Development of a systems-based in situ multiplex biomarker screening approach for the assessment of immunopathology and neural tissue plasticity in male rats after traumatic brain injury (original) (raw)
. Author manuscript; available in PMC: 2018 Oct 1.
Published in final edited form as: J Neurosci Res. 2017 May 2;96(4):487–500. doi: 10.1002/jnr.24054
Abstract
Traumatic brain injuries (TBI) pose a massive burden of disease and continue to be a leading cause of morbidity and mortality throughout the world. A major obstacle in developing effective treatments is the lack of comprehensive understanding of the underlying mechanisms that mediate tissue damage and recovery after TBI. As such, our work aims to highlight the development of a novel experimental platform capable of fully characterizing the underlying pathobiology that unfolds post-TBI. This platform encompasses an empirically optimized multiplex immunohistochemistry staining and imaging system customized to screen for a myriad of biomarkers required to comprehensively evaluate the extent of neuroinflammation, neural tissue damage and repair in response to TBI. Herein, we demonstrate that our multiplex biomarker screening platform is capable of evaluating changes in both the topographical location and functional states of resident and infiltrating cell types that play a role in neuropathology after controlled cortical impact injury to the brain in male Sprague-Dawley rats. Our results demonstrate that our multiplex biomarker screening platform lays the groundwork for the comprehensive characterization of changes that occur within the brain after TBI. Such work may ultimately lead to the understanding of the governing pathobiology of TBI, thereby fostering the development of novel therapeutic interventions tailored to produce optimal tissue protection, repair and/or regeneration with minimal side effects and may ultimately find utility in wide variety of other neurological injuries, diseases and disorders that share components of TBI pathobiology.
Keywords: traumatic brain injury (TBI), controlled cortical impact (CCI), immunopathology, neural plasticity, systems biology, multiplex immunohistology
Graphical Abstract
Traumatic brain injury (TBI) remains a leading cause of death and disability worldwide. Ten-color fluorescence immunohistochemistry using specific combinations of antibodies is a novel method capable of simultaneously identifying the changing topographical and spatiotemporal patterning of all major cell types within the brain in response to TBI. Such work may ultimately lead to the comprehensive understanding of the governing pathobiology of TBI, thereby fostering the development of novel therapeutic interventions tailored to produce optimal tissue protection, repair and/or regeneration.
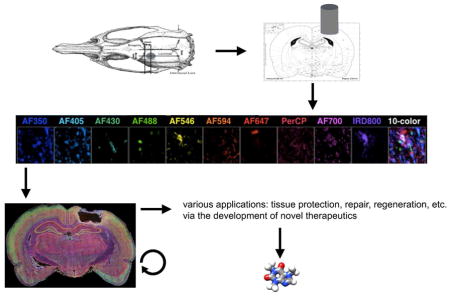
Introduction
Traumatic brain injury (TBI) remains a leading cause of both morbidity and mortality in people under the age 45 years and therefore represents a major public health burden throughout both the developing and developed world (Corrigan et al. 2010; Langlois et al. 2003). Of note, current epidemiological data continues to suggest that the incidence of TBI is likely to expand primarily due to the increased use of motor vehicles throughout the developing world as the rise of motorized transportation continues to outpace the development of an adequate safety infrastructure (Roozenbeek et al. 2013), (Park et al. 2008; Tagliaferri et al. 2006). Current estimates indicate that around 5.3 million people in the United States, and nearly 7.7 million people in Europe live with a TBI-related disability (Rubiano et al. 2015). Furthermore, mild TBI (mTBI) accounts for 70–90% of all cases (Levin and Diaz-Arrastia 2015) and it may also be associated with persistent symptoms and prolonged disability (Bazarian and Atabaki 2001). Despite such a public health burden, there are as of yet no specific treatment options with proven clinical efficacy available for those that suffer from TBI (Maas et al. 2010).
With the accelerating discovery of new brain injury mechanisms (Rhodes 2011; Zink et al. 2010), and continuing failure of clinical trials of therapies targeting single pathways (Maas et al. 2010), TBI has become more widely viewed as a highly complex, multifactorial process that involves interplay of many non-dominant effectors (Marklund and Hillered 2011; Povlishock and Katz 2005). TBI results from an initial primary injury induced by an external force to the brain (e.g. direct impact, acceleration/deceleration, and/or blast). The primary injury goes on to initiate a secondary pathophysiological cascade, that is characterized by excitotoxicity, the generation of free radicals, lipid peroxidation (Hall et al. 2010), mitochondrial dysfunction (Mustafa et al. 2011), the swelling and loss of astrocytes (Meaney et al. 2001), axonal (Pettus and Povlishock 1996), and neuronal injury (Clark et al. 2000). Secondary injury induced after TBI is associated with a marked inflammatory response and alterations in both cerebral blood flow and metabolism (Kochanek et al. 2000), which leads to the parenchymal accumulation of tau and amyloid beta protein (Johnson et al. 2012), demyelination (Ng et al. 1994) axonal degeneration (Smith et al. 1999), and apoptotic neuronal death via caspase-3 activation (Clark et al. 2000). Beyond driving secondary pathology, inflammatory cells have also been linked to endogenous attempts by the brain to engage in tissue remodeling and repair (Hinson et al. 2015; Woodcock and Morganti-Kossmann 2013); in line with such findings are reports highlighting angiogenesis and neurogenesis after TBI (Addington et al. 2015; Budinich et al. 2012; Hallbergson et al. 2003; Xiong et al. 2010). While various animal models have been developed to study the pathophysiology of TBI and to track the efficacy of potential intervention/therapeutics (Marklund and Hillered 2011; Morganti-Kossmann et al. 2010), the drivers of tissue injury (both spatial and temporal) remain poorly understood.
