratified seeds were transferred to growth chambers. MIR172, MIR173, MIR167, and MIR178 were expressed preferentially in some organs. The MIR172 transcript was present in inflorescences, leaves, and stems at similar levels but was barely detectable in siliques and was not detectable in roots. The MIR173 transcript was present in inflorescences and leaves but was barely detectable in roots. The MIR167 transcript was most abundant in inflorescences. The mature MIR178 transcript (20 nt) was only present in inflorescences, although the probe also detected two bands at 80–100 nt in all three tissues. In, inflorescences; L, leaves; R, roots; St, stems; and Si, siliques. The sense probe for MIR173 did not detect any signals. 5S rRNA was used as an internal control.
Figure 3. Requirement for CAF and HEN1 in miRNA Accumulation.
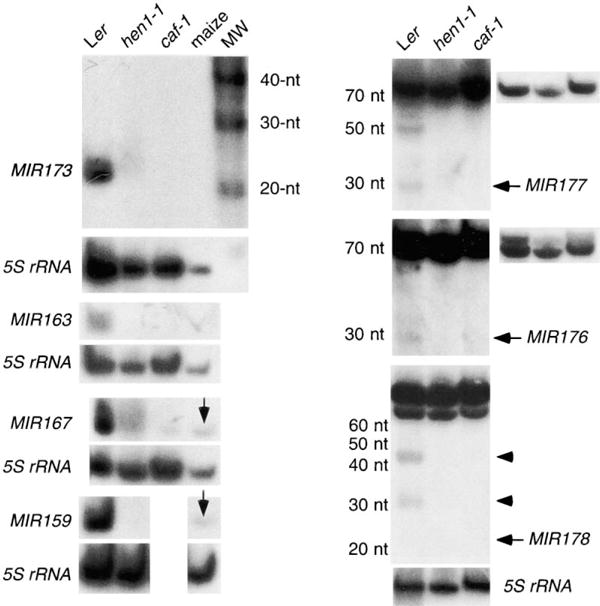
The accumulation of mature transcripts corresponding to MIR173, 163, 167, 159, 177, and 176 was undetectable in hen1-1 and caf-1 or was greatly reduced compared to wild-type. Note that MIR177 and MIR176 antisense probes detected an approximately 70-nt signal in all genotypes but detected a 27-nt signal only in wild-type. Lower exposures of the precursor-sized transcripts are shown on the right. The MIR178 panel is meant to show that the two transcripts of intermediate size (arrowheads) are not present in either hen1-1 or caf-1. The mature 20-nt transcript was not visible at this exposure. However, it was visibly present in wild-type but was absent from hen1-1 or caf-1 upon longer exposure. RNAs from the entire aerial portions of 1 month-old plants were used for the MIR173, 163, 167, and 159 hybridization, whereas RNAs from inflorescences were used for the MIR177, MIR176, and MIR178 hybridization. MW, molecular weight standards. Vertical arrows in MIR167 and MIR159 panels indicate signals from maize seedlings.
Among the 11 miRNAs confirmed by expression studies, 3 were identified by Reinhart et al. [37] in an independent study, in which the genes were designated MIR159, MIR163, and MIR167. To be consistent with the existing nomenclature, we named the eight new MIR genes MIR172_–_MIR179, so that the numbers were consecutive with those in the report by Reinhart et al. [37]. The isolation of many small RNAs from Arabidopsis has also recently been reported by Llave et al. [38].
The fold-back structures of the putative miRNA precursors were largely similar to those in metazoans (Figure 1A). However, there appeared to be more and maybe larger loops in the stems of the plant precursors, and this finding suggests that the requirement for double strandedness in the precursors is more relaxed in plants. Like metazoan miRNAs, the Arabidopsis miRNAs were found primarily in the double-stranded regions of the structures. The structure of MIR179 was unusual in that the miRNA was predicted to be located nearly symmetrically around the loop (Figure 1A). The MIR176 and MIR177 miRNAs were potentially derived from the same precursor (Figure 1A).
Genome Organization of MIR Genes
The MIR173, MIR174, and MIR178 miRNAs were each encoded by a single copy gene (Table 1). The other eight miRNAs, however, had multiple identical or related sequences scattered in the genome. We analyzed all the identical sequences and the related sequences that differed from the mature miRNAs by no more than four nucleotides to determine whether the putative precursors could form stem-loop structures. Indeed, all eight miRNAs had either identical copies or closely related homologs in the genome, the putative precursors of which were able to form stable stem-loop structures (Figure 1A, Table 1; data not shown). Since the nucleotide differences were unlikely to generate new base-pairing specificities with target genes, we gave the homologous sequences the same gene names and distinguished them from the original MIR genes by letters (b, c, etc.). Genes corresponding to the identical copies were distinguished from the original MIR genes by numbers after dashes, such as MIR176-2, MIR176-3, etc. Identical or related copies of MIR genes were not found in tandem repeats (Table 1). The precursors of identical copies of miRNAs were either identical (as for MIR176) or related (as for MIR172 and MIR175) in sequence (Figure 1A). Interestingly, the three homologs of MIR163 were all located in exons of potential protein-coding genes (Table 1 and data not shown).
