Enhancing cancer immunotherapy using antiangiogenics: opportunities and challenges (original) (raw)
. Author manuscript; available in PMC: 2018 May 1.
Published in final edited form as: Nat Rev Clin Oncol. 2018 Mar 6;15(5):325–340. doi: 10.1038/nrclinonc.2018.29
Abstract
Immunotherapy has emerged as a major therapeutic modality in oncology. Currently, however, the majority of patients with cancer do not derive benefit from these treatments. Vascular abnormalities are a hallmark of most solid tumours and facilitate immune evasion. These abnormalities stem from elevated levels of proangiogenic factors, such as VEGF and angiopoietin 2 (ANG2); judicious use of drugs targeting these molecules can improve therapeutic responsiveness, partially owing to normalization of the abnormal tumour vasculature that can, in turn, increase the infiltration of immune effector cells into tumours and convert the intrinsically immunosuppressive tumour microenvironment (TME) to an immunosupportive one. Immunotherapy relies on the accumulation and activity of immune effector cells within the TME, and immune responses and vascular normalization seem to be reciprocally regulated. Thus, combining antiangiogenic therapies and immunotherapies might increase the effectiveness of immunotherapy and diminish the risk of immune-related adverse effects. In this Perspective, we outline the roles of VEGF and ANG2 in tumour immune evasion and progression, and discuss the evidence indicating that antiangiogenic agents can normalize the TME. We also suggest ways that antiangiogenic agents can be combined with immune-checkpoint inhibitors to potentially improve patient outcomes, and highlight avenues of future research.
Immunotherapy has revolutionized the treatment of cancer, enabling durable control of previously incurable and highly aggressive cancers, such as melanoma, in a considerable proportion of patients. Most notably, immune-checkpoint inhibitors that reactivate dysfunctional and/or exhausted T cells have marked efficacy against a broad range of solid and haematological cancers1,2 (Supplementary Table S1). Moreover, in May 2017, the FDA granted an unprecedented approval to pembrolizumab, an inhibitory antibody that targets the immune-checkpoint receptor programmed cell death protein 1 (PD-1), for the treatment of adult and paediatric patients with unresectable or metastatic, microsatellite instability-high (MSI-H) or mismatch repair-deficient (dMMR) solid tumours that have progressed after prior treatment, independent of the tissue of origin, provided that no satisfactory alternative treatments are available3. This move by the FDA constitutes the first approval of an anticancer drug that is not based on cancer type, or in other words, is ‘histology agnostic’. However, 50–80% of patients with tumours for which immune-checkpoint inhibitors are indicated do not benefit from these drugs, and many patients experience severe adverse events (Supplementary Table S1). An insufficient abundance of tumour neoantigens — which are a prerequisite for recognition of tumour cells by T cells — alone cannot explain this lack of responsiveness in the majority of patients2,4, nor can the differential expression of immune-checkpoint proteins1. Indeed, no robust mechanistic data obtained to date accurately explain the unconventional pattern of clinical response to immune-checkpoint inhibitors5. Our hypothesis is that the tumour micro-environment (TME) — characterized by hypoxia, a low pH, and a high interstitial fluid pressure — can reduce the effectiveness of virtually all types of anticancer therapies, including immunotherapy6. Thus, normalizing the TME has the potential to improve the effectiveness of immunotherapy7.