Biomarkers have historically been critical in advancing therapies for a broad range of clinical conditions (Lesko and Atkinson 2001; Robb et al. 2016) and there remains a critical need to identify, develop, and validate new biomarkers if one seeks to advance putative TBI diagnostics or novel treatments. Accordingly, employing animal models allows the study of the pathophysiological processes that unfold in the brain after TBI. Currently, there are a number of lissencephalic models used to recapitulate the pathophysiology that unfolds in the brain after TBI, including controlled cortical impact (CCI) (Johnson et al. 2015), impact acceleration (Marmarou et al. 1994), fluid percussion (Zhao et al. 2003), blast (Yeoh et al. 2013) and CHIMERA (Closed-Head Impact Model of Engineered Rotational Acceleration) (Namjoshi et al. 2016; Namjoshi et al. 2014). Despite the existence of such models, challenges remain in advancing discoveries from the bench to the bedside (Xiong et al. 2013).
Accordingly, herein we describe the development of a novel multiplex immunohistochemistry (MP-IHC) staining and imaging platform capable of simultaneously identifying a myriad of relevant biomarkers of brain tissue damage, repair and regeneration in an effort to characterize these processes and ultimately obtain a better understanding of the cellular and molecular mechanisms underlying brain injury and remodeling after TBI. Via the use of specific combinations of antibodies designed to reflect core components of tissue pathology, including neuroinflammation, neuroplasticity and angiogenesis, this novel method allows one to begin to more fully elucidate brain cytoarchitecture and associated molecular signaling in response to TBI. Such work may ultimately lead to the elucidation of the governing pathobiology of TBI and may ultimately find utility in a wide variety of other neurological injuries, diseases and disorders that share components of TBI pathobiology (e.g. ischemic stroke, spinal cord injury, multiple sclerosis, etc.).
Material and Methods
Controlled cortical impact (CCI)
All animal procedures were reviewed and approved by the Uniformed Services University of the Health Sciences (USUHS) Institutional Animal Care and Use Committee (IACUC). Adult male Sprague-Datwley rats (10–12 weeks, weighting between 200–250g) were used for all experiments. The rats were subjected to a unilateral cortical impact via the use of an electronically-controlled impactor (Impact One, Leica, Wetzlar, Germany), with cortical compression being performed on the left parietoccipital cortex. Induction of rats was performed via spontaneous ventilation using between 3–5% isoflurane in 100% oxygen for between 3–5 minutes in a rodent volatile anesthesia box. After the application of protective ointment (Lacri-Lube) to the eyes, the head was shaved using electric clippers. Following hair removal, the head of the animal was placed in a standard stereotaxic frame and positioned using ear and incisor bars (Stoelting, Wood Dale, IL) and the skin was prepped with betadine ointment and 70% ethanol. Rectal temperature was maintained at 37°C with an isothermal heating pad and feedback controller (Stoelting, Wood Dale, IL). Anesthesia was maintained with 1.5–2% isoflurane. Unilateral controlled cortical impact (CCI) to the left cerebral hemisphere was performed using a modified technique as has been previously described (Budinich et al. 2012). In brief, a midline incision was made to expose the bregma and lambda sutures. Skin and fascia were reflected, and a craniectomy was performed over the left parietotemporal cortex using an electric rotary drill. A 4x microscope (Zeiss, Thornwood, NY) was used to visualize the skull during craniectomy, and intermittent saline irrigation was performed in an effort to reduce the intensity of heat generated during drilling. Care was taken to avoid mechanical injury to the dura mater, and dural integrity was examined prior to impactor positioning. The CCI device consisted of a computer-controlled, electromagnetically driven impactor fitted with a 3.0-mm-diameter steel tip mounted on a stereotaxic micromanipulator. The impactor tip was positioned at brain coordinates bregma −2.04mm and interaaural 6.96mm and then brought into contact with exposed dura and retracted prior to setting an impact depth of 2.0mm. Speed of impact was set to 5 meters/sec, and dwell time was 100ms. Following injury, the skull fragment was carefully replaced, and the incision was closed using interrupted 4.0 silk sutures. All surgical procedures were performed in a sterile manner. Rats were placed in a heated cage to maintain body temperature until fully awake, after which they were returned to their home cage. Due to the exploratory nature of our study, we used the contralateral hemisphere as a matched control. The contralateral side of rats that underwent CCI did not show any major alterations in tissue morphology as compared to shams via hematoxylin and eosin (H&E) staining (data not shown).
Tissue Collection and Processing
At 24 h and 72 h after CCI, the animals were deeply anesthetized and transcardially perfused with heparin infused saline followed by 4% paraformaldehyde (PFA) in phosphate-buffered saline (PBS) pH 7.4. Thereafter, the brains were promptly removed from the skull and post-fixed in 4% PFA/PBS at 4°C for 24 h, before sequentially cryoprotected in 15% and 30% sucrose in PBS. The brains were then embedded in OCT, snap-frozen in isopentane and 10 μm thick coronal sections cut in the areas immediately adjacent to CCI coordinates (Histoserv, Germantown, MD). The sections were stored at −80°C and air-dried at room temperature (RT) for 1 h prior to MP-IHC staining. Approximately 100 coronal sections completely covering area of injury were screened, for each brain with every 10th section in a series stained with H&E to evaluate tissue damage and select comparable areas for MP-IHC staining and analysis.
