Interacting models of cooperative gene regulation (original) (raw)
Abstract
Cooperativity between transcription factors is critical to gene regulation. Current computational methods do not take adequate account of this salient aspect. To address this issue, we present a computational method based on multivariate adaptive regression splines to correlate the occurrences of transcription factor binding motifs in the promoter DNA and their interactions to the logarithm of the ratio of gene expression levels. This allows us to discover both the individual motifs and synergistic pairs of motifs that are most likely to be functional, and enumerate their relative contributions at any arbitrary time point for which mRNA expression data are available. We present results of simulations and focus specifically on the yeast cell-cycle data. Inclusion of synergistic interactions can increase the prediction accuracy over linear regression to as much as 1.5- to 3.5-fold. Significant motifs and combinations of motifs are appropriately predicted at each stage of the cell cycle. We believe our multivariate adaptive regression splines-based approach will become more significant when applied to higher eukaryotes, especially mammals, where cooperative control of gene regulation is absolutely essential.
Keywords: cooperativity, correlation, expression data, transcription regulation
Regulation of gene transcription in eukaryotes is complex and is inherently combinatorial in nature (1, 2). Transcriptional synergy is a key element of such combinatorial control in gene regulation networks. It requires cooperative binding of multiple transcription factors (TFs) and is intrinsically nonlinear in nature (2). Taking adequate account of such synergy in computational models is extremely important to have an accurate view of the underlying biology.
Conventional computational methods (3) have focused on identifying motifs upstream of the clusters of coexpressed genes. However, many genes fail to cluster and, therefore, regulatory elements of a large number of genes are unknown. Recent work (4, 5) has attempted to overcome this problem by correlating the frequency of DNA motifs with the logarithm of expression levels by using multivariate linear regression. Despite the success in identifying many known important motifs, this method does not account for the synergistic effects and nonlinearities present during transcription regulation. When applied to the yeast cell-cycle data, we found that these methods can explain only 10% of the variations in the data on an average (noise level accounts for ≈50%; ref. 4).
More recently, models that account for cooperativity between TFs during transcription regulation have been developed (6-10). However, all of these models are limited by one or more of the following factors. Some of these methods (6-8), like expression coherence (EC) score approach (6, 7), require data from multiple time points, which are not always available. Methods based on regression trees (8), on the other hand, cannot take proper account of additive effects. In other cases (9, 10), we found either the known pairs of motifs are not correctly predicted or the accuracy of the regression model does not improve significantly (≈5-10%) when interacting pairs are introduced in the model, which is inconsistent with the biological notion of synergistic gene regulation.
Here, we discuss a computational method that overcomes these limitations. It finds potentially functional cis-regulatory elements given microarray expression data and a set of candidate motifs. Some of the key features of this method are that it (i) can be applied to expression data from a single time point, (ii) can find both individual motifs and cooperative pairs of motifs that are more likely to be functional under a particular condition, (iii) allows the user to rank the relative strengths of individual motifs and pairs, and (iv) works with higher precision than the current computational methods.
Our approach is based on the well known multivariate adaptive regression splines (MARS) algorithm (11, 12). MARS builds response function in terms of nonlinear component functions and their products. The component functions used are linear splines, which have the shape of a hockey stick, i.e., they are zero below (above) a threshold, termed knot, and increase linearly above (below) it (Fig. 1). Thus, MARS uses nonlinear functions with minimal number of parameters to model the data. The model-building procedure used by MARS is easiest understood by considering its analogy with stepwise linear regression used in reduce (4). In the latter, one starts with a model with a constant term. One then finds the motif that best explains the current variation in the expression data by using a linear model. Its predicted contribution is subtracted from the observed data, and this motif is removed from the set of all motifs. The process is then repeated until a preset significance level is reached. This procedure yields a set of basis functions, each of which is a line: (1, n _k_1, n _k_2,..., n k L), where n j = count of motif j, and k i values are a selection from the original motif indices. In MARS, by contrast, one selects a linear spline at each step that best explains the data. A second difference is that products of splines that already exist in the basis set are also considered. Thus, the set of basis functions here looks like (1, θ(n _k_1 - ξ_k_1,0, 0), θ(n _k_2 - ξ_k_2,0, 0), θ(n _k_1 - ξ_k_1,0, 0).θ(n k_2 - ξ_k_2,0,0),...), where θ's are linear splines (Eq. 1), ξi,j represents the knot j of the motif i. [Here, for simplicity, we have shown splines of only one type. However, the other type, i.e., θ(ξ_i,j - n i, 0), is also considered in actual model building.] The final prediction is an additive contribution from each such basis function (Eq. 2). The biggest concern in using this approach would be overfitting the data. This problem is avoided by finding the model that has the least generalized cross-validation score (Eq. 3), which seeks a balance between the residual sum of squares and the number of parameters introduced in the model. A simple example of the model building procedure used by MARS is discussed in Data Set 1, which is published as supporting information on the PNAS web site.
Fig. 1.

Basis functions in MARS. Two types of linear splines (Eq. 1) used as basis functions in MARS. n represents the predictor variable. The points ξ1 and ξ2 are the knots (see text for definition).
