An Eulerian path approach to DNA fragment assembly (original) (raw)
Abstract
For the last 20 years, fragment assembly in DNA sequencing followed the “overlap–layout–consensus” paradigm that is used in all currently available assembly tools. Although this approach proved useful in assembling clones, it faces difficulties in genomic shotgun assembly. We abandon the classical “overlap–layout–consensus” approach in favor of a neweuler algorithm that, for the first time, resolves the 20-year-old “repeat problem” in fragment assembly. Our main result is the reduction of the fragment assembly to a variation of the classical Eulerian path problem that allows one to generate accurate solutions of large-scale sequencing problems. euler, in contrast to the celera assembler, does not mask such repeats but uses them instead as a powerful fragment assembly tool.
Children like puzzles, and they usually assemble them by trying all possible pairs of pieces and putting together pieces that match. Biologists assemble genomes in a surprisingly similar way, the major difference being that the number of pieces is larger. For the last 20 years, fragment assembly in DNA sequencing mainly followed the “overlap–layout–consensus” paradigm (1–6). Trying all possible pairs of pieces corresponds to the overlap step, whereas putting the pieces together corresponds to the layout step of the fragment assembly. Our new euler algorithm is very different from this natural approach—we never even try to match the pairs of fragments, and we do not have the overlap step at all. Instead, we do a very counterintuitive (some would say childish) thing: we cut the existing pieces of a puzzle into even smaller pieces of regular shape. Although it indeed looks childish and irresponsible, we do it on purpose rather than for the fun of it. This operation transports the puzzle assembly from the world of a difficult Layout Problem into the world of the Eulerian Path Problem, with polynomial algorithms for puzzle assembly (in the context of DNA sequencing).
Although the classical approach culminated in some excellent programs (phrap, cap, tigr, andcelera assemblers among them), critical analysis of the “overlap–layout–consensus” paradigm reveals some weak points. The major difficulty is that there is still no polynomial algorithm for the solution of the notorious “repeat problem” that amounts to finding the correct path in the overlap graph (layout step). Unfortunately, this problem is difficult to overcome in the framework of the “overlap–layout–consensus” approach. All the programs we tested made errors (up to 19% misassembled contigs) while assembling shotgun reads from the bacterial sequencing projects (Table1; Fig. 1). These genomes were assembled despite the fact that there is no error-free fragment assembler today (by errors we mean incorrect assemblies rather than unavoidable base-calling errors). Biologists “pay” for these errors at the time-consuming finishing step (7).
Table 1.
Comparison of different software tools for fragment assembly
| ideal | euler | phrap | cap3 | tigr assembler | ||
|---|---|---|---|---|---|---|
| CJ | No. of contigs (no. of misassembled contigs) | 24 (5) | 29 (0) | 33 (2) | 54 (3) | >300 (>10) |
| Coverage by contigs | 99.5% | 96.7% | 94.0% | 92.4% | 90.0% | |
| Coverage by misassembled contigs | – | 0.0% | 16.1% | 13.6% | 1.2% | |
| NM | No. of contigs (no. of misassembled contigs) | 79 (126) | 149 (0) | 160 (17) | 163 (14) | >300 (9) |
| Coverage by the contigs | 99.8% | 99.1% | 98.6% | 97.2% | 87.4% | |
| Coverage by misassembled contigs | – | 0.0% | 10.5% | 9.2% | 1.3% | |
| LL | No. of contigs (no. of misassembled contigs) | 6 (61) | 58 (0) | 62 (10) | 85 (8) | 245 (2) |
| Coverage by the contigs | 99.9% | 99.5% | 97.6% | 97.0% | 90.4% | |
| Coverage by misassembled contigs | – | 0.0% | 19.0% | 11.4% | 0.7% |
Figure 1.

Comparative analysis of euler, phrap,cap, and tigr assemblers (NM sequencing project). Every box corresponds to a contig in NM assembly produced by these programs with colored boxes corresponding to assembly errors. Boxes in the ideal assembly correspond to islands in the read coverage. Boxes of the same color show misassembled contigs; for example, two identically colored boxes in different places show the positions of contigs that were incorrectly assembled into a single contig. In some cases, a single colored box shows a contig that was assembled incorrectly (i.e., there was a rearrangement within this contig). Repeats with similarity higher than 95% are indicated by numbered boxes at the solid line showing the genome. To check the accuracy of the assembled contigs, we fit each assembled contig into the genomic sequence. Inability to fit a contig into the genomic sequence indicates that the contig is misassembled. For example,phrap misassembles 17 contigs in the NM sequencing project, each contig containing from two to four fragments from different parts of the genome.