Multiplex Fluorescence Immunohistochemistry
The list of primary and secondary reagents used for the multiplex IHC study to characterize the complexity of brain tissue remodeling following TBI is presented as Supplemental Table 1 which contains Research Resource Identifiers (RRIDs). This table also lists specific antibody targets, host and immunoglobulin class/subclass, conjugated fluorophores and pertinent links to literature/data sheets. For multiplex IHC staining, 10 μm thick coronal rat brain sections were immunoreacted for 1 h at RT using a mixture of the following immunocompatible primary antibodies (refer to Supplemental Table 1 for full technical information for each primary antibody): 1:100 rat IgG2a anti-MBP (EMD Millipore, RRID: AB_94975), 1:200 mouse IgG2a anti-GAD67 (EMD Millipore, RRID: AB_2278725), 1:200 Isolectin anti-IB4-biotin (Thermo Fisher Scientific, Product# I21414), 1:200 mouse IgM anti-CD57 (BD Biosciences, RRID: AB_397184), 1:500 mouse IgG1 anti-Nestin (EMD Millipore, RRID: AB_94911), 1:500 chicken IgG (IgY) anti-GFAP (glial fibrillary acidic protein) (Abcam, RRID: AB_304558), 1:200 mouse IgG2b anti-APC [CC-1] (Abcam, RRID: AB_443473), 1:200 rabbit IgG anti-Iba1 (Wako Chemicals, RRID: AB_839504), 1:200 guinea pig IgG anti-Parvalbumin (PV) (Synaptic Systems, RRID: AB_2156476). These sections were then washed in PBS supplemented with 1 mg/ml bovine serum albumin (BSA) and immunoreacted using a 1 μg/ml mixture of the following spectrally compatible fluorochrome-conjugated secondary antibodies (refer to Supplemental Table 1 for full technical information for each secondary antibody): goat anti-rat IgG-Alexa Fluor 350 (Thermo Fisher Scientific, RRID: AB_2535748), goat anti-mouse IgG2a-DyLight 405 (Jackson Immuno Research, RRID: AB_2338800), Streptavidin-Alexa Fluor 430 (Thermo Fisher Scientific, Product# S11237), goat anti-mouse IgM-PerCP (Jackson Immuno Research, RRID: AB_2338639), goat anti-mouse IgG1-Alexa Fluor 546 (Thermo Fisher Scientific, RRID: AB_2535764), goat anti-chicken IgG (IgY)-Alexa Fluor 594 (Thermo Fisher Scientific, RRID: AB_2534097), goat anti-mouse IgG2b-Alexa Fluor 647 (Thermo Fisher Scientific, RRID: AB_2535774), goat anti-rabbit IgG-Alexa Fluor 700 (Thermo Fisher Scientific, RRID: AB_2535709), donkey anti-guinea pig IgG-IRDye 800CW (Li-Cor Biosciences, RRID: AB_1850024). After washing away excess secondary antibodies, the potentially unbound F(ab′)2 antigen binding sites on the goat anti-mouse IgG1-Alexa Fluor 546 secondary antibody were blocked using 30-minute incubation at RT in 5 μg/ml unlabeled mouse IgG1 antibody purified from non-immunized mouse serum (BioLegend, RRID: AB_326426) and the sections were then stained using 1 μg/ml Alexa Fluor 488-conjugated mouse IgG1 anti-NeuN (EMD Millipore, RRID: AB_2149209), a pan-neuronal marker. Slides were mounted using Immu-Mount medium (Thermo Fisher Scientific) and imaged using a multi-channel wide field epifluorescence microscope (see below). Cell types and corresponding combinations of antibodies used to identify these cells are presented in Supplemental Table 2.
Multiplex Fluorescence Immunohistochemistry Image Acquisition
Images were acquired from whole brain coronal sections using the Axio Imager.Z2 slide scanning fluorescence microscope (Zeiss) equipped with a 20X/0.8 Plan-Apochromat (Phase-2) non-immersion objective (Zeiss), a high resolution ORCA-Flash4.0 sCMOS digital camera (Hamamatsu), a 200W X-Cite 200DC broad band lamp source (Excelitas Technologies), and 10 customized filter sets (Semrock) optimized to detect the following fluorophores: Alexa Fluor 350, Alexa Fluor 405, Alexa Fluor 430, Alexa Fluor 488, Alexa Fluor 546, Alexa Fluor 594, Alexa Fluor 647, Alexa Fluor 700, Alexa Fluor 790 and PerCP. Image tiles (600 x 600 micron viewing area) were individually captured at 0.32 micron/pixel spatial resolution, and the tiles seamlessly stitched into whole brain images using the ZEN 2 image acquisition and analysis software program (Zeiss), with an appropriate color table having been applied to each image channel to either match its emission spectrum or to set a distinguishing color balance. Pseudocolored stitched images were then exported to Adobe Photoshop and overlaid as individual layers to create multicolored merged composites. It is prudent to note that the images/qualitative data presented are meant to highlight the development of our MP-IHC system. However, to confirm the utility of the system NeuN (representing neurons) and IBA1 (representing microglia) were manually counted in A1/A2 regions of ipsilateral hemisphere and matching B1/B2 regions of contralateral hemisphere at 24-hour time point post TBI by two independent researchers. The average values of these counts are reported as percent change relative to the contralateral hemisphere control in the Result section.
Results
24 hours post-controlled cortical impact (CCI)
At 24 h post-CCI, H+E staining revealed the presence of both subarachnoid and intraparenchymal hematomas (Figure 1a). In addition, moderate edema within the cortex and the adjacent white matter was noted on the ipsilateral side of injury (Figure 1a, 1b). Pathologic regions of interest were identified within both the ipsilateral (regions B1, B2 and B3) and contralateral (regions A1, A2, and A3) sides in an effort to develop internal controls (Figure 1b).
Figure 1.




(a) Evaluation of tissue damage 24 h post-CCI (left parietooccipital area) via H+E staining (coronal section at level of impact). (b) Bright field phase contrast images of enclosed areas in 1a comparing morphological changes in the cerebral cortex/matched areas within the ipsilateral and contralateral hemispheres, including the zoomed in areas showing the subarachnoid hematoma at the impact site (B1), as well as hematoma in the external pyramidal layer (B2) and white matter in corpus callosum (B3) on the ipsilateral side, with A1, A2, and A3 matching areas within the contralateral hemispheres serving as internal controls. (c) Evaluation of tissue damage 72 h post-CCI (left parietooccipital area) via H+E staining (coronal section at level of impact). (d) Bright field phase contrast images of enclosed areas in 1c comparing morphological changes in the cerebral cortex/matched areas within the ipsilateral and contralateral hemispheres, including the zoomed in areas showing the subarachnoid hematoma at the impact site (B1), as well as hematoma in the external pyramidal layer (B2) and white matter in corpus callosum (B3). A1, A2, and A3 matching areas within the contralateral hemispheres serving as internal controls. A and B regions are comparable to those at 24 h post-CCI.