Group I miRNAs
All group I miRNAs were only detectable as the mature transcripts under all experimental conditions (Figures 2 and 3; data not shown). The seven group I miRNAs were not uniformly expressed throughout the plant but instead showed preferential accumulation in some organs (Figure 2, Table 2). For example, MIR172 was not expressed detectably in roots, and MIR167 miRNA was most abundant in inflorescences (Figure 2). We also studied the temporal patterns of miRNA accumulation during development for five of the group I MIR genes (Table 2). All five showed an increase in RNA accumulation at specific developmental time points (Figure 2, Table 2).
Table 2.
Accumulation of miRNAs during Development and in Mutant Backgrounds
| miRNA Genes | Plant Organ | Days in Growth Chamber | Genotypea | Sense Probe | FLb Probe | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Leaf | Stem | Inc | Root | Silique | 7 | 14 | 21 | 25 | WTd | hen1-1 | caf-1 | |||
| Group I | ||||||||||||||
| MIR159 | ++ | ++ | + | +/− | ++ | ++ | ++ | ++ | − | |||||
| MIR163 | +++ | +/− | + | + | + | +++ | +++ | − | − | |||||
| MIR167 | ++ | +++ | + | + | ++ | ++ | ++ | ++ | +/− | +/− | ||||
| MIR172 | + | + | + | − | +/− | − | +/− | + | ++ | ++ | +/− | +/− | ||
| MIR173 | +++ | ++ | +++ | + | +++ | ++ | +++ | +++ | +++ | ++ | +/− | − | − | − |
| MIR174 | +/− | + | − | + | +/− | |||||||||
| MIR175 | ++ | + | ++ | + | + | ++ | − | − | ||||||
| Group II | ||||||||||||||
| MIR176 | +/− | + | + | + | − | − | − | L | ||||||
| MIR177 | +/− | +/− | + | + | − | − | − | +/− | + | − | − | − | L | |
| MIR178 | − | ++ | − | ++ | − | − | − | L | ||||||
| MIR179e | −31 | ++31 | −31 | ++31 | +31 | −31 | ||||||||
| −20 | +−20 | −20 | +20 | −20 | −20 |
Since Dicer is required for the generation of miRNAs from their precursors in C. elegans, we tested whether CAF, the Arabidopsis homolog of Dicer, has a similar function in miRNA metabolism in Arabidopsis. In fact, CAF is indeed required for the accumulation of miRNAs in Arabidopsis, since the accumulation of the five tested group I miRNAs was either nondetectable or greatly reduced in caf-1 plants as compared to wild-type (Figure 3, Table 2). Therefore, the mechanism of miRNA production may be conserved between plants and animals. Despite the absence or greatly reduced abundance of the mature miRNAs, accumulation of miRNA precursors was never detected in caf-1 (data not shown). In C elegans, however, miRNA precursors accumulated to a higher level in animals in which Dicer activity was abolished or reduced by mutations or by RNAi [22]. This suggests that certain steps or _trans_-acting functions in miRNA metabolism differ between Arabidopsis and C elegans.
The similar phenotypes exhibited by hen1-1 and caf-1 mutants prompted us to test whether HEN1 acts similarly to CAF in miRNA metabolism. The accumulation of all seven group I miRNAs was nondetectable or greatly reduced in abundance in hen1-1 plants as compared to wild-type (Figure 3; Table 2). As in caf-1, precursors of group I miRNAs were also not detected in hen1-1 (data not shown).
Group II miRNAs
In previous attempts to isolate miRNAs from metazoans, sequences corresponding to rRNAs, tRNAs, etc. often constituted a significant proportion of cloned sequences but were not analyzed further, presumably based on the assumption that these represented degradation products from abundant noncoding RNAs [17, 18]. We found that four of our cloned sequences related to known noncoding RNAs in our study likely corresponded to bona fide miRNAs. We were able to detect small RNA transcripts of the expected size for MIR176, 177, 178, and 179 genes (Figures 2 and 3; data not shown). In contrast to group I miRNAs, larger precursorsized transcripts were also detected for all four group II miRNAs (Figures 2 and 3; data not shown). In some cases, sense probes and antisense flanking sequence probes were used to demonstrate the identities of the small transcripts (Table 2). For example, an antisense oligonucleotide probe for MIR178 detected both a 20-nt signal and two precursor-sized (80–100-nt) signals in wild-type plants (Figure 2). A sense oligonucleotide probe failed to detect the 20-nt or the 80–100-nt signals. An antisense oligonucleotide probe complementary in sequence to the 5′ flanking region of MIR178 was able to detect the 80–100-nt signals, but not the 20-nt signal, confirming the identity of the 20-nt signal (Table 2).