In applying MARS to the microarray data, we treated the log ratio of gene expression levels, i.e., between a test sample and a control, as response variables and TF-binding motif occurrence scores (namely, occurrence frequencies, weight matrix scores, etc.) as predictor variables. We first analyzed the extent to which MARS can model expression data by applying it to the simulated data. We then built a program with MARS as the core regression tool to obtain functional motifs and their cooperative combinations from real gene expression data. This program is called marsmotif. The results of application of marsmotif to the yeast cell-cycle expression data are discussed below.
Methods
MARS. MARS (11, 12) is a nonparametric and adaptive regression method. It builds the model in terms of linear splines described by
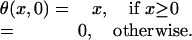 |
1 |
|---|
MARS builds the model by using stepwise forward addition of linear splines and their products. The fitted model has the functional form
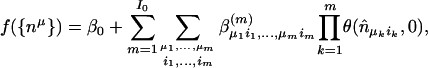 |
2 |
|---|
where n̂_μ_i = n_μ - ξ_i, or, ξ_i_ - _n_μ, _n_u is the motif count (or weight matrix score), ξi is a constant termed knot, and _I_0 is the maximum interactions allowed (denoted by the “int” parameter in the program). The product terms always involve distinct variables. Terms are then deleted sequentially to obtain a set of models _f_λ of different sizes λ. Optimal value of λ is obtained by minimizing the generalized cross-validation score GCV(λ), which is the residual sum of squares (RSS) times a factor that penalizes for model complexity
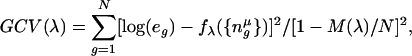 |
3 |
|---|
where e g = E g/E gC; E g is the expression level for gene g; M(λ) is the effective number of parameters; C is the control set; and N is the total number of genes. The GCV score is a generalization of leave-one-out cross-validation for least squares fit to N data points (12). M(λ) is obtained by cross-validation. The GCV-based optimization restricts the final model to a very small number of terms (Data Set 1). We used the mars program available from Salford Systems (San Diego) (13).
Percent Reduction in Variance. Percent reduction in variance (4), Δχ2, is defined by
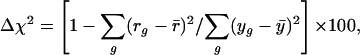 |
4 |
|---|
where y g = log(E g/EgC), residual 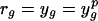 (p indicates the predicted value of y), and ȳ and r̄ are their corresponding means.
(p indicates the predicted value of y), and ȳ and r̄ are their corresponding means.
Simulated Data. For foreground genes, the log of expression level was obtained by using
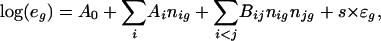 |
5a |
|---|
and for background genes
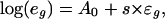 |
5b |
|---|
where e g = E g/E gC; ε_g_ is the N(0, 1) noise; s is a scale factor for the noise and is 0 or 1, unless otherwise mentioned; and n ig is the number of occurrences of the _i_th motif for the gene g. Each n ig for foreground genes ranges from 0 to 3. Linear model fitting was done with a multivariate linear regression model in R.
Cell Cycle Data. Motifs and expression data. We used the following sets for candidate motifs. (i) Motifs generated by using AlignACE by Pilpel et al. (6): we used the counts of motifs (PC) and Gibbs sampling scores (PW) separately. (ii) Counts of motifs (K) found by Kellis et al. (14). (iii) A manually curated set (CUR) of motifs (Table 6, which is published as supporting information on the PNAS web site). (iv) A 5-7mer word count with two different clustering methods: clustering by overlap (W57) and by using motifs from ref. 14 as reference templates (K57) (see Supporting Text, which is published as supporting information on the PNAS web site). We clustered the words to make sure that the input motifs in marsmotif are nonredundant. Nonredundancy is achieved in a linear model (4) by carrying out the regression in a stepwise manner. In the curated set (CUR), we also included the Mcm1 weight matrix motif from ref. 4 (Table 6). marsmotif is able to analyze a hybrid input of counts and weight matrix scores.
Kolmogorov-Smirnov (KS) test. KS test is a nonparametric test used to determine whether two samples are drawn from the same distribution. For one motif, we compared the distributions of expression values for the genes that have the motif with those that do not have the motif. For a pair of motifs, we compared genes that have that pair with those that have only one of the two motifs. This comparison potentially captures the synergistic pairs. KS test was implemented according to ref. 15.
marsmotif runs for individual motifs. For a set of candidate motifs, we first checked their association with expression by using the KS test. The top 100 motifs by KS P value were used in MARS with int = 1 setting to obtain the significant motifs.
marsmotif runs for interacting motifs. The pairs of motifs were first constructed from the top 100 motifs above and sorted by using the KS test. The top 200 motif pairs from the KS test were then used in MARS with int = 2 and int = 3 separately.
P values of motifs and motif pairs and model pruning. P values of motifs and motif pairs were computed based on an F test (12)
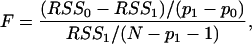 |
6 |
|---|
where _RSS_1 is the residual sum of squares of the final MARS model with _p_1 + 1 terms, and _RSS_0 is the residual sum of squares of the MARS model without a particular motif (or pair), which has _p_0 + 1 terms. N is the number of genes. The F statistic has a F distribution with _p_1 - _p_0 numerator degrees of freedom and N - _p_1 - 1 denominator degrees of freedom. P values were calculated in s-plus. Only motifs and motif pairs with P ≤ 0.01 (after multiple testing) were kept in the final MARS model, for which the Δχ2 is reported here. We invoke this P value cutoff for easier comparison with linear methods (4, 5). Overfitting in our technique is prevented by GCV minimization, as mentioned above.