Further evaluation a panel of neuronal markers revealed massive changes within the ipsilateral hemisphere (Figure 2). On the side of CCI, marked disruption of pyramidal neurons as evidenced by staining with neuronal markers NeuN+ (i.e. with an ~60% decrease in NeuN positive cells noted on the ipsilateral side vs the contralateral control via a scoring of regions A1/A2 vs B1/B2) and CD57+ and interneurons (PV+) was noted in all cortical layers (exemplified by regions B1 and B2 in Figure 2), as was the prominent loss of gamma-aminobutyric acid (GABA)ergic interneurons (glutamic acid decarboxylase 67 [GAD67+]). A decrease of GAD67 expression was also notable within the white matter of the ipsilateral corpus callosum (exemplified by region B3 in Figure 2).
Figure 2.

Changes in the neuronal cytoarchitecture adjacent to the site of cortical impact at 24 h post-CCI; NeuN (green), CD57 (red), parvalbumin (purple), GAD67 (cyan)
Glial cytoarchitecture was also massively affected at 24 h post-injury as evidenced by a marked disruption of oligodendrocyte processes (via staining with myelin basic protein [MBP+]) within the cortex (exemplified by regions B1 and B2 in Figure 3) and reduction in MBP+ staining within the ipsilateral white matter and corpus callosum (region B3 in Figure 3). In addition, adenomatous polyposis coli (APC+) staining –reflective of the oligodendrocyte cell body– demonstrated the loss of mature oligodendrocytes throughout the ipsilateral cortex (exemplified by B1 and B2 in Figure 3) and the possible DE NOVO differentiation of oligodendrocytes within the white matter and corpus callosum (region B3 in Figure 3). Changes in astrocyte cytoarchitecture and distribution on the ipsilateral side of the injury clearly demonstrated a redistribution of resident astrocytes (Nestin−/GFAP+) to areas proximal to the impact side (see regions B1, B2 in Figure 4), whereas local de-differentiation of some astrocytes was manifested by up-regulation of Nestin (Nestin+/GFAP+) near the impact site within the ipsilateral hemisphere (regions B1, B2 in Figure 4). Furthermore, possible DE NOVO generation of immature astrocytes (Nestin+/GFAP+) from neural precursors and their possible migration to the impact site via corpus callosum could also be observed in the region B3 of Figure 4.
Figure 3.

Changes in oligodendrocyte cytoarchitecture adjacent to the site of cortical impact at 24 h post-CCI; APC (red), MBP (blue)
Figure 4.

Changes in astrocyte cytoarchitecture adjacent to the site of cortical impact at 24 h post-CCI; nestin (yellow), GFAP (orange).
Alterations in microglial activation states and tissue distribution post-injury were also noted within the ipsilateral hemisphere (Figure 5). Morphologically these changes were characterized via a transition from an inactive and resting dendritic form (shown in regions A1–A3 of Figure 5) to rounded active form (shown in regions B1–B3 of Figure 5); this is supported by an ~56% increase in IBA1 positive cells noted on the ipsilateral side vs the contralateral control via a scoring of regions A1/A2 vs B1/B2). In addition, prominent activation of microglia was noted at the impact site as shown in part via an increase isolectin-IB4 (ILIB4+) staining. Interestingly, increased expression of activated microglia as evidenced by co-staining with ionized calcium binding adaptor molecule 1 (IBA1+) and ILIB4+ was noted around blood vessels proximal to the abovementioned hematomas (exemplified by regions B1 and B2 in Figure 5).
Figure 5.

Changes in microglial and endothelial morphology and activation in areas adjacent to the site of cortical impact at 24 h post-CCI; IB4 (aquamarine), Iba1 (magenta)
Figure 6 shows a merged 10-color composite of the MP-IHC imaging results using 10 select biomarkers described in Figures 2–5 and illustrates the myriad of changes in neuronal, macroglial and microglial cytoarchitecture and initial infiltration of neuro-inflammatory cells at 24 h post-CCI. Taken together these findings clearly reveal the requirement to use multiplex biomarker screening to better characterize the complex processes and mechanisms underlying the tissue remodeling after TBI.
Figure 6.

Merged composite of 10 relevant biomarkers shown in Figures 2–5 revealing changes to cortical cytoarchitecture 24 h post-CCI.
72 hours post-controlled cortical impact (CCI)
At 72 h post-CCI, severe edema was observed within the ipsilateral hemisphere notably displacing all cortical layers and white matter tracts as revealed by H&E staining (Figure 1c) and phase contrast bright field imaging (Figure 1d). Neuronal staining demonstrated heavy losses of both pyramidal neurons (NeuN+) and interneurons (PV+) in all cortical layers and a concordant increase in GABAergic neurons (GAD67+) within the white matter and corpus callosum (Figure 7). In addition, a marked increase in infiltrating natural killer (NK) cells (NeuN−CD57+) was observed proximal to the impact site (exemplified in regions B1 and B2 of Figure 7).
Figure 7.