How can one resolve these problems? Surprisingly enough, an unrelated area of DNA arrays provides a hint. Sequencing by Hybridization (SBH) is a 10-year-old idea that never became practical but (indirectly) created the DNA arrays industry. Conceptually, SBH is similar to fragment assembly; the only difference is that the “reads” in SBH are much shorter _l_-tuples. In fact, the first approaches to SBH (8, 9) followed the “overlap–layout–consensus” paradigm. However, even for error-free SBH data, the corresponding layout problem leads to the NP-complete Hamiltonian Path Problem. Pevzner (10) proposed a different approach that reduces SBH to an easy-to-solve Eulerian Path Problem in the de Bruijn graph.
Because the Eulerian path approach transforms a once difficult layout problem into a simple one, a natural question is: “Could the Eulerian path approach be applied to fragment assembly?” Idury and Waterman, mimicked fragment assembly as an SBH problem (11) by representing every read of length n as a collection of_n_ − l + 1 overlapping_l_-tuples (continuous short strings of fixed length_l_). At first glance, this transformation of every read into a collection of _l_-tuples (breaking the puzzle into smaller pieces) is a very short-sighted procedure, because information about the sequencing reads is lost. However, the loss of information is minimal for large l and is well paid for by the computational advantages of the Eulerian path approach. In addition, lost information can be restored at later stages.
Unfortunately, the Idury–Waterman approach, although very promising, did not scale up well. The problem is that sequencing errors transform a simple de Bruijn graph (corresponding to an error-free SBH) into a tangle of erroneous edges. Moreover, repeats pose serious challenges even in the case of error-free data. This paper abandons the “overlap–layout–consensus” approach in favor of an Eulerian superpath approach. This reduction led to the euler software that generated error-free solutions for all large-scale assembly projects that were studied.
The difficulties with fragment assembly led to the introduction of double-barreled (DB) DNA sequencing (15, 16) that was first used in 1995 in the assembly of Haemophilus influenzae (17). The published fragment assemblers ignore the DB data, with the exception only of the celera assembler (Myers et al., ref.18) and gigassembler (Kent and Haussler, ref. 19). Although a number of sequencing centers use heuristic procedures to utilize DB data, such approaches often fail for complex genomes.euler-db analyzes DB data in a new way by transforming mate-pairs “read–intermediate gap of length_d_-read2” (a roughly 2-fold increase in the effective read length) into mate-reads” read1-intermediate dna sequence of length “d_-read2” in most cases. Assuming the clone length is 5 kb, it transforms the original fragment assembly problem with N reads of length 500 bp into a new problem with about_N/2 reads of length 5,000 bp. From some perspective,euler-db provides an algorithmic shortcut for the still unsolved experimental problem of increasing the read length. This approach typically resolves all repeats except perfect repeats that are longer than the insert length.
New Ideas
The classical “overlap–layout–consensus” approach to fragment assembly is based on the notion of the overlap graph. The DNA sequence in Fig. 2a consists of four unique segments A, B, C, D, and one triple repeat R. Every read corresponds to a vertex in the overlap graph, and two vertices are connected by an edge if the corresponding reads overlap (Fig.2b). The fragment assembly problem is thus cast as finding a path in the overlap graph visiting every vertex exactly once, a Hamiltonian Path Problem. The Hamiltonian Path Problem is NP-complete, and the efficient algorithms for solving this problem are unknown. This is why fragment assembly of highly repetitive genomes is a notoriously difficult problem.
Figure 2.

(a) DNA sequence with a triple repeat R; (b) the layout graph; (c) construction of the de Bruijn graph by gluing repeats; (d) de Bruijn graph.
This paper suggests an approach to the fragment assembly problem based on the notion of the de Bruijn graph. In an informal way, one can visualize the construction of the de Bruijn graph by representing a DNA sequence as a “thread” with repeated regions covered by a “glue” that “sticks” them together (Fig. 2c). The resulting de Bruijn graph (Fig. 2d) consists of 4 + 1 = 5 edges (we assume that the repeat edge is obtained by gluing three repeats and has multiplicity three). In this approach, every repeat corresponds to an edge rather than a collection of vertices in the layout graph.
One can see that the de Bruijn graph (Fig. 2c) is a much simpler representation of repeats than the overlap graph (Fig.2a). What is more important is that the fragment assembly is now cast as finding a path visiting every edge of the graph exactly once, an Eulerian Path Problem. There are two Eulerian paths in the graph: one of them corresponds to the sequence reconstruction ARBRCRD, whereas the other one corresponds to the sequence reconstruction ARCRBRD. In contrast to the Hamiltonian Path Problem, the Eulerian path problem is easy to solve even for graphs with millions of vertices, because there exist linear-time Eulerian path algorithms (20). This is a fundamental difference between the euler algorithm and conventional approaches to fragment assembly.