Corrections for multiple testing. Corrections for multiple testing were done by using the false discovery rate (FDR) method (16). The F test P values were sorted: P(0) ≤ P(1) ≤···≤ P(M), where M denotes the total number of tests. The adjusted P value is then
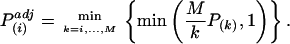 |
7 |
|---|
Further Details. For further details, see Supporting Methods, which is published as supporting information on the PNAS web site.
Results
Simulated Data. We first used simulation data to test the ability of MARS to correlate motif counts to expression data. The results obtained here generalize to the weight matrix scores. The simulation data consist of a set of foreground and background genes: the foreground genes have a nonzero number of binding motifs in their promoter DNA, and their log ratio of expression levels are generated by using a model with linear and pairwise terms in motif frequencies plus a noise term (Eq. 5). The background genes do not have any binding motif in their promoters, and their expression levels consist of base expression level and noise. For example, for a cell-cycle experiment, the foreground genes would represent the cell-cycle regulated genes and the background genes the non-cell-cycle genes.
Table 1 shows the results of the simulation for various parameter settings for linear regression and MARS runs with maximum allowable interactions (int) as 1 (no interactions between distinct motifs), 2 (pairwise interactions), and 3 (third-order interactions). The int = 1 model contains the linear effects as well as any self-interactions, i.e., interactions of the same motifs. The int > 1 models capture interactions between distinct motifs. The performance of any particular regression model is evaluated in terms of the percent reduction in variance (4) in residuals (Δχ2) (Eq. 4). For all parameter settings, we find that MARS with int = 2 consistently outperforms the linear model or MARS with int = 1.
Table 1. Summary of simulation results.
| % reduction in variance | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| MARS | ||||||||||
| Row number | Background genes | No. of motif clusters | Motif number per cluster | Noise scale factor(s) | Weight | Number of motifs not included in MARS input | Linear | int = 1 | int = 2 | int = 3 |
| 1 | 0 | 1 | 5 | 0 | 1 | 0 | 61.7 | 61.9 | 100 | 100 |
| 2 | 0 | 1 | 10 | 0 | 1 | 0 | 60.5 | 60.8 | 99.9 | 98.1 |
| 3 | 0 | 1 | 5 | 1 | 1 | 0 | 59.7 | 59.8 | 93.8 | 93.8 |
| 4 | 0 | 1 | 10 | 1 | 1 | 0 | 61.2 | 61.5 | 98.4 | 96.7 |
| 5 | 1,000 | 1 | 10 | 1 | 1 | 0 | 60.6 | 61.5 | 95.2 | 93.6 |
| 6 | 2,000 | 1 | 10 | 1 | 1 | 0 | 58.9 | 59.7 | 92.1 | 91 |
| 7 | 3,000 | 1 | 10 | 1 | 1 | 0 | 61.2 | 62 | 89.9 | 90.1 |
| 8 | 4,000 | 1 | 10 | 1 | 1 | 0 | 62.2 | 63 | 89.4 | 89.1 |
| 9 | 4,000 | 1 | 5 | 1 | 1 | 0 | 51.7 | 52.3 | 79.1 | 78.7 |
| 10 | 4,000 | 2 | 5 | 1 | 1 | 0 | 52.9 | 53.8 | 77 | 78.8 |
| 11 | 4,000 | 3 | 5 | 1 | 1 | 0 | 55.6 | 56.5 | 75.9 | 80.2 |
| 12 | 4,000 | 4 | 5 | 1 | 1 | 0 | 54.9 | 56.1 | 73.9 | 79.5 |
| 13 | 4,000 | 4 | 5 | 1.5 | 1 | 0 | 47.4 | 48.2 | 60.8 | 65.8 |
| 14 | 4,000 | 4 | 5 | 2 | 1 | 0 | 34.1 | 34.7 | 46 | 50 |
| 15 | 4,000 | 4 | 5 | 2.5 | 1 | 0 | 28.4 | 29.1 | 37.6 | 40.7 |
| 16 | 4,000 | 4 | 5 | 3 | 1 | 0 | 23.5 | 24 | 31.5 | 33.8 |
| 17 | 4,000 | 4 | 5 | 3.5 | 1 | 0 | 19.5 | 20 | 26.2 | 27.5 |
| 18 | 4,000 | 4 | 5 | 3.5 | 2 | 0 | 18.5 | 19.2 | 25.1 | 26.5 |
| 19 | 4,000 | 4 | 5 | 3.5 | 4 | 0 | 18.1 | 18.8 | 24.5 | 26.2 |
| 20 | 4,000 | 4 | 5 | 3.5 | 16 | 0 | 19.1 | 19.9 | 26.1 | 27.2 |
| 21 | 4,000 | 4 | 5 | 3.5 | 100 | 0 | 18.1 | 18.5 | 24.7 | 26.1 |
| 22 | 0 | 1 | 10 | 3.5 | 1 | 0 | 53 | 53.2 | 86.1 | 84.9 |
| 23 | 0 | 4 | 5 | 3.5 | 1 | 0 | 43.4 | 44.3 | 60.4 | 63.4 |
| 24 | 0 | 4 | 5 | 3.5 | 1 | 1 | 43.9 | 44.8 | 59.8 | 62 |
| 25 | 0 | 4 | 5 | 3.5 | 1 | 2 | 41.4 | 42 | 56.3 | 57.7 |
| 26 | 0 | 4 | 5 | 3.5 | 1 | 4 | 34.2 | 34.7 | 46.7 | 47.4 |
| 27 | 0 | 4 | 5 | 0 | 1 | 4 | 57 | 57.9 | 75.5 | 77.9 |
Rows 1-4 display the performance of MARS both without and with any noise in the absence of any background gene and provide a baseline for comparison for all other settings. Introduction of noise has marginal effect on the prediction accuracy in this case. We explored the effects of various parameters on the performance of MARS with int = 2. (i) For background genes, increasing their number from 0 to 4,000 decreases the accuracy of MARS by ≈9% (rows 4-8). (ii) One subgroup of genes is regulated by a certain set of motifs, whereas another subgroup is regulated by a different set of motifs. We call such disjoint motif sets motif clusters. As we increase the number of motif clusters from 1 to 4, the accuracy decreases by ≈5% (rows 9-12). (iii) When the strength of the noise is examined, as we increase the noise scale factor (in Eq. 5) from 1 to 3.5, MARS accuracy decreases by ≈48% (rows 12-17). This has, by far, the strongest effect. Putting extra weights on the foreground genes does not help MARS to recover the actual model (rows 18-21). The accuracy is much higher if there are no background genes and/or no heterogeneous motif clusters (rows 22 and 23), even if the noise level is very high. (iv) The true predictors of expression levels are binding affinities of various TFs to DNA motifs and TF concentrations. In the regression approach, motif frequencies and weight matrix scores are used as surrogates. To explore the effect of using incorrect predictors, we randomly removed some true motifs from the input to MARS. Increasing the number of true motifs not included in MARS input from 0 to 4 decreases the accuracy by ≈14% (rows 23-26). Accuracy improves significantly if there is no noise (row 27).