Changes in the neuronal cytoarchitecture adjacent to the site of cortical impact at 72 h post-CCI; NeuN (green), CD57 (red), parvalbumin (purple), GAD67 (cyan)
Oligodendrocyte staining revealed a major disruption of myelinating processes (MBP+) in all cortical layers (regions B1 and B2 in Figure 8) and a concordant increase in the intensity of MBP within the white matter and corpus callosum (region B3 in Figure 8). Furthermore, a near complete loss of APC+ oligodendrocytes was observed in the cortical layers (regions B1 and B2 in Figure 8) yet compensatory mechanisms may have driven DE NOVO oligodendrocyte differentiation within the white matter and corpus callosum (region B3 in Figure 8). Staining for astrocyte associated markers revealed a prominent loss of resident astrocytes (GFAP+) proximal to the area of cortical impact (region B1 in Figure 9). Interestingly, this staining also revealed the de-differentiation of mature astrocytes and/or DE NOVO generation of immature astrocytes from neural progenitors as characterized by increased numbers of Nestin+/GFAP+ cells (regions B2 and B3 in Figure 9). Microglial activation was still evident morphologically as evidenced by an abundance of active and rounded cells near the impact site (regions B2 and B3 in Figure 10). Furthermore, a massive infiltration of macrophages was noted at the site of CCI (region B1 in Figure 10). The merged composite of all 10 biomarkers screened reveals the complex changes in neuronal, macroglial and microglial cytoarchitecture as well as extensive tissue remodeling due to infiltration of neuroinflammatory cells at 72 h post-CCI (Figure 11).
Figure 8.

Changes in oligodendrocyte cytoarchitecture adjacent to the site of cortical impact at 72 h post-CCI; APC (red), MBP (blue)
Figure 9.

Changes in astrocyte cytoarchitecture adjacent to the site of cortical impact at 72 h post-CCI; nestin (yellow), GFAP (orange).
Figure 10.

Changes in microglial and endothelial morphology and activation in areas adjacent to the site of cortical impact at 72 h post-CCI; IB4 (aquamarine), Iba1 (magenta)
Figure 11.

Merged composite of 10 relevant biomarkers shown in Figures 7–10 revealing changes to cortical cytoarchitecture 72 h post-CCI.
Discussion
The accurate diagnosis of TBI and/or the development of targeted therapeutics capable of intervening in the aforementioned secondary injury cascade have yet to be realized. Such paucity of diagnostic and therapeutic options stands in stark contrast to the intensity of research efforts and number of clinical trials that have been performed to date (Maas et al. 2010).
Therapeutic interventions in animal models aimed at modulating major molecular pathways induced after TBI have limited the extent of injury and improved neurological recovery in the laboratory, yet all phase III clinical trials of these therapies in patients have failed to demonstrate efficacy. The restricted success of such a massive research investment demands a reevaluation of the pathobiology of evolving brain injury and a subsequent adjustment of experimental and therapeutic approaches (Dash et al. 2010; Liu et al. 2010; Lizhnyak and Ottens 2015).
Herein we have effectively demonstrated that our novel multiplex fluorescence immunohistochemistry (MP-IHC) technique is capable of simultaneously identifying 10 relevant biomarkers of damage, repair and/or regeneration in an effort to fully characterize and ultimately intervene in the evolving pathogenesis of brain injury after TBI. Such work sets the stage for the comprehensive description of CNS pathogenesis post-TBI and may ultimately be extended to other diseases/disorders or insults to the CNS that require one to comprehensively profile the tissues response to damage (i.e. thereby generating systems level information in the context of the injured tissue). It is prudent to note that iterate rounds of multiplex IHC staining may ultimately increase the number of screened biomarkers to hundreds or even thousands per tissue section. Currently, we are in the process of expanding our biomarker panels by developing novel combinations of validated antibodies capable of fully characterizing the complex response of CNS tissue after traumatic injury. In line with the aforementioned it is also important to note that our findings using MP-IHC validate previous observations made in a myriad of TBI models that have employed traditional staining techniques.
For example our study revealed prominent disruptions of NeuN-positive neurons on the ipsilateral side of the injury within all cortical layers at both 24 h and 72 h which is in line with the loss of NeuN immunoreactivity reported in the ipsilateral cortex in a fluid percussion model in rats at both 24 h and 14 days post-TBI ((Karklin Fontana et al. 2016)). Beyond NeuN, our staining for PV demonstrated a prominent disruption in PV-positive interneurons ipsilateral to CCI within all cortical layers at both at 24 h and 72 h post-CCI. Such a reduction in PV-positive neurons is in agreement with the previously observed loss of PV-positive dentate neurons within the hippocampus following entorhinal injury (Nitsch and Frotscher 1993). Further, our findings are also supported by the reduction in the number of PV-positive neurons within the reticular thalamic nucleus demonstrated 6 months after exposure to a fluid percussion model of TBI (Huusko and Pitkanen 2014).
Beyond the alterations shown to occur within the neuronal lineages, we effectively demonstrated vast changes within both the micro and macroglial compartments. Specifically, we demonstrated an obvious loss of mature resident astrocytes (Nestin−/GFAP+) in the area of CCI injury at 24 hours. These findings are in agreement with an acute TBI study that demonstrated the early loss of GFAP immunoreactivity in ipsilateral hippocampal CA3 in rats as fast as 30 minutes after exposure to a percussion based TBI model (Zhao et al. 2003). Furthermore, this study showed progressive loss of astrocytes with the most prominent decrease being noted at 24 hours (Zhao et al. 2003). Interestingly, we also note an increase in proliferating astrocytes (Nestin+/GFAP+) which again reflects what has been documented to occur within the literature (i.e. the significant increase noted in double positive staining for nestin and GFAP in the rat brain near glial scar 3–7 days after CCI) (Itoh et al. 2007). Beyond astrocytes, pronounced changes were noted in microglia. Iba1 is a classic marker for microglial cells and is expressed by all microglial cell subpopulations. Using Iba1, we definitively demonstrated both morphological and spatiotemporal changes that occur within the resident microglia population after traumatic brain injury. In line with such findings, a recent TBI study in a rat model found a significant increase in the number of Iba1+ cells in the ipsilateral frontal cortex as compared to the contralateral frontal cortex (Harvey et al. 2015) post-CCI.