The TME consists of blood and lymphatic vessels, various stromal cells, and resident and infiltrating immune cells, all of which are ensconced in an extracellular matrix. Our work and that of others has revealed that, in comparison with nonmalignant tissues, each component of the TME is abnormal and heterogeneous, and this abnormality fuels tumour progression and treatment resistance6,7. Thus, normalizing components of the TME could improve the outcomes of patients treated with standard and emerging anticancer therapies, including chemotherapeutics, radiation, molecularly targeted agents, and immunotherapeutics8. In this Perspective, we focus on one important component of the TME, the tumour vasculature, and discuss how normalizing the tumour vasculature with antiangiogenic agents (BOX 1) could potentially be used to improve the effectiveness of immunotherapy, particularly immune checkpoint blockade (ICB). We also discuss the direct effects of antiangiogenic agents on immune cells, both systemically and in the local TME, and conversely, discuss how immune cells and ICB can alter the tumour vasculature. Given that more than a dozen antiangiogenic drugs that inhibit the VEGF signalling pathway have been approved for the treatment of cancer (Supplementary Table S2), and that a number of antiangiogenic agents that target the angiopoietin 1 (ANG1), ANG2, and tyrosine-protein kinase receptor TIE2 (also known as angiopoietin 1 receptor) pathway are in clinical trials, we highlight the potential of these treatments to improve the outcomes of immunotherapy. We outline opportunities and challenges relating to this emerging strategy to enhance immunotherapy. These concepts and approaches are very timely, considering that patients with cancer will increasingly be treated with various immunotherapies and that biosimilar versions of the anti-VEGF antibody bevacizumab are beginning to be approved (Supplementary Table S3).
Box 1. From tumour starvation to vessel normalization.
The discovery that VEGF is a key mediator of angiogenesis prompted concerted efforts to target this pathway therapeutically, with the goal of inducing regression of blood vessels to starve tumours of oxygen, glucose, and other essential metabolites. Proof of principle for anti-VEGF therapy was demonstrated in preclinical tumour models, leading to rapid clinical translation of this approach and the development of many drugs targeting the VEGF pathway — with the anti-VEGF antibody bevacizumab being the most widely prescribed antiangiogenic drug7 (Supplementary Table S2). Bevacizumab alone does not confer a clinically or statistically significant overall survival benefit to patients with metastatic colorectal cancer or glioblastoma; however, the combination of bevacizumab or other VEGF pathway inhibitors with cytotoxic therapies has been demonstrated to result in clinically meaningful overall survival benefits for patients with advanced-stage colorectal cancer, non-small-cell lung cancer, mesothelioma, or cervical cancer, but not those with other cancers, such as breast cancer, prostate cancer, or glioblastoma127–134 (Supplementary Table S2). These results prompted fundamental mechanistic studies to elucidate how antiangiogenic therapies targeting VEGF and other angiogenic factors, initially developed to reduce the delivery of metabolites to tumours, somewhat paradoxically improve the effectiveness of systemic chemotherapy. In 2001, we proposed the vessel normalization hypothesis as an explanation to resolve this conundrum61, and this concept has been validated in a number of preclinical and clinical studies7.
The abnormal tumour vasculature
The growth and progression of tumours are facilitated by the recruitment of blood vessels. Tumour blood vessels not only supply oxygen and nutrients and remove waste products, but also sustain a favourable niche for so-called cancer stem cells and serve as a conduit for both the metastatic dissemination of tumour cells and the infiltration of immune cells9,10. One would expect a rapidly growing organ or tissue to have normally functioning vessels; however, tumour vessels are abnormal, both structurally and functionally, relative to those of nonmalignant tissues7. Perhaps counter-intuitively, this abnormality promotes tumour progression by impairing perfusion, which results in tumour hypoxia and a low pH intratumourally7. In addition, the leaky nature of tumour blood vessels, coupled with dysfunctional lymphatic drainage, leads to elevated interstitial fluid pressure in the TME6. Tumour cells adapt to overcome these conditions and even to gain a survival advantage over nonmalignant cells through several mechanisms7. The abnormal vessels and impaired perfusion can also restrict the entry of cytotoxic drugs and immune cells from the circulation into tumours, limiting their anticancer activity. Furthermore, the abnormal TME might modulate the activity and, thus, the effectiveness of these drugs and immune cells after they accrue in the tumour. For example, an adequate level of oxygen is required for many drugs and immune cells to kill cancer cells7. Additionally, various cytokines produced by diverse cell types in response to hypoxia and the low pH in the TME can cause both local (FIGS 1,2) and — after entering the circulation — systemic immunosuppression (FIG. 3). Finally, the abnormal blood and lymphatic vessels also facilitate tumour invasion and metastasis via multiple mechanisms6. For example, fluid leaking out of a tumour, owing to insufficient drainage via the defective intratumoural lymphatic system, can carry cancer cells to nearby peri-tumour lymphatic vessels, thereby promoting lymphatic metastasis11. Abnormal leaky vessels might also facilitate intravasation and shedding of cancer cells into the blood; after entering the systemic circulation, some of these tumour cells can disseminate to distal organs and form metastases. Moreover, the cytokines, such as VEGF, that seep from the TME into the systemic circulation can also potentiate the extravasation of metastatic cancer cells out of the blood vessels within distant organs12. By contrast, while CD8+ cytotoxic T lymphocytes (CTLs) can also home to tumours, multiple factors present in the TME often render these cells dysfunctional and/or limit their penetration into the tumour (FIGS 1,2). Other adverse consequences of abnormal tumour vasculature and the resulting conditions in the TME include inflammation, fibrosis, genomic instability, DNA hypermethylation, activation of the unfolded protein response, a switch to anaerobic metabolism, resistance to autophagy and apoptosis, induction of a cancer stem cell programme, epithelial-to-mesenchymal transition, and tumour cell invasion and metastasis7. In the following sections, we elaborate on the roles of angiogenic factors in mediating two of these consequences: immunosuppression and metastasis.