Apart from the fact that int = 2 MARS performs much better than the linear and int = 1 MARS, a couple of aspects are clear from the simulations. First, comparison between int = 2 and int = 3 MARS runs (last two columns in Table 1) shows that overfitting by MARS is minimal and typically happens if there is a large number of motif clusters. For instance, the accuracy sometimes can decrease with int = 3 compared to int = 2. Second, MARS (int = 2) can capture the full underlying model except for the random noise (Table 7, which is published as supporting information on the PNAS web site). This is true even when the noise is the strongest.
Yeast Cell Cycle. Following the success of MARS in the simulations, we built the program marsmotif with MARS as the core regression tool to analyze real biological data. marsmotif starts with a large set of candidate motifs and prioritizes the motifs and motif pairs by using a KS test, which is nonparametric. It then runs MARS with int = 1, 2, and 3, with this prioritized set of motifs and pairs. Of these three runs, the one with the maximum Δχ2 is considered as the representative model. The third-order interactions in the int = 3 model are built from the component pairs obtained from KS test. Because the number of candidate motifs and motif pairs can be very large, filtering by a method like KS test is necessary to make optimal use of MARS (for details, see Methods and Fig. 3, which is published as supporting information on the PNAS web site).
We ran marsmotif on yeast cell-cycle data spanning 77 experiments (3, 17). Because the simulations suggest that a large number of background genes may lead to a lower accuracy of MARS, we applied marsmotif only to the expression data of the cell-cycle regulated genes (≈800 genes; ref. 3). For candidate motifs, we used 5-7mer word counts and motifs reported in the literature, as obtained by Gibbs sampling (6) and cross-species conservation (14) on the yeast promoters. A curated set of motifs (Table 6) and a set obtained by combining 5-7mer word count and cross-species conservation were also used. The description of the various motif sets and their corresponding notation are detailed in Methods.
Table 2 shows the performance of marsmotif for all of these data sets. Like in simulations, the performance is measured in terms of the percent reduction in variance of residuals (Eq. 4), averaged over 77 experiments (termed average reduction in variance, 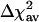 ). In comparison with linear regression (reduce) (4), where the
). In comparison with linear regression (reduce) (4), where the 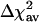 is 9.6%, for various data sets, we find the marsmotif
is 9.6%, for various data sets, we find the marsmotif 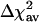 varying between 13.9% and 32.9%. Thus, the marsmotif accuracy is ≈1.5-3.5 times that of reduce. Because word counts were used as predictor variables in reduce, we believe that the true improvement lies toward the upper end of this range. Even if we do not consider the int = 1 case in our analysis, the
varying between 13.9% and 32.9%. Thus, the marsmotif accuracy is ≈1.5-3.5 times that of reduce. Because word counts were used as predictor variables in reduce, we believe that the true improvement lies toward the upper end of this range. Even if we do not consider the int = 1 case in our analysis, the 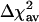 does not change much in most cases. For most data sets, we find an improvement when interactions between distinct motifs are included (int > 1) over no interactions (int = 1) in ≈69-88% of the experiments. The average increase in Δχ2 in these cases over int = 1 case is in the range of 47-96%. This finding is consistent with the notion that synergy plays a key role in transcriptional regulation (2). In the data set with word counts (W57), most of the interactions are accounted for by self-interactions (due to the clustering of motifs, see Methods) and, therefore, the number of experiments showing improvement with interactions is smaller.
does not change much in most cases. For most data sets, we find an improvement when interactions between distinct motifs are included (int > 1) over no interactions (int = 1) in ≈69-88% of the experiments. The average increase in Δχ2 in these cases over int = 1 case is in the range of 47-96%. This finding is consistent with the notion that synergy plays a key role in transcriptional regulation (2). In the data set with word counts (W57), most of the interactions are accounted for by self-interactions (due to the clustering of motifs, see Methods) and, therefore, the number of experiments showing improvement with interactions is smaller.