Although de Bruijn graphs have algorithmic advantages over overlap graphs, it is not clear how to construct de Bruijn graphs from collections of sequencing reads. The described “gluing” idea requires knowledge of the finished DNA sequence that is not available until the last step of the assembly, a Catch-22. Below we show how to construct de Bruijn graphs from the read data without knowing the finished DNA sequence.
The existing assembly algorithms postpone the consensus step (error correction in reads) until the end of the fragment assembly.euler reverses this practice by making consensus the first step of fragment assembly.
Let s be a sequencing read (with errors) derived from a genome G. If the sequence of G is known, then the error correction in s can be done by aligning the read_s_ against the genome G. In real life, the sequence of G is not known until the last “consensus” step of the fragment assembly. It is another Catch-22: to assemble a genome, it is highly desirable to correct errors in reads first, but to correct errors in reads, one has to assemble the genome first. To bypass this Catch-22, let us assume that, although the sequence of_G_ is unknown, the set G l of all_l_-tuples present in G is known. Of course,G l is also unknown, but_G_ l can be reliably approximated without knowing the sequence of G. Our error correction idea uses this approximation to correct errors in reads. In contrast to existing algorithms based on pairwise alignments, it utilizes the multiple alignments of short substrings to modify the original reads and to create a new instance of the fragment assembly problem with a greatly reduced number of errors.
Imagine an ideal situation where error correction has eliminated all errors, and we deal with a collection of error-free reads. Is there an algorithm to reliably assemble such error-free reads in a large-scale sequencing project? At first glance, the problem looks simple but, surprisingly enough, the answer is no: we are unaware of any algorithm that solves this problem. For example, phrap,cap3, and tigr assemblers (with default settings) make 17, 14, and 9 assembly errors, respectively, while assembling real reads from the NM genome. All these algorithms still make errors while assembling the error-free reads from the NM genome (although the number of errors reduces to 5, 4, and 2, respectively). euler made no assembly errors and produced fewer contigs with real data than other programs produced with error-free data.
To achieve such accuracy, euler has to restore information about sequencing reads that was lost in the construction of the de Bruijn graph. Our Eulerian Superpath idea addresses this problem. Every sequencing read corresponds to a path in the de Bruijn graph called a read-path, and the fragment assembly problem corresponds to finding an Eulerian path that is consistent with all read-paths, an Eulerian Superpath Problem.
Error Correction and Data Corruption
Sequencing errors make implementation of the SBH-style approach to fragment assembly difficult. To bypass this problem, we reduce the error rate by a factor of 35–50 and make the data almost error-free by solving the Error Correction Problem. We use the NM sequencing project (13) as an example. NM is one of the most “difficult-to-assemble” and “repeat-rich” bacterial genomes completed so far. It has 126 long nearly perfect repeats up to 3,832 bp in length (not to mention many imperfect repeats). The length of the genome is 2,184,406 bp. The sequencing project resulted in 53,263 reads (coverage is 9.7), with 255,631 errors distributed over these reads. It results in 4.8 errors per read (an error rate of 1.2%).
Our error correction procedure uses an approximation of_G_ l (the set of all l_-tuples in genome_G) rather than the sequence G to correct sequencing errors. An _l_-tuple is called solid if it belongs to more than M reads (where M is a threshold) and weak otherwise. A natural approximation for G l is the set of all solid _l_-tuples from a sequencing project.
Let T be a collection of _l_-tuples called a spectrum. A string s is called a _T_-string if all its _l_-tuples belong to T. Our approach to error correction leads to the following.
Spectral Alignment Problem.
Given a string s and a spectrum T, find the minimum number of mutations in s that transform s into a _T_-string.
A similar problem was considered by Pe'er and Shamir (21) in a different context of resequencing by hybridization. In the context of error corrections, the solution of the Spectral Alignment Problem makes sense only if the number of mutations is small. In this case, the Spectral Alignment Problem can be efficiently solved by dynamic programming even for large l.
Spectral alignment of a read against the set of all solid_l_-tuples from a sequencing project suggests the error corrections that may change the sets of weak and solid_l_-tuples. Iterative spectral alignments with the set of all reads and all solid _l_-tuples gradually reduce the number of weak _l_-tuples, increase the number of solid_l_-tuples, and lead to elimination of many errors in bacterial sequencing projects. Although the Spectral Alignment Problem helps to eliminate errors (and we use it as one of the steps ineuler), it does not adequately capture the specifics of DNA sequencing.
The Error Correction Problem described below is a better model for fragment assembly that leads to elimination of ≈97% of errors in a typical bacterial project.
We found that some reads from the NM project have very poor spectral alignment. These reads are likely to represent contamination, vector, isolated reads, or an error in the sequencing pipeline. It is a common practice in sequencing centers to discard such “poor-quality” reads, and we adopt this approach. Another important advantage of spectral alignment is an ability to identify chimeric reads. Such reads are characterized by good spectral alignments of the prefix and suffix parts (6), which, however, cannot be extended to a good spectral alignment of the entire read.