Figure 1. Direct effects of angiogenic factors on various immune cells.
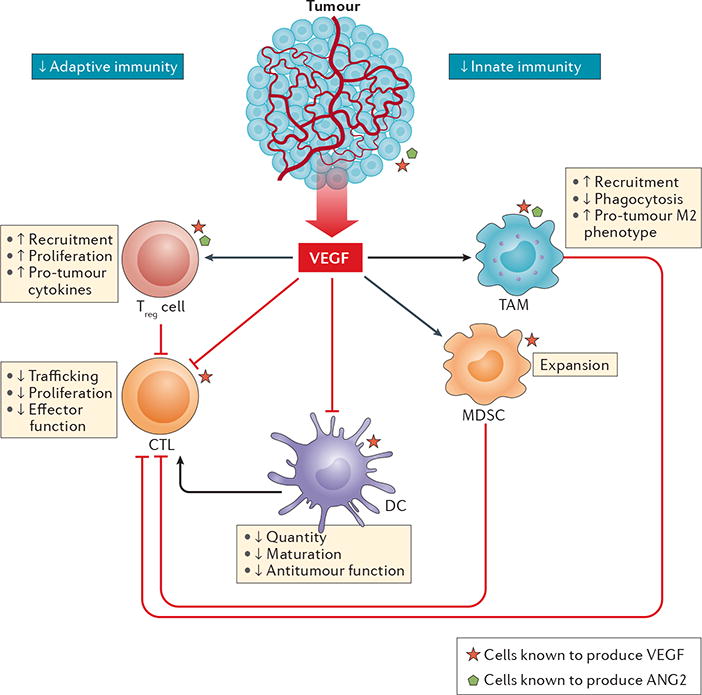
VEGF and other angiogenic factors, such as angiopoietin 2 (ANG2), modulate the functions of innate and adaptive immune cells towards immunosuppression. VEGF can be produced by tumour cells and immune cells (red stars) as well as by endothelial cells and stromal cells. VEGF creates a pro-tumour microenvironment by increasing the number and enhancing the suppressive functions of regulatory T (Treg) cells and tumour-associated macrophages (TAMs) and/or monocytes. These cell populations also produce VEGF (red stars) and ANG2 (green pentagons) and thereby engage in paracrine and autocrine VEGF (and/or ANG2) signalling. Among immune cells, Treg cells have been identified as the major source of VEGF in the tumour microenvironment using in vivo models17. This finding was further confirmed in a different study89. While VEGF directly inhibits the development and/or activation of cytotoxic T lymphocytes (CTLs), CTL-produced VEGF can induce angiogenesis and promote tumour progression97. Furthermore, VEGF has been shown to increase the numbers of CD4+ memory T cells (Supplementary Table S4). VEGF can promote the expansion of myeloid-derived suppressor cells (MDSCs) and improve their immunosuppressive function in the tumour microenvironment (TME)23. Importantly, dendritic cells (DCs), which are required for priming of CTLs, lose their ability to mature and present antigens following VEGF exposure in vitro. Consequently, the anticancer activity of CTLs in the TME might be compromised. In this context, CTLs have a decreased capacity to traffic to the tumour, proliferate, and produce cytokines that are integral to the anticancer immune response, such as IFNγ and tumour necrosis factor172. Collectively, Treg cells, TAMs, MDSCs, and immature DCs suppress the activity of CTLs, resulting in a pro-tumour microenvironment. The roles of ANG2 (and ANG1) in modulating various immune cells are not as well understood as those of VEGF; thus, the effects of ANG2 are not illustrated in this figure.