Table 2.
Summary of marsmotif results on the yeast cell-cycle data
| Algorithm | Data set | Motif discovery method | Average reduction in variance (best of int = 1, 2, and 3), % | Average reduction in variance with int > 1 only (best of int = 2 and 3), % | % Cases that have an increase in Δ_X_2 with interactions (int > 1) over int = 1 | Average percent increase in Δ_X_2 with int > 1 over int = 1 for cases in the previous column |
|---|---|---|---|---|---|---|
| reduce | 1-7mer nucleotides | World count | 9.6 | − | − | − |
| marsmotif | Motif counts from Pilpel et al. (6) (PC) | Gibbs sampling | 20.0 | 19.0 | 80.5 (62 of 77) | 95.7 |
| marsmotif | Motif scores from Pilpel et al. (6) (PW) | Gibbs sampling | 19.9 | 19.2 | 87 (67 of 77) | 59.8* |
| marsmotif | Motif counts from Kellis et al. (14) (K) | Cross-species conservation | 13.9 | 12.6 | 70.1 (54 of 77) | 52.8* |
| marsmotif | Motif counts from curated data set (CUR) | Curation | 21.7 | 21.2 | 88.3 (68 of 77) | 51.8 |
| marsmotif | 5-7mer nucleotides (W57) | Word count | 32.9 | 23.6 | 33.8 (26 of 77) | 46.5 |
| marsmotif | Counts of 5-7mers clustered by using motifs from Kellis et al. (14) (K57) | Word count and cross-species conservation | 29.7 | 26.8 | 68.8 (53 of 77) | 69.2 |
Significant Motifs and Motif Pairs. We now turn to the significant motifs and motif combinations predicted by marsmotif. Let us consider the 49-min time point of the α-arrest series of experiments which lies in the G2/M phase. Table 3 shows the marsmotif predictions using the data set PC as predictor variables. Mcm1 and Fkh1/2 are two key regulators in this phase: they cooperatively drive the transcription of the genes in the CLB2 cluster (18). Ste12 and Swi5 play an important role in early M phase (18). We find the motifs of all these factors with high significance. The P values were calculated by using F test (Eq. 6), adjusted for multiple testing (Eq. 7). The interaction between Mcm1 and Fkh1/2 (motif SFF′, ref. 6) is also found to be significant. Previous regression models (4, 9) failed to identify this cooperative interaction. MCB element is typically functional in the G1/S phase. The fact that we find this element during the G2/M phase might be due to the secondary processes going on with the cell-cycle where this element is active. MCB-MCM1 and SFF′-STE12 are among the other significant pairs found in this phase. The MCB-MCM1 pair was found significant in the EC score approach (6). The SFF′-STE12 pair has not been characterized experimentally. However, each TF works via a common partner, MCM1, to influence cell cycle and mating response in G2/M phase. During pseudohyphal differentiation, Ste12 is critical for the cell cycle shift to G2/M (19). So the discovery of the SFF′-STE12 pair is not unwarranted. The other motifs and motif pairs at this time point involve one or more of the motifs discovered from the upstream regions of the genes in the MIPS functional categories (6) (Table 8, which is published as supporting information on the PNAS web site).
Table 3.
Significant motif and motif pairs for α49 experiment (3)
| Motifs and motif pairs | P value |
|---|---|
| MCM1 | 4.8_E_-15 |
| SFF′ | 4.8_E_-15 |
| STE12 | 4.33_E_-11 |
| SWI5 | 1.17_E_-10 |
| MCB | 5.32_E_-09 |
| MCM1*SFF′ | 4.8_E_-15 |
| MCB*MCM1 | 4.8_E_-15 |
| SFF′*STE12 | 4.8_E_-15 |
We have found several other motif pairs as significant at different stages of the cell-cycle in α-arrest experiments (Table 4). Some of these have already been characterized. Examples include Mcm1-Ste12 and _Ace_2-Swi5 pairs found in M and M/G1 phases, respectively. Mcm1 and Ste12 coordinately regulate the transcription of several genes involved in mating, which peak at the G1 phase (20), whereas _Ace_2-Swi5 pair regulates the M/G1 transcription of genes in SIC1 cluster (21). ECB-SFF pair, which emerges significant in the G1/S phase, is strongly implicated in several experimental findings (22, 23).