Given a collection of reads (strings) S = {s_1, …, s n} from a sequencing project and an integer_l, the spectrum of S is a set_S_ l of all l_-tuples from the reads_s_1, …, s n and s̄1, …, s̄_n, where s̄ denotes a reverse complement of read s. Let Δ be an upper bound on the number of errors in each DNA read. A more adequate approach to error correction motivates the following.
Error Correction Problem.
Given S, Δ, and l, introduce up to Δ corrections in each read in S in such a way that |S l| is minimized.
An error in a read s affects at most_ll_-tuples in s and ll_-tuples in s̄ and usually creates 2_l erroneous_l_-tuples that point to the same sequencing error (2_d_ for positions within a distance d < l from the endpoint of the reads). A greedy approach for the Error Correction Problem is to look for error corrections in the reads that reduce the size of S l by 2_l_ (or 2_d_ for positions close to the endpoints). This simple procedure already eliminates 86.5% of the errors in sequencing reads.euler uses a more involved approach that eliminates 97.7% of sequencing errors and transforms the original sequencing data with 4.8 errors per read on average into almost error-free data with 0.11 errors per read on average (22).
A word of caution is in place. Our error-correction procedure is not perfect while deciding which nucleotide, among, let us say, A or T is correct in a given _l_-tuple within a read. If the correct nucleotide is A, but T is also present in some reads covering the same region, the error-correction procedure may assign T instead of A to all reads, i.e., to introduce an error rather than to correct it. Because our algorithm sometimes introduces errors, data corruption is probably a more appropriate name for this approach! Introducing an error in a read is not such a bad thing, as long as the errors from overlapping reads covering the same position are consistent (i.e., they correspond to a single mutation in a genome). An important insight is that, at this stage of the algorithm, we do not care much whether we correct or introduce errors in the sequencing reads. From an algorithmic perspective, introducing a consistent error that simply corresponds to changing a nucleotide in a final assembly is not a big deal. It is much more important to make sure that we eliminate a competition between A and T at this stage, thus reducing the complexity of the de Bruijn graph. In this way, we eliminate false edges in our graph and deal with this problem later: the correct nucleotides are easily reconstructed either by a majority rule or by a variation of the Churchill–Waterman algorithm (23). For the NM sequencing project, orphan elimination corrects 234,410 errors and introduces 1,452 errors.
Eulerian Superpaths
Given a set of reads S = {s_1, …, s n}, define the de Bruijn graph_G(S l) with vertex set_S_ _l_−1 (the set of all (l − 1)-tuples from S) as follows. An (l − 1)-tuple v ∈ S l_−1 is joined by a directed edge with an (l − 1)-tuple w ∈_S _l_−1, if S l contains an_l_-tuple for which the first l − 1 nucleotides coincide with v and the last l − 1 nucleotides coincide with w. Each _l_-tuple from S l corresponds to an edge in G. If S contains the only sequence s_1, then this sequence corresponds to a path visiting each edge of the de Bruijn graph, a Chinese Postman path (20). The Chinese Postman Problem is closely related to the problem of finding a path visiting every edge of a graph exactly once, an Eulerian Path Problem (24). One can transform the Chinese Postman Problem into the Eulerian Path Problem by introducing multiplicities of edges in the de Bruijn graph. For example, one can substitute every edge in the de Bruijn graph by_k parallel edges, where k is the number of times the edge is used in the Chinese Postman path. If S contains the only sequence s_1, this operation creates_k “parallel” edges for every _l_-tuple repeating k times in _s_1 (23). Finding Eulerian paths is a well known problem that can be efficiently solved in linear time. We assume that S contains a complement of every read and that the de Bruijn graph can be partitioned into two subgraphs (the “canonical” one and its reverse complement).
With real data, the errors hide the correct path among many erroneous edges. The graph corresponding to the error-free data from the NM project has 4,039,248 vertices (roughly twice the length of the genome), whereas the graph corresponding to real sequencing reads has 9,474,411 vertices (for 20-tuples). After the error-correction procedure, this number is reduced to 4,081,857.
A vertex v is called a source if indegree(v) = 0, a sink if outdegree(v) = 0, and a branching vertex if indegree(v)⋅ outdegree(v) > 1. For the NM genome, the de Bruijn graph has 502,843 branching vertices for original reads (for _l_-tuple size 20). Error corrections simplify this graph and lead to a graph with 382 sources and sinks and 12,175 branching vertices. Because the de Bruijn graph gets very complicated even in the error-free case, taking into account the information about which _l_-tuples belong to the same reads (that was lost after the construction of the de Bruijn graph) helps us to untangle this graph.