Again, it is prudent to note that our study was designed as a proof-of-principle in which we sought to describe methodology associated with multiplex immunohistochemistry. Accordingly, computational solutions are being devised to leverage this technology in order to comprehensively and robustly quantitate the histological changes mediating complex cellular processes in TBI and other neuropathological disorders (ischemic stroke, spinal cord injury, multiple sclerosis, brain tumors, etc.) using existing quantitative algorithms for image segmentation and analysis (e.g. the FARSIGHT toolkit) (Bjornsson et al. 2008). More advanced computational solutions are also being developed that will employ machine learning algorithms customized to our imaging MP-IHC paradigm, ultimately enabling easily scalable image data analysis to support systems biology and translational research into devising optimal treatment strategies to reduce the burden of neurological disease and disorders and/or traumatic insults.
Limitations of our study include its exploratory nature and the use of descriptive analysis in the evaluation of differences with regard to the changes noted post-CCI. Further, the work only profiled the response of male rats post-CCI and is therefore unable highlight differences attributable to biologic sex in acute and subacute responses to CCI. This is important to highlight as studies have in fact emerged to indicate that sex may influence recovery after TBI with female rats having significantly smaller cortical contusions than male rats in a fluid percussion model of TBI (Bramlett and Dietrich 2001; Mannix et al. 2014). Future work will therefore be sure to include animals of both sexes in an effort to fully elucidate TBI pathophysiology. Further we will seek to expand our antibody panels and iterative rounds of staining to maximize the systems level data analysis that computational solutions promise to provide.
In summary, our novel MP-IHC staining and imaging platform is a useful modality capable of providing a comprehensive analysis of the complex changes that occur in brain cytoarchitecture after insults and injuries. It is the authors’ contention that this method and future iterations thereof may ultimately aid in the development of successful therapeutic interventions for TBI (and other related injuries that share core components of TBI pathobiology) via an insightful description of the molecular and cell-based mechanisms involved in brain tissue repair and/or regeneration. Of note, this method can be tailored to test any given experimental question via the selection of specific combinations of biomarkers to fully characterize and evaluate tissue pathology after TBI. It is also the authors’ contention that this method may aid in development of effective therapeutic interventions aimed at restoring normal brain function and fostering regeneration and repair after traumatic insults. If successful, this systematic approach may find utility in wide variety of other neurological injuries, diseases and disorders that share components of TBI pathobiology (e.g. ischemic stroke, spinal cord injury, multiple sclerosis, amyotrophic lateral sclerosis, etc.).
Supplementary Material
Supplemental Table 1
Supplemental Table 2
Significance Statement.
Herein, we describe the development and the application of a system capable of evaluating changes in both the topographical location and functional states of resident and infiltrating cell types that play a role in neuropathology after TBI. Our screening platform lays the groundwork for the comprehensive characterization of changes that occur within the brain after TBI. Further, the multiplex system may readily be adapted thereby allowing screening to unfold in other translational scenarios in which one would need to identify biomarkers and/or develop novel therapeutics.
Acknowledgments
Funding
This work was supported by the CNRM/USUHS and NINDS/NIH intramural research programs.
Footnotes
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Author Contribution statement
TB and JDB contributed to the study via data analysis and interpretation and via the writing of the manuscript. GB and SG contributed to the study experimentally. BMC and JMH contributed via data analysis/interpretation and via the editing of the manuscript. DM contributed via experimental planning, data analysis and interpretation and via the editing of the manuscript.
References
- Addington CP, Roussas A, Dutta D, Stabenfeldt SE. Endogenous repair signaling after brain injury and complementary bioengineering approaches to enhance neural regeneration. Biomark Insights. 2015;10(Suppl 1):43–60. doi: 10.4137/BMI.S20062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bazarian JJ, Atabaki S. Predicting postconcussion syndrome after minor traumatic brain injury. Academic emergency medicine : official journal of the Society for Academic Emergency Medicine. 2001;8(8):788–795. doi: 10.1111/j.1553-2712.2001.tb00208.x. [DOI] [PubMed] [Google Scholar]
- Bjornsson CS, Lin G, Al-Kofahi Y, Narayanaswamy A, Smith KL, Shain W, Roysam B. Associative image analysis: a method for automated quantification of 3D multi-parameter images of brain tissue. J Neurosci Methods. 2008;170(1):165–178. doi: 10.1016/j.jneumeth.2007.12.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bramlett HM, Dietrich WD. Neuropathological protection after traumatic brain injury in intact female rats versus males or ovariectomized females. J Neurotrauma. 2001;18(9):891–900. doi: 10.1089/089771501750451811. [DOI] [PubMed] [Google Scholar]