Table 4. Selective cooperative motif pairs for the alpha-arrest experiments.
| Motif1/T F1 | Motif2/TF2 | Motif set | Time point | Phase |
|---|---|---|---|---|
| ALPHA2 | MCM1 | PW | 7 | G1 |
| ACE2 | XBP1_(HSF 1-coocuring) | K | 56 | G1 |
| SFF | SWI5 | PW | 70 | G1/S |
| ECB | SFF | PW | 70 | G1/S |
| GCR1 | ACE2 | CUR | 84 | S |
| SMP1 | RAP1 | K57 | 84 | S/G2 |
| MCM1_Reduce | DIG1/STE12 | CUR | 42, 56 | M |
| ACE2 | FKH1/2 | CUR | 105 | M |
| ACE2 | SWI5 | CUR, K | Mult | M/G1 |
| GCR1 | SWI4 | CUR | 119 | M/G1 |
| Reb1 | GCR1 | K57 | 119 | M/G1 |
The second class of synergistic pairs discovered by marsmotif involves regulators that are known to participate in processes secondary to cell cycle. Examples are Alpha2-Mcm1, _Ace_2-Hsf1, and SFF-Swi5 found at the G1, G1, and G1/S phases, respectively. Alpha2-Mcm1 pair binds DNA as a heterodimer to regulate transcription of mating-type-specific genes in yeast (24), whereas _Ace_2-Hsf1 and SFF-Swi5 have been implicated as active under stress-related conditions (7).
The third class of significant pairs contains motif combinations predicted de novo by marsmotif. GCR1-SWI4 and GCR1-ACE2 are two such examples. Recent studies show that Gcr1 plays a critical role in glucose-dependent stimulation of CLN-dependent processes in the M and G1 phases (25). Gcr1 involvement in cell-cycle regulation was studied by constructing gcr1Δcln3Δ and gcr1Δcln1Δcln2Δ strains. All gcr1Δ strains have a cell-cycle delay that predominates in G1 or M phase. Given this scenario, we suggest that Swi4, a G1-specific regulator, and _Ace_2, an M-specific regulator, partner with Gcr1 in a phase-specific manner giving rise to the significant motif combinations.
Several pairs of regulators that were predicted as significant in ref. 7 are also found by our method. Examples are _Ace_2-Fkh1/2, Smp1-Rap1, Mbp1-Ste12, and Fkh1/2-Sok2. We have also been able to verify several pairs found significant in ref. 6 by using the same data sets, i.e., PC and/or PW:MCB-SFF′ (G1 phase; PC and PW), MCB-MCM1′ (time point 63; PW), and ECB-SFF (time point 70; PW) are examples. The advantage of using marsmotif over these methods is that we are able to assign a well defined phase/time points to these pairs where they are active. However, there are some pairs found in ref. 6 that we could not validate with our method. One such example is PAC-mRRPE pair. When we evaluated the EC score of this pair by using only the cell-cycle related genes, we found that the EC score of this motif pair is much lower than that of any one of the motifs taken by itself (Supporting Text). Therefore, the PAC-mRRPE pair may not be a true cell-cycle regulator. In fact, in a recent study (26), PAC and mRRPE have been mainly implicated in rRNA transcription and processing.
marsmotif is able to confirm many of the classical individual motifs (18) for cell-cycle regulation that have been predicted at correct phases in the previous computational analyses (3, 4, 9). For instance, if we consider the curated data set (CUR), we find the motifs for regulators Mbp1 and Swi4 significant in the G1/S phase (e.g., time points 14 and 21), motifs for Fkh1/2 and Mcm1 significant in G2/M phase (e.g., time points 35 and 42), and motifs for _Ace_2, Ste12, and Swi5 significant in the M/G1 phase (e.g., time point 56). Like other regression approaches (4), we find these motifs significant at some of the other phases as well. We address this issue of varying phase specificity below. Besides the classic motifs, we also uncover some of the motifs that have been characterized as important in yeast cell-cycle regulation or transcription regulation in general in this and other data sets as significant (Table 5). For example, Rme1 is responsible for activating some of the cyclins in the G1 phase and can act as a substitute for the factor SBF (27). We find its binding motif significant at the G1/S time point 21. The proteins Abf1, Reb1, Adr1, and Rap1 have been associated with chromosomal domain barrier function (28). Their corresponding motifs were determined to be functional at multiple time points near G2 and S/G2 phases. Also, the motifs corresponding to Rlm1, Sok2, Hsf1, and Msn1/2 emerge significant at multiple time points. The results of our marsmotif analysis for all of the experiments and across all of the candidate motif sets are available on our web site (http://rulai.cshl.edu/MARSMotif).
Table 5. Select set of significant motifs for the alpha-arrest experiments.
| Motif/TF | Motif set | Time point | Phase caps |
|---|---|---|---|
| CIN5/YAP1 | K57 | 63 | G1 |
| RME1 | K | 21 | G1/S |
| RAP1 | K57 | Mult | S/G2 |
| ABF1 | PC, CUR, K, K57 | 28, 42 | G2 |
| REB1 | PW | 35 | G2 |
| Novel_SOK2_2 | CUR | Mult | G2/M |
| HSF1 | CUR, K | Mult | M/G1 |
| RLM1 | K | Mult | M/G1 |
| ADR1 | CUR, K, K57 | Mult | - |
| MSN1/2 | CUR | Mult | - |
Periodic Regulation of Cell Cycle. Concentrations of many TFs vary periodically throughout the cell cycle (18). Correspondingly, one would expect that the significance of their binding motifs and combinations thereof will vary periodically. However, when an algorithm like marsmotif or reduce (4) is applied to a large collection of candidate motifs, this periodicity may not be apparent. Several factors, such as P value cutoff, strength of biochemical signal, and ongoing secondary processes, are responsible for this (Supporting Text). To see whether marsmotif can truly capture the cell-cycle-related periodicity, one needs to consider one motif, or motif pair, at a time.