A path _v_1 … v n in the de Bruijn graph is called a repeat if indegree(_v_1) > 1, outdegree(v n) > 1, and indegree (_v_1) = outdegree(v i) = 1 for 1 ≤ i ≤ n − 1 (Fig.3). Edges entering the vertex_v_1 are called entrances into a repeat, whereas edges leaving the vertex v n are called exits from a repeat. An Eulerian path visits a repeat a few times, and every such visit defines a pairing between an entrance and an exit. Repeats may create problems in fragment assembly, because there are a few entrances in a repeat and a few exits from a repeat, but it is not clear which exit is visited after which entrance in the Eulerian path. A read-path covers a repeat if it contains an entrance into and an exit from this repeat. Every covering read-path reveals some information about the correct pairings between entrances and exits. A repeat is called a tangle if there is no read-path containing this repeat (Fig.3). Tangles create problems in fragment assembly, because pairings of entrances and exits in a tangle cannot be resolved via the analysis of read-paths. To address this issue, we formulate the following generalization of the Eulerian Path Problem:
Figure 3.

A repeat _v_1 … v n and a system of paths overlapping with this repeat. The uppermost path contains the repeat and defines the correct pairing between the corresponding entrance and exit. If this path were not present, the repeat_v_1 … v n would become a tangle.
Eulerian Superpath Problem.
Given an Eulerian graph and a collection of paths in this graph, find an Eulerian path in this graph that contains all these paths as subpaths.
To solve the Eulerian Superpath Problem, we transform both the graph_G_ and the system of paths 𝒫 in this graph into a new graph_G_1 with a new system of paths 𝒫1. Such transformation is called equivalent if there exists a one-to-one correspondence between Eulerian superpaths in (𝒢, 𝒫) and (𝒢1, 𝒫1). Our goal is to make a series of equivalent transformations
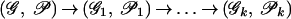
that lead to a system of paths 𝒫k, with every path being a single edge. Because all transformations on the way from (𝒢, 𝒫) to (𝒢k, 𝒫k) are equivalent, every solution of the Eulerian Path Problem in (𝒢k, 𝒫k) provides a solution of the Eulerian Superpath Problem in (𝒢, 𝒫).
Below, we describe a simple equivalent transformation that solves the Eulerian Superpath Problem in the case when the graph G has no multiple edges. Let x = (v in,v mid) and y = (v mid,v out) be two consecutive edges in graph G, and let 𝒫x,y be a collection of all paths from 𝒫 that include both these edges as a subpath. Informally,x,y_-detachment bypasses the edges x and_y via a new edge z and directs all paths in 𝒫x,y through z, thus simplifying the graph. However, this transformation affects other paths and needs to be defined carefully. Define 𝒫→x as a collection of paths from 𝒫 that end with x and 𝒫y_→ as a collection of paths from 𝒫 that start with y. The x,y_-detachment is a transformation that adds a new edge z = (v in, v out) and deletes the edges_x and y from G (Fig.4a). This detachment alters the system of paths 𝒫 as follows: (i) substitute_z for x, y in all paths from 𝒫x,y, (ii) substitute z for_x_ in all paths from 𝒫→x, and (iii) substitute z for y in all paths from 𝒫_y_→. Because every detachment reduces the number of edges in G, the detachments will eventually shorten all paths from 𝒫 to single edges and will reduce the Eulerian Superpath Problem to the Eulerian Path Problem.
Figure 4.

Equivalent transformations: (a) x, _y_-detachment and (b) _x_-cut.
However, in the case of graphs with multiple edges, the detachment procedure may lead to errors, because “directing” all paths from the set 𝒫→x through a new edge z may not be an equivalent transformation. In this case, the edge_x_ may be visited many times in the Eulerian path, and it may or may not be followed by the edge y on some of these visits.
For illustration purposes, let us consider a simple case when the vertex v mid has the only incoming edge_x_ = (v in, v mid) with multiplicity 2 and two outgoing edges _y_1 = (v mid, v _out_1) and _y_2 = (v mid, v _out_2), each with multiplicity 1. In this case, the Eulerian path visits the edge x twice; in one case, it is followed by _y_1, and in another case, it is followed by _y_2. Consider an x,_y_1-detachment that adds a new edge z = (v in,v _out_1) after deleting the edge y_1 and one of two copies of the edge x. This detachment (i) shortens all paths in 𝒫_x,y_1 by substitution of x, y_1 by a single edge z, and (ii) substitutes z for y_1 in every path from 𝒫_y_1→. This detachment is an equivalent transformation if the set 𝒫→_x is empty. However, if 𝒫→_x is not empty, it is not clear whether the last edge of a path P ∈ 𝒫→_x should be assigned to the edge z or to the (remaining copy of) edge x.