Figure 2. The abnormal tumour vasculature contributes to immunosuppression in the tumour microenvironment.
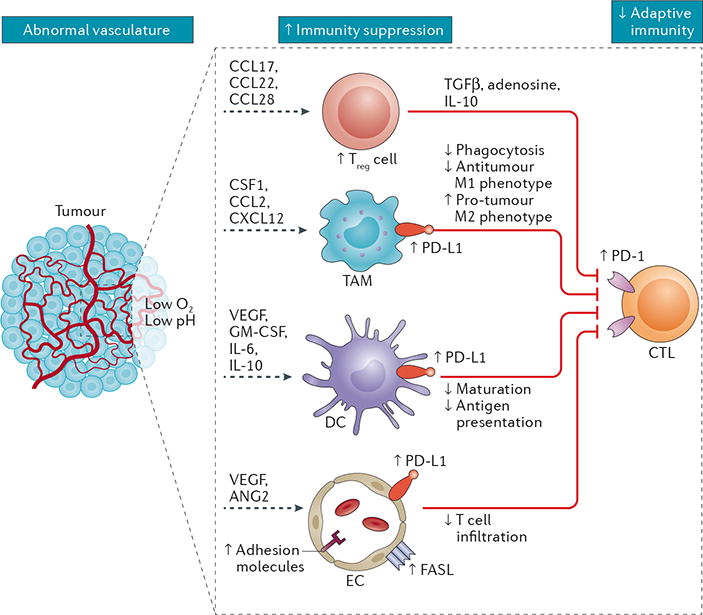
Abnormalities in the tumour vasculature result in hypoxia and acidosis of the tumour microenvironment (TME), which in turn contribute to immunosuppression via several mechanisms. These mechanisms include increased accumulation, activation, and expansion of immunosuppressive regulatory T (Treg) cells; recruitment of inflammatory monocytes and tumour-associated macrophages (TAMs) and reprogramming of TAMs from an anticancer M1-like phenotype towards the pro-tumour M2 phenotype; suppression of dendritic cell (DC) maturation, which results in impaired antigen presentation and activation of tumour-specific cytotoxic T lymphocytes (CTLs); and expansion of abnormal endothelial cells (ECs) with immunosuppressive phenotypes. Importantly, the programmed cell death protein 1 (PD-1)–programmed cell death 1 ligand 1 (PD-L1) pathway is often activated in the TME as a mechanism to evade anticancer immune responses, with upregulation of PD-L1 expression on TAMs, DCs, and ECs, as well as on tumour cells. In addition, tumour-infiltrating CTLs typically upregulate PD-1, marking them as dysfunctional or ‘exhausted’ and limiting their cytotoxic potential against tumour cells. Overall, the consequence of aberrant tumour angiogenesis and vascular abnormality is an immunosuppressive TME. ANG2, angiopoietin 2; CCL, C-C-motif chemokine ligand; CXCL12, C-X-C-motif chemokine ligand 12; CSF1, macrophage colony-stimulating factor 1; FASL, FAS antigen ligand; GM-CSF, granulocyte–macrophage colony-stimulating factor; TGFβ, transforming growth factor β.
Figure 3. Tumours secrete factors that cause systemic immunosuppression.