The accumulation of the small transcripts from the group II miRNA genes appeared to be under spatial control such that mature miRNAs were present at different levels in different organs or at different developmental time points (Figure 2, Table 2), although the precursor-sized transcripts were present at similar levels in all organs or at all time points tested (Figure 2, data not shown). For example, the 20-nt MIR178 signal was only present in inflorescences, whereas the larger 80–100-nt transcripts were present in roots, leaves, and inflorescences at similar levels (Figure 2). Furthermore, the small transcripts from all four group II miRNA genes did not accumulate in caf-1 or hen1-1 plants or accumulated at lower levels in caf-1 or hen1-1 plants than in wild-type plants (Figure 3, Table 2). These data argue against the assumption that the small transcripts were random degradation products from noncoding RNAs. The larger precursor-sized transcripts for all four group II genes were present in hen1-1, caf-1, and wild-type plants at similar levels (Figure 3 and data not shown). These precursor-sized transcripts may be the true precursors of the miRNAs. Alternatively, they could be derived from other noncoding RNA genes with sequence similarity to the MIR genes. Although group II miRNA transcripts were clearly found, we cannot, without functional studies, rule out the possibility that these transcripts are merely products of noncoding RNA processing and that they themselves do not serve any functions in the organism.
MIR176 and MIR177 were particularly interesting in that the two mature miRNA sequences were found only 6 nt apart in presumably the same precursor sequence (Figure 1A). This and the nearly identical spatial patterns of accumulation of the two miRNAs (Table 2) suggest that they are processed from the same precursor. Consistent with this assumption, homologs of MIR176 and MIR177 found throughout the genome are always arranged in pairs and are 6 bp apart (Table 1).
Complementarity to mRNAs
The absence of many miRNAs and the presence of a wide array of developmental defects in hen1-1 and caf-1 mutants indicate that miRNAs serve regulatory roles in plant development. miRNAs are thought to regulate the expression of protein-coding genes at the posttranscriptional level by base pairing with mRNAs of the target genes. We found that 8 of the 11 miRNAs were complementary in at least 15 nucleotides to sequences in known or putative protein-coding genes (Figure 1B, Table 1). Some of these potential targets are transcription factors (Table 1).
Similarity or Complementarity to Potential Promoter Sequences
Both HEN1 and CAF have putative nuclear localization signals [30, 32], suggesting that aspects of miRNA metabolism happen in the nucleus. We wondered whether miRNAs may be present in the nucleus and regulate gene expression at the DNA level. We determined whether the miRNAs were similar or complementary in at least 15 nucleotides to potential promoter sequences, which we arbitrarily set to be sequences 0.1–1 kb upstream of the start codon. Indeed, microRNAs 163, 172, 173, 175, 176, 177, and 179 were similar to promoter sequences in either the sense or antisense orientation (see Table S1 in the Supplementary Material available with this article online).
miRNAs in Other Plant Species
miRNAs do not appear to be unique to Arabidopsis in the plant kingdom. We were able to detect 21-nt transcripts from tobacco with probes complementary to MIR159 MIR167, and MIR172 (Table 1) and maize with probes complementary to MIR159 and MIR167 (Figure 3, Table 1), and this suggests that homologs of these genes are present in tobacco and maize. In addition, the rice genome has sequences that are identical to, or only one nucleotide different from, seven of the MIR genes (Table 1).
HEN1 Homologs in Other Species
Given the universal presence of miRNAs and the potentially conserved mechanism in the generation of miRNAs in plants and animals, we wondered whether HEN1 may be part of an miRNA metabolic pathway common among plants and animals. HEN1 codes for a novel protein of 942 amino acids without motifs of known function [32]. A stretch of approximately 230 amino acids at the C terminus of the protein shows 40%–50% similarity to protein sequences predicted from cDNAs or from genome annotation of a number of metazoan, fungal, and bacterial species (Figure 4). Interestingly, the two hen1 mutants, hen1-1 and hen1-2, both contain mutations in this region of the protein [32], and these mutations suggest the functional significance of this domain. The HEN1 homologs in these other species are considerably shorter than HEN1 and are all of unknown function. However, it is tempting to hypothesize that HEN1 homologs in metazoans and perhaps in Schizosaccharomyces pombe also act in miRNA metabolism. Although it is not known if Schizosaccharomyces pombe is able to perform RNAi or generate miRNAs, it has a Dicer homolog in its genome [8].
Figure 4. Potential HEN1 Homologs.

(A) A Clustal W alignment of a conserved domain shared among HEN1 homologs (predicted proteins from cDNAs or from genome annotation) from a few species. The overall amino acid similarity within this domain among these proteins is 40%–50%. Positions at which at least five amino acids are identical are shown in a darker gray color, whereas positions at which at least five amino acids are similar are shown in a lighter gray color. Hs, Homo sapiens; Mm, Mus musculus; Dm, Drosophila melanogaster; Ce, Caenorhabditis elegans; Sp, Schizosaccharomyces pombe; and Sc, Streptomyces coelicolor. Nostoc, Nostoc sp. PCC 7120, a cyanobacterium. GenBank accession numbers of the proteins are shown after the species abbreviation.