To resolve this dilemma, one has to analyze every path P ∈ 𝒫→x and decide whether it “relates” to 𝒫x,y_1 (in this case, it should be directed through z) or to 𝒫_x,y_2 (in this case, it should be directed through x). By “relates” to 𝒫_x,y_1 (𝒫_x,_y_2), we mean that every Eulerian superpath visits _y_1 (_y_2) immediately after visiting P.
Two paths are called consistent if their union is a path again. A path_P_ is consistent with a set of paths 𝒫 if it is consistent with all paths in 𝒫 and inconsistent otherwise (i.e., if it is inconsistent with at least one path in 𝒫). There are three possibilities (Fig. 5): (i) P is consistent with exactly one of the sets 𝒫x,y_1 and 𝒫_x,y_2, (ii) P is inconsistent with both 𝒫_x,y_1 and 𝒫_x,y_2, and (iii) P is consistent with both 𝒫_x,y_1 and 𝒫_x,_y_2.
Figure 5.

(a) P is consistent with_P_ x,y_1 but inconsistent with_P x,y_2; (b) P is inconsistent with both_P x,y_1 and_P x,y_2; (c) P is consistent with both_P x,y_1 and_P x,_y_2.
In the first case, the path P is called resolvable, because it can be unambiguously related to either 𝒫x,y_1 or 𝒫_x,y_2. An edge x is called resolvable if all paths in 𝒫→_x are resolvable. If the edge x is resolvable, then the described_x_, y_-detachment is an equivalent transformation after the correct assignments of last edges in every path from 𝒫→_x. In our analysis of the NM project, we found that 18,026 among 18,962 edges in the de Bruijn graph are resolvable.
The second condition implies that the Eulerian Superpath Problem has no solution, because P, 𝒫x,y_1, and 𝒫_x,_y_2 impose three different scenarios for just two visits of the edge x. After discarding the poor-quality and chimeric reads, we did not encounter this condition in the NM project.
The last condition (P is consistent with both 𝒫x,y_1 and 𝒫_x,y_2) corresponds to the most difficult situation. If this condition holds for at least one path in 𝒫→_x, the edge x is called unresolvable, and we postpone analysis of this edge until all resolvable edges are analyzed. We observed that equivalent transformation of other resolvable edges often resolves previously unresolvable edges. However, some edges cannot be resolved even after the detachments of all resolvable edges are completed. Such situations usually correspond to tangles, and they have to be addressed by another equivalent transformation called a cut.
Consider a fragment of graph G with 5 edges and 4 paths_y_3 − x, _y_4 − x, x − _y_1, and x − _y_2 (Fig. 4b). In this symmetric situation,x is a tangle, and there is no information available to relate any of paths _y_3 − x and _y_4 − x to any of paths x − y_1 and x − y_2. An edge x = (v, w) is removable if (i) it is the only outgoing edge for v and the only incoming edge for_w, and (ii) x is either the initial or the terminal edge for every path P ∈ 𝒫 containing_x. An x_-cut transforms 𝒫 into a new system of paths by simply removing x from all paths in 𝒫→_x and 𝒫_x_→ without affecting the graph G itself (Fig. 4b). Obviously, an x_-cut is an equivalent transformation if_x is a removable edge.
Detachments and cuts proved to be powerful techniques to untangle the de Bruijn graph and to reduce the fragment assembly to the Eulerian Path Problem for all studied bacterial genomes. However, there is still a gap in the theoretical analysis of the Eulerian Superpath Problem in the case when the system of paths is not amenable to either detachments or cuts.
euler with Clone-End Data
One hundred twenty-six long nearly perfect repeats in the NM genome tangle the de Bruijn graph and make it difficult to analyze.euler-db untangles this graph by using the clone-end DB data euler-db maps every read into some edge(s) of the de Bruijn graph. After this mapping, most mate-pairs of reads correspond to paths that connect the positions of these reads in the de Bruijn graph (provided the distance between these positions in the graph is approximately equal to the estimated distance between reads from the mate-pairs). euler-db views such paths as long artificial mate–reads and analyzes them with the same Eulerian Superpath algorithm that is used for the analysis of standard reads.
Mapping reads into the de Bruijn graph allows one to identify errors in the DB data. In most cases, both reads _r_1 and_r_2 from the mate-pair (_r_1, _r_2) are mapped to the same graph component. In this case, one can find a path between these reads and compare the length d(_r_1, r_2) of this path with an estimated distance_l(_r_1, _r_2) between clone ends. In most cases, such a path is unique and its length approximately matches the clone length [d(_r_1, r_2) ≈_l(_r_1, r_2)]. In this case, we reconstruct the intermediate sequence between reads and transform the mate-pair into mate path. In case the difference between_d(_r_1, r_2) and_l(_r_1, _r_2) is beyond the acceptable variation in the clone length, it is most likely an error in the DB data. In the case of multiple paths between _r_1 and r_2 in the de Bruijn graph, we transform the mate-pair into the corresponding mate-read if the clone length_l(_r_1, _r_2) matches the length of exactly one path between _r_1 and_r_2.