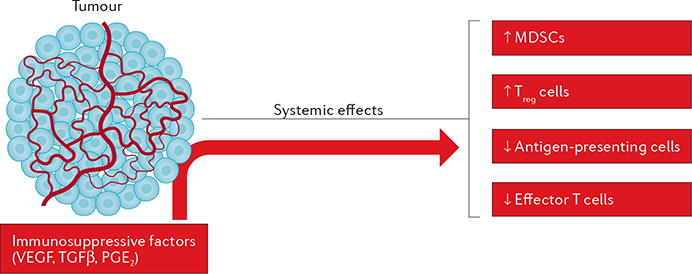
The cells of the tumour microenvironment exert systemic immunosuppressive effects by releasing factors such as VEGF, transforming growth factor β (TGFβ), and prostaglandin E2 (PGE2) into the circulation. Collectively, these cytokines reduce the ability of antigen-presenting cells to prime T cells and thus reduce the anticancer responses of effector T cells. In addition, tumour-derived factors increase the presence and function of myeloid-derived suppressor cells (MDSCs) and regulatory T (Treg) cells, which suppress anticancer immunity.
Immunosuppression
Anticancer immunity is conferred by both tissue-resident and blood-borne, tumour-infiltrating immune cells. To infiltrate a tumour and integrate into the TME, immune cells must enter the tumour blood vessels, adhere to the endothelium, and transmigrate across the vessel wall13. Angiogenic molecules present in the TME, such as VEGF (FIG. 1), can control the trafficking of immune cells to the tumour by altering the expression of adhesion molecules, including the integrin ligands intercellular adhesion molecule 1 (ICAM1) and vascular cell adhesion protein 1 (VCAM1), on endothelial cells (ECs) and immune cells10,14,15. Tumour-associated ECs can also express the immune-checkpoint protein programmed cell death 1 ligand 1 (PD-L1), which binds to PD-1 expressed on T cells and thereby suppresses their anticancer activity16 (FIG. 2). In addition, tumours secrete soluble factors, such as C-C-motif chemokine ligand 2 (CCL2), CCL28, C-X-C-motif chemokine ligand 8 (CXCL8), CXCL12, ANG2, VEGF, PlGF, and adenosine, all of which promote the recruitment of immunosuppressive cells7,17–20 (FIG. 2). These cells include immature dendritic cells (DCs), regulatory T (Treg) cells, myeloid-derived suppressor cells (MDSCs), and tumour-associated macrophages (TAMs) with pro-tumour phenotypes. In turn, through the production of various cytokines and growth factors, such as VEGF, ANG2, IL-10, and transforming growth factor β (TGFβ), these leukocytes work in concert with tumour cells and ECs lining abnormal tumour blood vessels to promote angiogenesis and both local and systemic immunosuppression7 (FIGS 1–3).
Among various angiogenic molecules, VEGF is known to have diverse activities in the TME (BOX 2). In particular, the roles of VEGF in systemic and local immunosuppression have been extensively investigated in tumour models7,15,21. Excessive levels of VEGF in the TME induce tumour-associated immunosuppression via at least four distinct mechanisms (FIG. 1; Supplementary Table S4). First, increased VEGF directly inhibits CTL trafficking, proliferation, and effector function22. Second, VEGF inhibits DC maturation and antigen presentation, thus hampering T cell activation and, consequently, diminishing the T cell-mediated anticancer immune response23,24. Third, high levels of VEGF promote the recruitment and proliferation of immunosuppressive cells, including Treg cells, MDSCs, and pro-tumour, M2-like TAMs25,26. Fourth, VEGF promotes angiogenesis that results in an aberrant tumour vasculature, leading to hypoxia and a low pH in the TME, which in turn fosters immunosuppression both locally (FIG. 2) and systemically (FIG. 3), as discussed above21,27,28.
Box 2. The diverse functions of VEGF signalling in the tumour microenvironment.
The VEGF family of growth factors includes VEGFA, VEGFB, VEGFC, VEGFD, and PlGF135. VEGFA (referred to as VEGF) signals mainly through VEGF receptor 2 (VEGFR2; also known as KDR)9. VEGF, which was initially identified as a vascular permeability factor136, was cloned in 1989 and subsequently established as a crucial survival factor and mitogen for endothelial cells (ECs)137, and as the central regulator of angiogenesis in physiological and pathological contexts136,138,139. Both cancer cells and stromal cells in the tumour microenvironment (TME) can produce VEGF140. ECs and tumour cells respond to autocrine and paracrine VEGF signalling141, and other cell types present in the TME, such as immune cells, can also express VEGFRs9,142–147 (Supplementary Table S4). Indeed, VEGF not only modulates the expression of adhesion molecules on ECs, thereby regulating leukocyte recruitment, but also functions as a chemokine for subsets of immune cells and thus guides their recruitment to, and regulates their activity in, the TME. In general, VEGF signalling has been implicated in fostering immunosuppression, both systemically and in the local TME15 (FIGS 1,2,3; Supplementary Table S4). Thus, VEGF signalling has diverse functions, all of which might influence the biological and clinical outcomes of antiangiogenic therapies, and should therefore be considered in efforts to optimize the use of such agents.