(B) Diagrams of HEN1 and potential HEN1 homologs, showing the positions of the conserved domain (hatched boxes) in these proteins.
Discussion
miRNAs Are Evolutionarily Ancient
Our studies and those by Reinhart et al. [37] clearly demonstrated the existence of microRNAs in Arabidopsis and probably in tobacco, maize, and rice. Altogether, 24 distinct Arabidopsis miRNAs were identified from the two studies. The fact that only three common miRNA genes were identified from the two independent studies and that most miRNA sequences in our study were only isolated once suggests that the search for miRNAs in plants is far from complete. In fact, the isolation of more than 100 small RNAs has recently been reported by Llave et al. [38]. In addition, we also demonstrated that group II miRNAs that are related in sequence to other noncoding RNAs, such as rRNAs, tRNAs, or snRNAs, are present in Arabidopsis. Given that the great majority of clones from miRNA isolation correspond to known noncoding RNAs in both Arabidopsis and metazoans, it is possible that group II miRNAs are also present in metazoans and that their prevalence is greater than is currently appreciated.
As in animals, the putative plant miRNA precursors are also able to form stable stem-loop structures. Furthermore, CAF, the Arabidopsis homolog of Dicer, is required for the accumulation of miRNAs in Arabidopsis. Therefore, it appears that miRNAs, their synthesis, and presumably their functions existed before the divergence of the plant and animal kingdoms. HEN1, a novel protein, is a new player in miRNA metabolism in Arabidopsis. Since HEN1 homologs are found in bacterial as well as fungal, plant, and metazoan genomes, it is even possible that miRNAs were part of an ancient, RNA-based gene regulatory network that existed in bacteria.
HEN1 in miRNA Metabolism
Studies in metazoan species have revealed a role of only Dicer and PPD proteins, such as ALG-1, ALG-2, and eIF2C2, in miRNA synthesis or action. We have demonstrated that HEN1, a novel protein without sequence motifs of potential functions, is required for miRNA accumulation in Arabidopsis. HEN1 may act in the synthesis or stabilization of the miRNA precursors, or in the processing or the stabilization of the miRNAs. Given that HEN1 homologs are present in metazoans, it is possible that the action of HEN1 in miRNA metabolism is conserved in plants and animals. Alternatively, HEN1 may be required for miRNA metabolism only in plants. One hypothesis is that the more relaxed secondary structures of the plant miRNA precursors require additional proteins to facilitate their processing.
Potential Functions of miRNAs in Plants
Like animal miRNAs, some miRNAs in Arabidopsis accumulate in a temporally or spatially restricted manner, suggesting that they play regulatory roles in development. This is consistent with the developmental defects exhibited by the caf-1 and hen1-1 mutants. In addition, mutations in two Arabidopsis PPD family members, ARGONAUTE1 and PINHEAD/ZWILLE, result in multifaceted developmental defects [39–41]. ARGONAUTE1 and PINHEAD/ZWILLE are more closely related to human eIF2C2, a component of a miRNP [21], than many metazoan PPD proteins [42]. Therefore, it is likely that the developmental abnormalities in argonaute1 and pinhead/zwille mutants are caused by defects in miRNA metabolism. While argonaute1 mutants displaying either severe or weak developmental abnormalities are both defective in PTGS [12, 43], zwille-3, which displays strong developmental defects, is capable of PTGS of a 35S-GUS transgene [43]. PPD proteins may play differential roles in siRNA and miRNA metabolism.
The only two miRNAs of known function, lin-4 and let-7, regulate developmental timing in C. elegans by base pairing with target mRNAs and inhibiting the translation of the mRNAs. Although the functions of plant miRNAs remain to be deciphered, many are potentially able to base pair with protein-coding RNAs. Some may also play a negative role in the regulation of gene expression. For example, 20 of the 21 nucleotides in MIR172 are complementary to a stretch of sequence in the RNA of the floral homeotic gene APETALA2 (AP2) (Figure 1B, Table 1; [44, 45]). If MIR172 negatively regulates the expression of AP2, the decreased accumulation of miRNA172 in hen1-1 (Table 2) would cause increased AP2 expression in hen1-1. This may be the molecular basis of the floral homeotic phenotypes exhibited by the hua1-1 hua2-1 hen1-1 mutants. In fact, this would be consistent with our previous genetic studies showing that ap2-2, a severe mutation in AP2 [44], was able to partially rescue the floral homeotic defects of the hua1-1 hua2-1 hen1-1 mutant [32].