- Budinich CS, Chen H, Lowe D, Rosenberger JG, Bernstock JD, McCabe JT. Mouse brain PSA-NCAM levels are altered by graded-controlled cortical impact injury. Neural plasticity. 2012;2012:378307. doi: 10.1155/2012/378307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark RS, Kochanek PM, Watkins SC, Chen M, Dixon CE, Seidberg NA, Melick J, Loeffert JE, Nathaniel PD, Jin KL, Graham SH. Caspase-3 mediated neuronal death after traumatic brain injury in rats. Journal of neurochemistry. 2000;74(2):740–753. doi: 10.1046/j.1471-4159.2000.740740.x. [DOI] [PubMed] [Google Scholar]
- Corrigan JD, Selassie AW, Orman JA. The epidemiology of traumatic brain injury. J Head Trauma Rehabil. 2010;25(2):72–80. doi: 10.1097/HTR.0b013e3181ccc8b4. [DOI] [PubMed] [Google Scholar]
- Dash PK, Zhao J, Hergenroeder G, Moore AN. Biomarkers for the diagnosis, prognosis, and evaluation of treatment efficacy for traumatic brain injury. Neurotherapeutics. 2010;7(1):100–114. doi: 10.1016/j.nurt.2009.10.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall ED, Vaishnav RA, Mustafa AG. Antioxidant therapies for traumatic brain injury. Neurotherapeutics. 2010;7(1):51–61. doi: 10.1016/j.nurt.2009.10.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hallbergson AF, Gnatenco C, Peterson DA. Neurogenesis and brain injury: managing a renewable resource for repair. The Journal of clinical investigation. 2003;112(8):1128–1133. doi: 10.1172/JCI20098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harvey LD, Yin Y, Attarwala IY, Begum G, Deng J, Yan HQ, Dixon CE, Sun D. Administration of DHA Reduces Endoplasmic Reticulum Stress-Associated Inflammation and Alters Microglial or Macrophage Activation in Traumatic Brain Injury. ASN neuro. 2015;7(6) doi: 10.1177/1759091415618969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hinson HE, Rowell S, Schreiber M. Clinical evidence of inflammation driving secondary brain injury: a systematic review. J Trauma Acute Care Surg. 2015;78(1):184–191. doi: 10.1097/TA.0000000000000468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huusko N, Pitkanen A. Parvalbumin immunoreactivity and expression of GABAA receptor subunits in the thalamus after experimental TBI. Neuroscience. 2014;267:30–45. doi: 10.1016/j.neuroscience.2014.02.026. [DOI] [PubMed] [Google Scholar]
- Itoh T, Satou T, Nishida S, Hashimoto S, Ito H. Immature and mature neurons coexist among glial scars after rat traumatic brain injury. Neurol Res. 2007;29(7):734–742. doi: 10.1179/016164107X208086. [DOI] [PubMed] [Google Scholar]
- Johnson VE, Meaney DF, Cullen DK, Smith DH. Animal models of traumatic brain injury. Handbook of clinical neurology. 2015;127:115–128. doi: 10.1016/B978-0-444-52892-6.00008-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson VE, Stewart W, Smith DH. Widespread tau and amyloid-beta pathology many years after a single traumatic brain injury in humans. Brain pathology. 2012;22(2):142–149. doi: 10.1111/j.1750-3639.2011.00513.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karklin Fontana AC, Fox DP, Zoubroulis A, Valente Mortensen O, Raghupathi R. Neuroprotective Effects of the Glutamate Transporter Activator (R)-(-)-5-methyl-1-nicotinoyl-2-pyrazoline (MS-153) following Traumatic Brain Injury in the Adult Rat. J Neurotrauma. 2016;33(11):1073–1083. doi: 10.1089/neu.2015.4079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kochanek PM, Clark RS, Ruppel RA, Adelson PD, Bell MJ, Whalen MJ, Robertson CL, Satchell MA, Seidberg NA, Marion DW, Jenkins LW. Biochemical, cellular, and molecular mechanisms in the evolution of secondary damage after severe traumatic brain injury in infants and children: Lessons learned from the bedside. Pediatric critical care medicine : a journal of the Society of Critical Care Medicine and the World Federation of Pediatric Intensive and Critical Care Societies. 2000;1(1):4–19. doi: 10.1097/00130478-200007000-00003. [DOI] [PubMed] [Google Scholar]
- Langlois JA, Kegler SR, Butler JA, Gotsch KE, Johnson RL, Reichard AA, Webb KW, Coronado VG, Selassie AW, Thurman DJ. Traumatic brain injury-related hospital discharges. Results from a 14-state surveillance system, 1997. MMWR Surveill Summ. 2003;52(4):1–20. [PubMed] [Google Scholar]
- Lesko LJ, Atkinson AJ., Jr Use of biomarkers and surrogate endpoints in drug development and regulatory decision making: criteria, validation, strategies. Annual review of pharmacology and toxicology. 2001;41:347–366. doi: 10.1146/annurev.pharmtox.41.1.347. [DOI] [PubMed] [Google Scholar]
- Levin HS, Diaz-Arrastia RR. Diagnosis, prognosis, and clinical management of mild traumatic brain injury. The Lancet Neurology. 2015;14(5):506–517. doi: 10.1016/S1474-4422(15)00002-2. [DOI] [PubMed] [Google Scholar]
- Liu MC, Akinyi L, Scharf D, Mo J, Larner SF, Muller U, Oli MW, Zheng W, Kobeissy F, Papa L, Lu XC, Dave JR, Tortella FC, Hayes RL, Wang KK. Ubiquitin C-terminal hydrolase-L1 as a biomarker for ischemic and traumatic brain injury in rats. Eur J Neurosci. 2010;31(4):722–732. doi: 10.1111/j.1460-9568.2010.07097.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lizhnyak PN, Ottens AK. Proteomics: in pursuit of effective traumatic brain injury therapeutics. Expert Rev Proteomics. 2015;12(1):75–82. doi: 10.1586/14789450.2015.1000869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maas AI, Roozenbeek B, Manley GT. Clinical trials in traumatic brain injury: past experience and current developments. Neurotherapeutics. 2010;7(1):115–126. doi: 10.1016/j.nurt.2009.10.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mannix R, Berglass J, Berkner J, Moleus P, Qiu J, Jantzie LL, Meehan WP, 3rd, Stanley RM, Robinson S. Sex differences in the effect of progesterone after controlled cortical impact in adolescent mice: a preliminary study. Journal of neurosurgery. 2014;121(6):1337–1341. doi: 10.3171/2014.8.JNS14715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marklund N, Hillered L. Animal modelling of traumatic brain injury in preclinical drug development: where do we go from here? Br J Pharmacol. 2011;164(4):1207–1229. doi: 10.1111/j.1476-5381.2010.01163.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marmarou A, Foda MA, van den Brink W, Campbell J, Kita H, Demetriadou K. A new model of diffuse brain injury in rats. Part I: Pathophysiology and biomechanics. Journal of neurosurgery. 1994;80(2):291–300. doi: 10.3171/jns.1994.80.2.0291. [DOI] [PubMed] [Google Scholar]