In contrast to VEGF, the immunomodulatory roles of ANG2, which is another key regulator of angiogenesis (BOX 3), are not well understood. Nevertheless, in experimental models, activated ANG2 signalling has been shown to promote immunosuppression in tumours through multiple mechanisms (Supplementary Table S4). First, ANG2 increases leukocyte–endothelial interactions via upregulation of adhesion molecules, thereby facilitating the recruitment of MDSCs29, Treg cells30, and TIE2-expressing monocytes that induce immunosuppression31. Second, ANG2 disrupts EC–pericyte contacts, thus facilitating the migration of immune cells out of the vasculature and into the TME32,33. Finally, ANG2 also modulates the function of monocytes by suppressing the secretion of tumour necrosis factor (TNF), thus restricting their anticancer activity34.
Box 3. The angiopoietin–TIE2 pathway.
Angiopoietin 1 (ANG1) and ANG2 are key angiogenic cytokines that signal through the ANG1 and ANG2 receptor (TIE2), which is expressed primarily on vascular endothelial cells (ECs). ANG1 is expressed by pericytes148 and maintains the stability of quiescent mature vessels through its agonistic effects on TIE2 by promoting cell–cell adhesion between ECs149,150, desensitizing ECs to apoptotic signals151–153, maintaining pericyte–EC contacts154, and downregulating ANG2 expression by ECs150. The crucial role of pericyte-derived ANG1 is underlined by the fact that this cytokine, when overexpressed in transgenic mouse models, could restore the functional integrity of leaky blood vessels that were induced by transgenic overexpression of VEGF in non-tumour-bearing mice155.
ANG2 is released by ECs upon their activation156, in the course of inflammation157 and in response to hypoxia158. However, tumour cells can be an alternative source of ANG2 (REFS 159–161). ANG2 can be either an agonistic or an antagonistic ligand of TIE2 in a context-dependent manner: in mice, the antagonistic effects of ANG2 on TIE2 can destabilize quiescent vessels162, enabling VEGF-induced angiogenesis to occur163, although ANG2 has also been shown to act as a partial TIE2 agonist in mouse tumour models164. Finally, ANG2 has also been implicated as a crucial regulator of the development and quiescence of lymphatic vessels162.
Experimental data indicate that VEGF and ANG2 might cooperate to induce tumour immunosuppression and that this effect might be dependent on the relative levels of these cytokines in the TME35–37. Importantly, however, preclinical evidence indicates that high-dose and/or long-term antiangiogenic therapy causes massive vessel pruning and increases immunosuppression in tumour models37–40, suggesting that an optimum level of VEGF and ANG2 blockade will be needed to alleviate immunosuppression in the TME. Interestingly, emerging data suggest that immunosuppression, in turn, contributes to resistance to antiangiogenesis treatments37–39,41.
Metastasis
As outlined above, overexpression of angiogenic factors in the TME might facilitate the metastatic process by causing structural and biochemical abnormality of tumour blood and lymphatic vessels. In fact, targeting of VEGF with bevacizumab has been shown to reduce the incidence of brain metastasis in a mouse model of non-small-cell lung cancer (NSCLC), and when combined with carboplatin and paclitaxel, is associated with a reduced frequency of brain metastases as a site of first relapse in patients with this disease42. Similarly, treatment with an antibody targeting VEGF receptor 2 (VEGFR2) increased the efficacy of HER2-targeted therapies against established brain metastases from HER2-positive breast cancer in a mouse xenograft model43. In animal models, VEGF also exerts pro-metastatic effects through activation of the calcineurin–nuclear factor of activated T cells (NFAT) pathway via upregulation of ANG2 expression in ECs44. In line with these preclinical data, high levels of ANG2 expression determined by transcriptomic analysis of tumour samples correlate with an increased metastatic burden in patients with breast cancer45.