Although lin-4 and let-7 regulate protein-coding genes at the level of translation, miRNAs may also act at other levels to regulate gene expression. Considering the structural resemblance of the siRNA and miRNA pathways, it is possible that some miRNAs regulate the stability of target mRNAs. In plants, siRNAs not only trigger RNA degradation, presumably in the cytoplasm, but they also direct DNA methylation in the nucleus in both post-transcriptional and transcriptional gene silencing [46–48]. Given that both HEN1 and CAF have putative nuclear localization signals and that some miRNAs are similar or complementary to potential promoter sequences, one cannot rule out the possible role of miRNAs in the regulation of gene expression at the DNA level, such as DNA methylation or transcription.
Experimental Procedures
Plant Material and Growth Conditions
Arabidopsis plants used in this study are Landsberg erecta (L_er_) wild-type, hen1-1, and caf-1. While hen1-1 is in the Landsberg ecotype, caf-1 was originally isolated in Ws and introgressed into Landsberg by five backcrosses to L_e_r by Dr. Steve Jacobsen. Arabidopsis plants were grown as described by Chen et al. [32].
Seedlings of Zea mays inbred line B73 were grown at 23°C with 16 hr light/8 hr dark cycles. Nicotiana tabacum cv. Petit Havana plants were grown in a greenhouse at 30°C with 16 hr light/8 hr dark cycles.
miRNA Cloning
The aerial portions of 9-day and 21-day Arabidopsis plants were used for miRNA isolation. Approximately 500 μg total RNA was extracted from liquid nitrogen-frozen plant tissues by using Tri Reagent (Molecular Research Center) according to the manufacturer’s instructions. Small RNAs between approximately 18 and 28 nt were recovered from polyacrylamide gel fractionation, ligated to adaptors, and amplified by RT-PCR according to the procedures described by Elbashir et al. [34]. Subsequently, the amplified sequences were concatamerized and cloned, also according to the procedures described by Elbashir et al. [34]. A total of 127 clones were sequenced with the BigDye terminator cycle sequencing kit (PE applied Biosystems) and were analyzed on an ABI3700 capillary DNA sequencer, and this resulted in 292 potential miRNA sequences, among which 230 were unique. After eliminating 176 sequences that corresponded to known Arabidopsis noncoding RNAs and 15 that had 2-nt or more mismatches with the Arabidopsis genome, the remaining 39 sequences were further analyzed to determine if the putative precursors were able to form stem-loop structures. Secondary structures were predicted with m-fold (http://www.bioinfo.math.rpi.edu/~m-fold/rna/form1.cgi) by using longer genomic sequences containing 60–110 nt on both ends of the cloned sequences, or 60–110 nt on one end and 10–30 nt on the other. Then, the putative precursors were reduced to 60–110 nt in size and were used to predict the secondary structures again with m-fold.
RNA Filter Hybridization
RNA preparations enriched for small-sized RNAs were obtained according to a protocol from Drs. Natalie Doetsch and Richard Jorgensen (University of Arizona, Tucson, AZ). Briefly, 400 μl (1–2mg) total RNA was combined with 50 μl each of 50% PEG8000 and 5 M NaCl, incubated on ice for 2 hr, and centrifuged at 15,000 × g for 10 min. After adding 1/10 volume of 3 M sodium acetate and 2 volumes of 95% ethanol to the supernatant, small-sized RNA was spun down at 15,000 × g following incubation at −20°C for 2 hr, washed with 75% ethanol, dried briefly, and resuspended in RNase-free water.
For RNA filter hybridization, 50 μg total RNA or 15 μg small-sized RNA enriched as above was separated on a denaturing 15% polyacryamide gel (15 × 15 cm) containing 8 M urea at 100V for 10 hr. RNA was electrophoretically transferred to Zeta-probe GT membranes (BioRad) by using a Trans-Blot Electrophoretic Transfer Cell (BioRad). An end-labeled 10-nt RNA ladder (Ambion) was also electrophoreased in the same gel, and the gel strip containing the size marker was excised before transfer and was exposed to film directly for size comparison with expression data.
After electroblotting, RNAs were fixed to the membrane by UV crosslinking and by baking in a vacuum oven at 80°C for 1 hr. Membranes were hybridized with 32P-end-labeled oligonucleotide probes at 40°C with Ultrahyb-oligo hybridization buffer (Ambion) and were washed twice at 40°C with 2× SSC/0.5% SDS. 5S rRNA hybridization was carried out at 42°C in 7% SDS/0.25 M sodium phosphate with an oligonucleotide (5′-GAGGGATGCAACACGAGGACTT-3′) that is complementary to Arabidopsis 5S rRNA as the probe.
Supplementary Material
supplement
Acknowledgments
We are grateful to Drs. Natalie Doetsch, Richard Jorgensen, and Vicki Chandler for protocols on small RNA isolation and hybridization. We thank Dr. Steve Jacobsen for providing caf-1 seeds and Dr. Jon Suzuki for providing tobacco floral tissue. We thank Drs. Bonnie and David Bartel for communicating work that is in press and Dr. Hugo Dooner, Dr. Ruth Steward, Dr. Wenming Wang, and Yulan Cheng for comments on the manuscript. We acknowledge Torrey Mesa Research Institute for providing public access to the rice genome. This work was supported by a grant from the National Institutes of Health (1 R01 GM61146-01) to X.C. and a grant from the Department of Energy (DE-F605-95ER20194) to J.M.