We emphasize important differences between our approach and other DB assemblers. The celera assembler masks repeats, generates a large set of contigs, and pieces these contigs together by using the DB data. There are two types of contigs subject to the DB step of the celera's algorithm: G-contigs flanked by gaps in read coverage (on at least one side) and R-contigs flanked by repeats (on both sides). In a genome with many repeats, the number of R-contigs may significantly exceed the number of G-contigs. For example, in the NM project, phrap generates 160 contigs, and only half of them are G-contigs, whereas in the L. lactis project, phrap generates 62 contigs, and only 7 of them are G-contigs.
The celera assembler provides an excellent solution for assembly of G-contigs, but it does not distinguish between two types of contigs. After masking repeats, the celera assembler generates a large number of contigs; only a small portion of them are G-contigs. Because the resulting contigs are short, it leads to a rather complicated DB step (Myers et al., ref. 18) that may cause disassembly for short contigs with limited DB data.
In contrast to other DB assemblers, euler-db distinguishes between G-contigs and R-contigs. euler-db does not mask repeats but instead uses them (combined with DB data) as a powerful tool for resolving R-contigs. The NM sequencing project illustrates the advantages of using (rather than masking) repeats in fragment assembly.euler-db reduces the number of contigs from 149 to 117 if only mate-pairs from nonrepeated regions are used. Use of mate-pairs that partially overlap the repeats further reduces the number of contigs to 91 (Fig. 1). euler-db typically resolves all tangles except tangles that are longer than the length of the insert. In the NM project (with insert length up to 1,800), euler left only 5 unresolved tangles of length 3,610, 3,215, 2,741, 2,503, and 1,831.
After completing euler-db, we build scaffolds by using DB data as “bridges” between different contigs (euler-sf). euler-sf combines the 91 contigs into 60 scaffolds, thus closing most gaps that are shorter than the insert length and further simplifying the finishing step (Fig. 1). Myers et al. (18) described an excellent solution of the scaffolding problem. Recently, Kent and Haussler (19) described a different greedy approach to the scaffolding problem that resulted in a successful draft assembly of the human genome. euler-sf uses a similar strategy, but it scaffolds a smaller set of longer contigs (mainly G-contigs). See ref. 25 for details.
Conclusions
Finishing is a bottleneck in large-scale DNA sequencing. Of course, finishing is an unavoidable step to extend the islands and close the gaps in read coverage. However, existing programs produce many more contigs than the number of islands, thus making finishing more complicated than necessary. What is worse, these contigs are often assembled incorrectly, thus leading to the time-consuming contig verification step. Even a single misassembly forces biologists to conduct total genome screening for assembly errors. euler bypasses the “repeat problem,” because the Eulerian Superpath approach transforms imperfect repeats into different paths in the de Bruijn graph. As a result, euler does not even notice repeats unless they are long perfect repeats.
Difficulties in resolving repeats led to the introduction of DB DNA sequencing and the breakthrough sequencing efforts reported by the Human Genome Program (26) and celera (27) Thecelera assembler is a two-stage procedure that includes masking repeats at the overlap–layout–consensus stage with further ordering of contigs via DB data. The DB step of the celera assembler is influenced by the previous “overlap–layout–consensus” step. All “overlap–layout–consensus” algorithms are “afraid” of repeats, and the celera assembler has no choice but to mask the repeats. euler does not require masking the repeats but instead provides a clear view of repeats (tangles) in the genome. These tangles may be resolved by DB information (euler-db). In addition, euler has excellent scaling potential for eukaryotic genomes, because there exist linear-time algorithms for the Eulerian Path Problem.
Acknowledgments
We are grateful to Herbert Fleischner, Michael Fonstein, Dmitry Frishman, Eugene Goltsman, David Harper, Alexander Karzanov, Zufar Mulyukov, Julian Parkhill, Steven Salzberg, and Alexei Sorokine for useful discussions and providing sequencing data.