Mechanistically, ANG2 might facilitate the metastatic process indirectly by disrupting pericyte attachment to the endothelial wall, structurally and functionally compromising the vasculature, and thereby causing hypoxia46. Hypoxia in the TME leads to cytoskeletal changes in tumour cells that are essential for migration and invasion and thus the metastatic process. The lack of pericyte fortification might also facilitate intravasation of tumour cells into blood and lymphatic vessels. Indeed, transgenic mice with EC-specific overexpression of ANG2 have decreased endothelial integrity, resulting in increased metastatic foci formation in the Lewis lung carcinoma model32. Interestingly, metastatic dissemination could be prevented by treatment of the mice with an ANG2-neutralizing antibody32. In other mouse tumour models, anti-ANG2 therapy has been shown to prevent metastasis through the inhibition of tumour cell dissemination to tumour-draining lymph nodes and distant tissues47,48, and has therapeutic inhibitory effects on the growth of established metastatic lesions47. Similarly, inhibition of vascular endothelial protein tyrosine phosphatase (VEPTP; also known as PTPRβ), which sustains endogenous activation of TIE2, also inhibited extravasation of metastatic breast cancer cells into the lungs and subsequent metastatic foci formation, and thus extended survival durations, in animal models of mammary cancer when added to adjuvant doxorubicin chemotherapy49. These findings are in line with those of studies in which a high level of ANG2 pathway activation led to lymph node invasion and metastasis in breast cancer models (Supplementary Table S5), and of studies showing that high levels of ANG2 expression in patients with breast cancer correlate with the presence of metastatic disease50–52.
The key role of VEGF and ANG2 in promoting metastasis is further illustrated by studies using models of resistance to anti-VEGF therapy. In this setting, ANG2 promoted the upregulation of endothelial CCL2 and ICAM1 expression in metastases and, in turn, promoted the recruitment and adhesion of tumour-promoting TIE2− CCR2+ macrophages53. Dual VEGF–ANG2 inhibition was also more efficacious in breast cancer models than anti-VEGF therapy alone, with a reduce frequency of metastatic dissemination and thus decreased formation of micrometastatic nodules54. Of note, while anti-VEGF therapy in combination with chemotherapy has been associated with improved patient survival in the metastatic setting across a number of solid tumour types, such treatment was found to be inefficacious in all phase III trials performed in the adjuvant setting to date6. This disparity is, in part, attributed to VEGF-independent nonangiogenic mechanisms of vascularization (such as vessel co-option) that are exploited by cancer cells at the early stages of metastasis formation6,9,55,56. By contrast, ICB has shown promise in the adjuvant setting in patients with melanoma or NSCLC57–59. Thus, a better understanding of the interactions between VEGF and ANG2 signalling might offer novel strategies to prevent and treat metastasis and increase the effectiveness of other therapeutic approaches such as immunotherapy, including ICB.
Strategies to normalize tumour vessels
Multiple molecular and physical mechanisms contribute to vascular dysfunction in tumours7,60; chief among them is the imbalance of signalling mediated by proangiogenic versus antiangiogenic molecules28. In nonmalignant tissues, this balance is exquisitely maintained to ensure the normal morphology and function of blood vessels. During carcinogenesis, however, this balance typically tips towards angiogenesis, and the vessels become chronically immature and abnormal. In 2001, we proposed to restore this balance using judicious doses of antiangiogenic agents and suggested that this strategy would transiently give rise to tumour vessels with structural and functional phenotypes closer to those of the vessels of nonmalignant tissues — a state we referred to as ‘normalized’ (REF. 61), because the vessels would not be perfectly normal (BOX 1). We also suggested that the use of other therapies during this ‘window of normalization’ (REF. 61) would result in better outcomes than if the same agents were given before or after this window.