- Meaney DF, Margulies SS, Smith DH. Diffuse axonal injury. Journal of neurosurgery. 2001;95(6):1108–1110. doi: 10.3171/jns.2001.95.6.1108. [DOI] [PubMed] [Google Scholar]
- Morganti-Kossmann MC, Yan E, Bye N. Animal models of traumatic brain injury: is there an optimal model to reproduce human brain injury in the laboratory? Injury. 2010;41(Suppl 1):S10–13. doi: 10.1016/j.injury.2010.03.032. [DOI] [PubMed] [Google Scholar]
- Mustafa AG, Wang JA, Carrico KM, Hall ED. Pharmacological inhibition of lipid peroxidation attenuates calpain-mediated cytoskeletal degradation after traumatic brain injury. Journal of neurochemistry. 2011;117(3):579–588. doi: 10.1111/j.1471-4159.2011.07228.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Namjoshi DR, Cheng WH, Carr M, Martens KM, Zareyan S, Wilkinson A, McInnes KA, Cripton PA, Wellington CL. Chronic Exposure to Androgenic-Anabolic Steroids Exacerbates Axonal Injury and Microgliosis in the CHIMERA Mouse Model of Repetitive Concussion. PLoS One. 2016;11(1):e0146540. doi: 10.1371/journal.pone.0146540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Namjoshi DR, Cheng WH, McInnes KA, Martens KM, Carr M, Wilkinson A, Fan J, Robert J, Hayat A, Cripton PA, Wellington CL. Merging pathology with biomechanics using CHIMERA (Closed-Head Impact Model of Engineered Rotational Acceleration): a novel, surgery-free model of traumatic brain injury. Mol Neurodegener. 2014;9:55. doi: 10.1186/1750-1326-9-55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ng HK, Mahaliyana RD, Poon WS. The pathological spectrum of diffuse axonal injury in blunt head trauma: assessment with axon and myelin strains. Clin Neurol Neurosurg. 1994;96(1):24–31. doi: 10.1016/0303-8467(94)90025-6. [DOI] [PubMed] [Google Scholar]
- Nitsch R, Frotscher M. Transneuronal changes in dendrites of GABAergic parvalbumin-containing neurons of the rat fascia dentata following entorhinal lesion. Hippocampus. 1993;3(4):481–490. doi: 10.1002/hipo.450030409. [DOI] [PubMed] [Google Scholar]
- Park E, Bell JD, Baker AJ. Traumatic brain injury: Can the consequences be stopped? Can Med Assoc J. 2008;178(9):1163–1170. doi: 10.1503/cmaj.080282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pettus EH, Povlishock JT. Characterization of a distinct set of intra-axonal ultrastructural changes associated with traumatically induced alteration in axolemmal permeability. Brain research. 1996;722(1–2):1–11. doi: 10.1016/0006-8993(96)00113-8. [DOI] [PubMed] [Google Scholar]
- Povlishock JT, Katz DI. Update of neuropathology and neurological recovery after traumatic brain injury. J Head Trauma Rehabil. 2005;20(1):76–94. doi: 10.1097/00001199-200501000-00008. [DOI] [PubMed] [Google Scholar]
- Rhodes J. Peripheral immune cells in the pathology of traumatic brain injury? Curr Opin Crit Care. 2011;17(2):122–130. doi: 10.1097/MCC.0b013e3283447948. [DOI] [PubMed] [Google Scholar]
- Robb MA, McInnes PM, Califf RM. Biomarkers and Surrogate Endpoints: Developing Common Terminology and Definitions. Jama. 2016;315(11):1107–1108. doi: 10.1001/jama.2016.2240. [DOI] [PubMed] [Google Scholar]
- Roozenbeek B, Maas AI, Menon DK. Changing patterns in the epidemiology of traumatic brain injury. Nat Rev Neurol. 2013;9(4):231–236. doi: 10.1038/nrneurol.2013.22. [DOI] [PubMed] [Google Scholar]
- Rubiano AM, Carney N, Chesnut R, Puyana JC. Global neurotrauma research challenges and opportunities. Nature. 2015;527(7578):S193–197. doi: 10.1038/nature16035. [DOI] [PubMed] [Google Scholar]
- Smith DH, Chen XH, Nonaka M, Trojanowski JQ, Lee VM, Saatman KE, Leoni MJ, Xu BN, Wolf JA, Meaney DF. Accumulation of amyloid beta and tau and the formation of neurofilament inclusions following diffuse brain injury in the pig. J Neuropathol Exp Neurol. 1999;58(9):982–992. doi: 10.1097/00005072-199909000-00008. [DOI] [PubMed] [Google Scholar]
- Tagliaferri F, Compagnone C, Korsic M, Servadei F, Kraus J. A systematic review of brain injury epidemiology in Europe. Acta Neurochir (Wien) 2006;148(3):255–268. doi: 10.1007/s00701-005-0651-y. discussion 268. [DOI] [PubMed] [Google Scholar]
- Woodcock T, Morganti-Kossmann MC. The role of markers of inflammation in traumatic brain injury. Front Neurol. 2013;4:18. doi: 10.3389/fneur.2013.00018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiong Y, Mahmood A, Chopp M. Angiogenesis, neurogenesis and brain recovery of function following injury. Curr Opin Investig Drugs. 2010;11(3):298–308. [PMC free article] [PubMed] [Google Scholar]
- Xiong Y, Mahmood A, Chopp M. Animal models of traumatic brain injury. Nat Rev Neurosci. 2013;14(2):128–142. doi: 10.1038/nrn3407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yeoh S, Bell ED, Monson KL. Distribution of blood-brain barrier disruption in primary blast injury. Annals of biomedical engineering. 2013;41(10):2206–2214. doi: 10.1007/s10439-013-0805-7. [DOI] [PubMed] [Google Scholar]
- Zhao X, Ahram A, Berman RF, Muizelaar JP, Lyeth BG. Early loss of astrocytes after experimental traumatic brain injury. Glia. 2003;44(2):140–152. doi: 10.1002/glia.10283. [DOI] [PubMed] [Google Scholar]
- Zink BJ, Szmydynger-Chodobska J, Chodobski A. Emerging concepts in the pathophysiology of traumatic brain injury. Psychiatr Clin North Am. 2010;33(4):741–756. doi: 10.1016/j.psc.2010.08.005. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Table 1
Supplemental Table 2