Fig. 2 shows the percent reduction of variance by using marsmotif and linear models for a single motif (SCB element) and a motif pair (MCM1-SFF pair). In both cases, marsmotif can clearly capture the periodicity. Because there are two cell cycles and percent reduction in variance is a positive semidefinite quantity, the time course has four peaks. Although marsmotif and linear models are almost identical for a single motif, marsmotif model provides a better description for the pair. Obviously, interactions for which a linear model cannot account are modelled in the latter (Supporting Text). Some more examples are shown in Fig. 4, which is published as supporting information on the PNAS web site. The exact periodic behavior ultimately depends on the motif or motif pair under consideration, experimental set up, and the quality of motifs being used.
Fig. 2.

Periodic time courses. Percent reduction in variance (%RIV) for SCB element (CRCGAAA) (a) and MCM1-SFF motif pair (data set PC) (b) using the marsmotif and linear models for the alpha-arrest experiments.
Discussion
In this paper, we have demonstrated that marsmotif goes beyond linear regression and can successfully model the cooperative effects of synergistic motif pairs along with linear and self-interaction effects of the individual motifs present during transcription regulation. It can achieve much higher quantitative accuracy than the currently available computational methods. At the same time, it can provide further insight into the underlying biology. marsmotif allows an easy feature selection, i.e., by selecting and prioritizing correct motifs and motif pairs from an input set of motifs. Periodic regulation of cell-cycle can also be seen clearly in this framework.
As we have shown, the marsmotif approach to gene regulation can work very well for single time points. If there are data from multiple time points, one would bypass the step involving the KS test and construct a prioritized set of motif pairs by using a method such as EC scores (6, 7), for instance, for input to MARS.
Here, we have primarily focused on pairs of interacting motifs because very little is known about higher-order combinations beyond pairing, and, therefore, higher-order combinations are difficult to compare. However, this method can easily be extended to obtain higher-order combinations.
There are several reasons why a MARS based method like marsmotif can improve significantly on the other existing methods. First, the linear splines used in MARS can capture the switch-like behavior intrinsic to synergistic control of transcription (2). Second, the basis functions used in MARS, in a sense, can faithfully model the energetics of the underlying biochemical process as follows. The transcription rate can be written as _d_[E _g_]/dt = _K_A - _K_D[E _g_], where [E _g_] is the mRNA expression level corresponding to gene _g, K_A is the activation rate, and K D is the mRNA decay rate. Under the steady-state approximation, _d_[E _g_]/dt ≈ 0, i.e., log([E _g_]) = log(_K_A) - log(_K_D). Because _K_A ∝ _p_bind, the binding probability of a TF to the DNA, which has the form of a sigmoidal function (to be more precise, a Fermi-Dirac distribution) (29), the log of _p_bind mimics hockey stick functions used as basis functions in MARS. We think this is one of the key reasons why a MARS-based tool can improve significantly over a similar method that uses linear regression. Third, the true predictors of expression levels, i.e., activator concentrations and their affinities for binding to DNA, are being approximately represented by motif occurrences (or scores). Therefore, true binding and transcriptional activation does not possibly happen unless the word count is above a nonzero threshold. Use of linear splines can rectify such noise present in the predictor variables.
A few other potential applications of this method are quite clear. First, because of its high predictive accuracy, marsmotif can be used to judge the quality of a motif data set. In yeast, counts of individual words seem to be the best set of predictor variables. However, if we consider the ease of interpretation along with performance, combination of cross-species conservation and word counts (K57) is the optimal choice. It is clear from both the simulations and yeast cell-cycle analysis that performance of MARS is critically dependent on the use of correct predictor variables. Secondly, marsmotif can also be used with the chromatin immunoprecipitation chip data to discover functional motifs and motif combinations. Finally, we have established the role of marsmotif in discovering functional elements rather than as an ab initio motif discovery tool. However, with some simple modifications, it can be easily extended to create an ab initio motif discovery tool as can be seen from application of marsmotif using the 5-7mer word counts.
In higher eukaryotes, especially in mammals, transcriptional regulation mechanism is much more complex (1). Our analysis suggests that both the degenerate motifs and complex combinatorial interactions, which are strongly characteristic of higher eukaryotes, are well handled by marsmotif. Furthermore, marsmotif can analyze weight matrix scores of motifs equally well as the motif frequencies (Table 2). Weight matrix scores must be used in higher eukaryotes. Therefore, we think the impact of this MARS-based discovery method will be much greater when applied to _cis_-regulatory element discovery in more complex organisms.
Supplementary Material
Supporting Information
Acknowledgments
We thank Gengxin Chen for several useful discussions during the course of this work and Pavel Sumazin for a careful reading of the manuscript. This work is supported by National Institutes of Health Grants GM060513 and HG001696 (to M.Q.Z.).