Footnotes
Supplementary Material
Supplementary Material on the similarity or complementarity of miRNA sequences to potential promoter sequences and on the modified secondary structures of some of the miRNA precursors is available at http://images.cellpress.com/supmat/supmatin.htm.
Note Added in Proof
Upon suggestions by Dr. David Bartel, we reanalyzed by m-fold prediction the secondary structures of the putative precursors for all miRNAs by including more flanking genomic sequences. We found that miRNAs 159, 163, and 167, but not other miRNAs, were present in better duplexed regions (see Figure S1 in the Supplementary Material). Therefore, the precursors of some plant miRNAs may be larger than metazoan miRNA precursors.
References
- 1.Hamilton AJ, Baulcombe DC. A species of small antisense RNA in posttranscriptional gene silencing in plants. Science. 1999;286:950–952. doi: 10.1126/science.286.5441.950. [DOI] [PubMed] [Google Scholar]
- 2.Zamore PD. Ancient pathways programmed by small RNAs. Science. 2002;296:1265–1269. doi: 10.1126/science.1072457. [DOI] [PubMed] [Google Scholar]
- 3.Plasterk RH. RNA silencing: the genome’s immune system. Science. 2002;296:1263–1265. doi: 10.1126/science.1072148. [DOI] [PubMed] [Google Scholar]
- 4.Zamore PD, Tuschl T, Sharp P, Bartel D. RNAi: double-stranded RNA directs the ATP-dependent cleavage of mRNA at 21 to 23 nucleotide intervals. Cell. 2000;101:25–33. doi: 10.1016/S0092-8674(00)80620-0. [DOI] [PubMed] [Google Scholar]
- 5.Bernstein E, Caudy AA, Hammond SM, Hannon GJ. Role for a bidentate ribonuclease in the initiation step of RNA interference. Nature. 2001;409:363–366. doi: 10.1038/35053110. [DOI] [PubMed] [Google Scholar]
- 6.Hammond SM, Bernstein E, Beach D, Hannon GJ. An RNA-directed nuclease mediates post-transcriptional gene silencing in Drosophila cells. Nature. 2000;404:293–296. doi: 10.1038/35005107. [DOI] [PubMed] [Google Scholar]
- 7.Nykänen A, Haley B, Zamore PD. ATP requirements and small interfering RNA structure in the RNA interference pathway. Cell. 2001;107:309–321. doi: 10.1016/s0092-8674(01)00547-5. [DOI] [PubMed] [Google Scholar]
- 8.Zamore PD. RNA interference: listening to the sound of silence. Nat Struct Biol. 2001;8:746–750. doi: 10.1038/nsb0901-746. [DOI] [PubMed] [Google Scholar]
- 9.Hammond SM, Boettcher S, Caudy AA, Kobayashi R, Hannon GJ. Argonaute2, a link between genetic and biochemical analyses of RNAi. Science. 2001;293:1146–1150. doi: 10.1126/science.1064023. [DOI] [PubMed] [Google Scholar]
- 10.Tabara H, Sarkissian M, Kelly WG, Fleenor J, Grishok A, Timmons L, Fire A, Mello CC. The rde-1 gene, RNA interference, and transposon silencing in C. elegans. Cell. 1999;99:123–132. doi: 10.1016/s0092-8674(00)81644-x. [DOI] [PubMed] [Google Scholar]
- 11.Catalanotto C, Azzalin G, Macino G, Cogoni C. Transcription: gene silencing in worms and fungi. Nature. 2000;404:245. doi: 10.1038/35005169. [DOI] [PubMed] [Google Scholar]
- 12.Fagard M, Boutet S, Morel JB, Bellini C, Vaucheret H. AGO1, QDE-2, and RDE-1 are related proteins required for post-transcriptional gene silencing in plants, quelling in fungi, and RNA interference in animals. Proc Natl Acad Sci USA. 2000;97:11650–11654. doi: 10.1073/pnas.200217597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lee RC, Feinbaum RL, Ambros V. The C. elegans heterochronic gene lin-4 encodes small RNAs with antisense complementarity to lin-14. Cell. 1993;75:843–854. doi: 10.1016/0092-8674(93)90529-y. [DOI] [PubMed] [Google Scholar]
- 14.Olsen PH, Ambros V. The lin-4 regulatory RNA controls developmental timing in Caenorhabditis elegans by blocking LIN-14 protein synthesis after the initiation of translation. Dev Biol. 1999;216:671–680. doi: 10.1006/dbio.1999.9523. [DOI] [PubMed] [Google Scholar]
- 15.Reinhart BJ, Slack FJ, Basson M, Pasquinelli AE, Bet-tinger JC, Rougvie AE, Horvitz HR, Ruvkun G. The 21-nucleotide let-7 RNA regulates developmental timing in Caenorhabditis elegans. Nature. 2000;403:901–906. doi: 10.1038/35002607. [DOI] [PubMed] [Google Scholar]