Abbreviations
SBH
Sequencing by Hybridization
DB
double-barreled
NM
N. meningitidis
References
- 1.Green P. Documentation for phrap. 1994. (http://www.genome.washington.edu/UWGC/analysis-tools/phrap.htm). [Google Scholar]
- 2.Bonfield J K, Smith K F, Staden R. Nucleic Acids Res. 1995;23:4992–4999. doi: 10.1093/nar/23.24.4992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sutton G, White O, Adams M, Kerlavage A. Genome Sci Technol. 1995;1:9–19. [Google Scholar]
- 4.Kececioglu J, Myers E. Algorithmica. 1995;13:7–51. [Google Scholar]
- 5.Myers E M. J Comput Biol. 1995;2:275–290. doi: 10.1089/cmb.1995.2.275. [DOI] [PubMed] [Google Scholar]
- 6.Huang X, Madan A. Genome Res. 1999;9:868–877. doi: 10.1101/gr.9.9.868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gordon D, Abajian C, Green P. Genome Res. 1998;8:195–202. doi: 10.1101/gr.8.3.195. [DOI] [PubMed] [Google Scholar]
- 8.Drmanac R, Labat I, Brukner I, Crkvenjakov R. Genomics. 1989;4:114–128. doi: 10.1016/0888-7543(89)90290-5. [DOI] [PubMed] [Google Scholar]
- 9.Lysov Y, Florent'ev V, Khorlin A, Khrapko K, Shik V, Mirzabekov A. Doklady Acad Nauk USSR. 1988;303:1508–1511. [PubMed] [Google Scholar]
- 10.Pevzner P. J Biomol Struct Dyn. 1989;7:63–73. doi: 10.1080/07391102.1989.10507752. [DOI] [PubMed] [Google Scholar]
- 11.Idury R, Waterman M. J Comput Biol. 1995;2:291–306. doi: 10.1089/cmb.1995.2.291. [DOI] [PubMed] [Google Scholar]
- 12.Parkhill J, Wren B, Mungall K, Ketley J M, Churcher C, Basham D, Chillingworth T, Davies R M, Feltwell T, Holroyd S, et al. Nature (London) 2000;403:665–668. doi: 10.1038/35001088. [DOI] [PubMed] [Google Scholar]
- 13.Parkhill J, Achtman M, James K D, Bentley S D, Churcher C, Klee S R, Morelli G, Basham D, Brown D, Chillingworth T, et al. Nature (London) 2000;404:502–506. doi: 10.1038/35006655. [DOI] [PubMed] [Google Scholar]
- 14.Bolotin A, Mauger S, Malarme K, Ehrlich S D, Sorokin A. Antonie van Leeuwenhoek. 1999;76:27–76. [PubMed] [Google Scholar]
- 15.Roach J, Boysen C, Wang K, Hood L. Genomics. 1995;26:345–353. doi: 10.1016/0888-7543(95)80219-c. [DOI] [PubMed] [Google Scholar]
- 16.Weber J, Myers G. Genome Res. 1997;7:401–409. doi: 10.1101/gr.7.5.401. [DOI] [PubMed] [Google Scholar]
- 17.Fleischmann R D, Adams M D, White O, Clayton R A, Kirkness E F, Kerlavage A R, Bult C J, Tomb J F, Dougherty B A, Merrick J M, et al. Science. 1995;269:496–512. doi: 10.1126/science.7542800. [DOI] [PubMed] [Google Scholar]
- 18.Myers E W, Sutton G G, Delcher A L, Dew I M, Fasulo D P, Flanigan M J, Kravitz S A, Mobarry C M, Reinert K H, Remington K A, et al. Science. 2000;287:2196–2204. doi: 10.1126/science.287.5461.2196. [DOI] [PubMed] [Google Scholar]
- 19.Kent, W. & Haussler, D. (2001) Genome Res., in press. [DOI] [PMC free article] [PubMed]
- 20.Fleischner H. Eulerian Graphs and Related Topics. London: Elsevier Science; 1990. [Google Scholar]
- 21.Pe'er I, Shamir R. Proceedings of the Eighth International Conference on Intelligent Systems for Molecular Biology. CA: San Diego; 2000. pp. 260–268. [PubMed] [Google Scholar]
- 22.Pevzner P A, Waterman M S. Proceedings of the Fifth International Conference on Computational Biology. Montreal: RECOMB 2001; 2001. pp. 256–265. [Google Scholar]
- 23.Churchill G, Waterman M. Genomics. 1992;14:89–98. doi: 10.1016/s0888-7543(05)80288-5. [DOI] [PubMed] [Google Scholar]
- 24.Pevzner P A. Computational Molecular Biology: An Algorithmic Approach. Cambridge, MA: MIT Press; 2000. [Google Scholar]
- 25.Pevzner, P. A. & Tang, H. (2001)Bioinformatics17, in press. [DOI] [PubMed]
- 26.Lander E S, Linton L M, Birren B, Nusbaum C, Zody M C, Baldwin J, Devon K, Dewar K, Doyle M, FitzHugh W, et al. Nature (London) 2001;409:860–921. doi: 10.1038/35057062. [DOI] [PubMed] [Google Scholar]
- 27.Venter J C, Adams M D, Myers E W, Li P W, Mural R J, Sutton G G, Smith H O, Yandell M, Evans C A, Holt R A, et al. Science. 2001;291:1304–1351. doi: 10.1126/science.1058040. [DOI] [PubMed] [Google Scholar]