Abbreviations: TF, transcription factor; EC, expression coherence; MARS, multivariate adaptive regression splines; KS, Kolmogorov-Smirnov.
References
- 1.Levine, M. & Tjian R. (2003) Nature 424**,** 147-151. [DOI] [PubMed] [Google Scholar]
- 2.Carey, M. (1998) Cell 92**,** 5-8. [DOI] [PubMed] [Google Scholar]
- 3.Spellman, P. T., Sherlock, G., Zhang M. Q., Iyer, V. R., Anders, K., Eisen, M. B., Brown, P. O., Botstein, D. & Futcher, B. (1998) Mol. Biol. Cell 9**,** 3273-3297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bussemaker, H. J., Li, H. & Siggia, E. D. (2001) Nat. Genet. 27**,** 167-171. [DOI] [PubMed] [Google Scholar]
- 5.Conlon, E. M., Liu, X. S., Lieb, J. D. & Liu, J. S. (2003) Proc. Natl. Acad. Sci. USA 100**,** 3339-3344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Pilpel, Y., Sudarsanam, P. & Church, G. M. (2001) Nat. Genet. 29**,** 153-159. [DOI] [PubMed] [Google Scholar]
- 7.Banerjee, N. & Zhang, M. Q. (2003) Nucleic Acids Res. 31**,** 7024-7031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Phuong, T. M., Lee, D. & Lee, K. H. (2004) Bioinformatics 20**,** 750-757. [DOI] [PubMed] [Google Scholar]
- 9.Keles, S., van der Laan, M. & Eisen, M. B. (2002) Bioinformatics 18**,** 1167-1175. [DOI] [PubMed] [Google Scholar]
- 10.Chiang, D. Y., Moses, A. M., Kellis, M., Lander, E. S. & Eisen, M. B. (2003) Genome Biol. 4**,** R43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Friedman, J. H. (1991) Ann. Stat. 19**,** 1-67. [Google Scholar]
- 12.Hastie, T., Tibshirani, R. & Friedman, J. H. (2001) The Elements of Statistical Learning (Springer, New York).
- 13.Steinberg, D. & Colla, P. (1999) MARS: An Introduction (Salford Systems, San Diego).
- 14.Kellis, M., Patterson, N., Endrizzi, M., Birren, B. & Lander, E. S. (2003) Nature 423**,** 241-254. [DOI] [PubMed] [Google Scholar]
- 15.Press, W. H., Flannery, B. P., Teukolsky, S. A. & Vetterling, W. T. (1992) Numerical Recipes in C: The Art of Scientific Computing (Cambridge Univ. Press, Cambridge, U. K.).
- 16.Benjamini, Y. & Hochberg, Y. (1995) J. R. Stat. Soc. B 57**,** 289-300. [Google Scholar]
- 17.Cho, R. J., Campbell, M. J., Winzeler, E. A., Steinmetz, L., Conway, A., Wodicka, L., Wolfsberg, T. G., Gabrielian, A. E., Landsman, D., Lockhart, D. J., et al. (1998) Mol. Cell 2**,** 65-73. [DOI] [PubMed] [Google Scholar]
- 18.Simon, I., Barnett, J., Hannett, N., Harbison, C. T., Rinaldi, N. J., Volkert, T. L., Wyrick, J. J., Zeitlinger, J., Gifford, D. K., Jaakkola, T. S., et al. (2001) Cell 106**,** 697-708. [DOI] [PubMed] [Google Scholar]
- 19.Ahn, S. H., Acurio, A. & Kron, S. J. (1999) Mol. Biol. Cell 10**,** 3301-3316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Oehlen, L. J., McKinney, J. D. & Cross, F. R. (1996) Mol. Cell. Biol. 16**,** 2830-2837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zhu, G., Spellman, P. T., Volpe, T., Brown, P. O., Botstein, D., Davis, T. N. & Futcher, B. (2000) Nature 406**,** 90-94. [DOI] [PubMed] [Google Scholar]
- 22.Pramila, T., Miles, S., GuhaThakurta, D., Jemiolo, D. & Breeden, L. L. (2002) Genes Dev. 16**,** 3034-3045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Mai, B., Miles, S. & Breeden, L. L. (2002) Mol. Cell. Biol. 22**,** 430-441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zhong, H., McCord, R. & Vershon, A. K. (1999) Genome Res. 9**,** 1040-1047. [DOI] [PubMed] [Google Scholar]
- 25.Willis, K. A., Barbara, K. E., Menon, B. B., Moffat, J., Andrews, B. & Santangelo, G. M. (2003) Genetics 165**,** 1017-1029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sudarsanam, P., Pilpel, Y. & Church, G. M. (2002) Genome Res. 12**,** 1723-1731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Toone, W. M., Johnson, A. L., Banks, G. R., Toyn, J. H., Stuart, D., Wittenberg, C. & Johnston, L. H. (1995) EMBO J. 14**,** 5824-5832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Yu, Q., Qiu, R., Foland, T. B., Griesen, D., Galloway, C. S., Chiu, Y. H., Sandmeier, J., Broach, J. R. & Bi, X. (2003) Nucleic Acids Res. 31**,** 1224-1233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Djordjevic, M., Sengupta, A. M. & Shraiman, B. I. (2003) Genome Res. 13**,** 2381-2390. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information