- 16.Slack FJ, Basson M, Liu Z, Ambros V, Horvitz HR, Ruvkun G. The lin-41 RBCC gene acts in the C. elegans heterochronic pathway between the let-7 regulatory RNA and the LIN-29 transcription factor. Mol Cell. 2000;5:659–669. doi: 10.1016/s1097-2765(00)80245-2. [DOI] [PubMed] [Google Scholar]
- 17.Lagos-Quintana M, Rauhut R, Lendeckel W, Tuschl T. Identification of novel genes coding for small expressed RNAs. Science. 2001;294:853–858. doi: 10.1126/science.1064921. [DOI] [PubMed] [Google Scholar]
- 18.Lau NC, Lim LP, Weinstein EG, Bartel DP. An abundant class of tiny RNAs with probable regulatory roles in Caenorhabditis elegans. Science. 2001;294:858–862. doi: 10.1126/science.1065062. [DOI] [PubMed] [Google Scholar]
- 19.Lee RC, Ambros V. An extensive class of small RNAs in Caenorhabditis elegans. Science. 2001;294:862–864. doi: 10.1126/science.1065329. [DOI] [PubMed] [Google Scholar]
- 20.Lagos-Quintana M, Rauhut R, Yalcin A, Meyer J, Len-deckel W, Tuschl T. Identification of tissue-specific microRNAs from mouse. Curr Biol. 2002;12:735–739. doi: 10.1016/s0960-9822(02)00809-6. [DOI] [PubMed] [Google Scholar]
- 21.Mourelatos Z, Dostie J, Paushkin S, Sharma AK, Charroux B, Abel L, Rappsilber J, Mann M, Dreyfuss G. miRNPs: a novel class of ribonucleoproteins containing numerous microRNAs. Genes Dev. 2002;16:720–728. doi: 10.1101/gad.974702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Grishok A, Pasquinelli AE, Conte D, Li N, Parrish S, Ha I, Baillie DL, Fire A, Ruvkun G, Mello CC. Genes and mechanisms related to RNA interference regulate expression of the small temporal RNAs that control C. elegans developmental timing. Cell. 2001;106:23–34. doi: 10.1016/s0092-8674(01)00431-7. [DOI] [PubMed] [Google Scholar]
- 23.Hutvágner G, McLachlan J, Pasquinelli AE, Balint É, Tuschl T, Zamore PD. A cellular function for the RNA-interference enzyme Dicer in the maturation of the let-7 small temporal RNA. Science. 2001;293:834–838. doi: 10.1126/science.1062961. [DOI] [PubMed] [Google Scholar]
- 24.Ketting RF, Fischer SE, Bernstein E, Sijen T, Hannon GJ, Plasterk RH. Dicer functions in RNA interference and in synthesis of small RNA involved in developmental timing in C. elegans. Genes Dev. 2001;15:2654–2659. doi: 10.1101/gad.927801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Dalmay T, Hamilton A, Rudd S, Angell S, Baulcombe DC. An RNA-dependent RNA polymerase gene in Arabidopsis is required for posttranscriptional gene silencing mediated by a transgene but not by a virus. Cell. 2000;101:543–553. doi: 10.1016/s0092-8674(00)80864-8. [DOI] [PubMed] [Google Scholar]
- 26.Hutvágner G, Zamore PD. RNAi: nature abhors a double-strand. Curr Opin Genet Dev. 2002;12:225–232. doi: 10.1016/s0959-437x(02)00290-3. [DOI] [PubMed] [Google Scholar]
- 27.Schwartz BW, Yeung EC, Meinke DW. Disruption of morphogenesis and transformation of the suspensor in abnormal suspensor mutants of Arabidopsis. Development. 1994;120:3235–3245. doi: 10.1242/dev.120.11.3235. [DOI] [PubMed] [Google Scholar]
- 28.McElver J, Tzafrir I, Aux G, Rogers R, Ashby C, Smith K, Thomas C, Schetter A, Zhou Q, Cushman MA, et al. Insertional mutagenesis of genes required for seed development in Arabidopsis thaliana. Genetics. 2001;159:1751–1763. doi: 10.1093/genetics/159.4.1751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Robinson-Beers K, Pruitt RE, Gasser CS. Ovule development in wild-type Arabidopsis and two female sterile mutants. Plant Cell. 1992;4:1237–1250. doi: 10.1105/tpc.4.10.1237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Jacobsen SE, Running M, Meyerowitz EM. Disruption of an RNA helicase/RNAse III gene in Arabidopsis causes unregulated cell division in floral meristems. Development. 1999;126:5231–5243. doi: 10.1242/dev.126.23.5231. [DOI] [PubMed] [Google Scholar]
- 31.Ray A, Lang JD, Golden T, Ray S. SHORT INTEGUMENT (SIN1), a gene required for ovule development in Arabidopsis, also controls flowering time. Development. 1996;122:2631–2638. doi: 10.1242/dev.122.9.2631. [DOI] [PubMed] [Google Scholar]
- 32.Chen X, Liu J, Cheng Y, Jia D. HEN1 functions pleiotropically in Arabidopsis development and acts in C function in the flower. Development. 2002;129:1085–1094. doi: 10.1242/dev.129.5.1085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Chen X, Meyerowitz EM. HUA1 and HUA2 are two members of the floral homeotic AGAMOUS pathway. Mol Cell. 1999;3:349–360. doi: 10.1016/s1097-2765(00)80462-1. [DOI] [PubMed] [Google Scholar]
- 34.Elbashir SM, Lendeckel W, Tuschl T. RNA interference is mediated by 21- and 22-nucleotide RNAs. Genes Dev. 2001;15:188–200. doi: 10.1101/gad.862301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. Basic local alignment search tool. J Mol Biol. 1990;215:403–410. doi: 10.1016/S0022-2836(05)80360-2. [DOI] [PubMed] [Google Scholar]
- 36.Mathews DH, Sabina J, Zuker M, Turner DH. Expanded sequence dependence of thermodynamic parameters improves prediction of RNA secondary structure. J Mol Biol. 1999;288:911–940. doi: 10.1006/jmbi.1999.2700. [DOI] [PubMed] [Google Scholar]
- 37.Reinhart BJ, Weinstein EG, Rhoades MW, Bartel B, Bartel D. microRNAs in plants. Genes Dev. 2002;16:1616–1626. doi: 10.1101/gad.1004402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Llave C, Kasschau KD, Rector MA, Carrington JC. Endogenous and silencing-associated small RNAs in plants. Plant Cell. 2002;14:1605–1619. doi: 10.1105/tpc.003210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Bohmert K, Camus I, Bellini C, Bouchez D, Caboche M, Benning C. AGO1 defines a novel locus of Arabidopsis controlling leaf development. EMBO J. 1998;17:170–180. doi: 10.1093/emboj/17.1.170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Moussian B, Schoof H, Haecker A, Jorgens G, Laux T. Role of the ZWILLE gene in the regulation of central shoot meristem cell fate during Arabidopsis embryogenesis. EMBO J. 1998;17:1799–1809. doi: 10.1093/emboj/17.6.1799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lynn K, Fernandez A, Aida M, Sedbrook J, Tasaka M, Masson P, Barton MK. The PINHEAD/ZWILLE gene acts pleiotropically in Arabidopsis development and has overlapping functions with the ARGONAUTE1 gene. Development. 1999;126:469–481. doi: 10.1242/dev.126.3.469. [DOI] [PubMed] [Google Scholar]
- 42.Schwarz DS, Zamore PD. Why do miRNAs live in the miRNP? Genes Dev. 2002;16:1025–1031. doi: 10.1101/gad.992502. [DOI] [PubMed] [Google Scholar]
- 43.Morel JB, Godon C, Mourrain P, Béclin C, Boutet S, Feuerbach F, Proux F, Vaucheret H. Fertile hypomorphic argonaute (ago1) mutants impaired in post-transcriptional gene silencing and virus resistance. Plant Cell. 2002;14:629–639. doi: 10.1105/tpc.010358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Bowman JL, Smyth DR, Meyerowitz EM. Genetic interactions among floral homeotic genes of Arabidopsis. Development. 1991;112:1–20. doi: 10.1242/dev.112.1.1. [DOI] [PubMed] [Google Scholar]
- 45.Jofuku KD, den Boer BGW, Montagu MV, Okamuro JK. Control of Arabidopsis flower and seed development by the homeotic gene APETALA2. Plant Cell. 1994;6:1211–1225. doi: 10.1105/tpc.6.9.1211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Baulcombe DC. RNA as a target and an initiator of post-transcriptional gene silencing in transgenic plants. Plant Mol Biol. 1996;32:79–88. doi: 10.1007/BF00039378. [DOI] [PubMed] [Google Scholar]
- 47.Sijen T, Vijn I, Recocho A, van Blokland R, Roelofs D, Mol JNM, Kooter JM. Transcriptional and posttranscriptional gene silencing are mechanistically related. Curr Biol. 2001;11:436–440. doi: 10.1016/s0960-9822(01)00116-6. [DOI] [PubMed] [Google Scholar]
- 48.Mette MF, Aufsatz W, van der Winden J, Matzke MA, Matzke AJM. Transcriptional silencing and promoter methylation triggered by double-stranded RNA. EMBO J. 2000;19:5194–5201. doi: 10.1093/emboj/19.19.5194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Goff SA, Ricke D, Lan TH, Presting G, Wang R, Dunn M, Glazebrook J, Sessions A, Oeller P, Varma H, et al. A draft sequence of the rice genome (Oryza sativa L. ssp japonica) Science. 2002;296:92–100. doi: 10.1126/science.1068275. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
supplement