We initially tested our vascular normalization hypothesis in mice using agents that target the central regulator of angiogenesis: VEGF (BOX 2). Our work provided compelling evidence in support of this hypothesis. For example, radiation therapy given during the window of normalization, especially when hypoxia was alleviated in tumours, had outcomes superior to those achieved when radiation was delivered outside this window62. A number of preclinical and clinical studies from our own institution and others have also provided evidence in support of the validity of this hypothesis for a number of malignancies16,63. Our initial efforts focused on cytotoxic therapies64, but we subsequently showed in preclinical studies that vascular-normalizing doses of anti-VEGF agents also convert the immunosuppressive TME to an immunosupportive one and improve the outcome of vaccine-based anticancer immunotherapy27. The findings of multiple other groups provide further evidence of these effects in preclinical models using different anti-VEGF agents, as well as different immunotherapies7,13 (TABLE 1), and such combinations are currently being tested in a number of clinical trials (TABLE 2). In fact, the results of a phase II trial demonstrate the benefit of such an approach for patients with advanced-stage renal cell carcinoma (RCC)65, and the results of a follow-up phase III trial comparing anti-PD-1 therapy plus axitinib with standard sunitinib monotherapy in the first-line treatment of metastatic RCC are forthcoming (TABLE 2). While RCC is a highly VEGF-dependent tumour, these early results point to the multiple roles of VEGF pathway inhibition in anticancer immunity.
Table 1.
Preclinical studies testing combinations of antiangiogenic agents and immunotherapies
| Immunotherapy | Antiangiogenic therapy | Tumour model | Key resultsa | Refs |
|---|---|---|---|---|
| Vaccination studies | ||||
| Tumour-antigen-specific picornaviral vaccination | Aflibercept (VEGF trap) | Glioblastomab,c | Aflibercept normalizes tumour vasculature (reduces vessel permeability and diameter) Trend towards ↑ CD8+ T cells Tumour progression ↓ and animal survival ↑ | 173 |
| Whole tumour cell vaccine (mitomycin treated and GM-CSF secreting) | DC101 (anti-mouse VEGFR2 mAb) | Breast cancerb,c,d | CD4+ and CD8+ T cells ↑ MDSCs and Treg cells ↓ Tumour regression and animal survival ↑ | 27,174 |
| Whole tumour cell vaccine (GM-CSF secreting) | Adenoviral-based expression of sVEGFR1 and sVEGFR2 | Colon cancerc,dMelanomab,c | CD4+ and CD8+ T cells ↑ MDSCs and Treg cells ↓ Animal survival ↑ | 175 |
| Pox virus vaccine expressing CEA and three co-stimulatory molecules | Sorafenib (VEGFR TKI) | Colon cancerc,d | Intratumoural CD8+ T cells ↑ and Treg cells ↓ MDSCsf and Treg cellsf ↓ Tumour volume ↓ Animal survival ↑ | 176 |
| HER2-derived B cell epitope vaccine | VEGF peptide mimic (associated with antagonistic effects) | Breast cancerc | Anticancer and antiangiogenic effects ↑ Tumour growth ↓ by combined immunization with HER2 and VEGF peptides | 177 |
| Whole tumour cell vaccine (irradiated) and CD86–IgG fusion protein | SU 6668 (VEGFR TKI) | Breast cancerc,d | CD8+ T cells ↑ (by immunohistochemistry only) Tumour growth ↓ | 178 |
| Cell therapy studies | ||||
| Anti-PMEL T cells, PMEL vaccine, and IL-2 | DC101 (anti-VEGFR2 mAb) and B20 (anti-VEGF mAb) | Melanomab,c | Immune cell infiltration ↑ Tumour growth ↓ Animal survival ↑ | 96 |
| Anti-VEGFR1 chimeric antigen receptor T cells | NA | Lung cancerd,e | Endothelial tube formation in vitro ↓ Tumour growth and metastasis ↓ | 179 |
| Tumour-associated peptide-pulsed DCs | Anti-VEGF mAb | Sarcomac,d | DC numbers and functionf ↑ Tumour growth ↓ | 180 |
| Immune checkpoint blockade studies | ||||
| Anti-PD-1 mAb (clone RMPI-14; 10 mg/kg three times per week) | Vanucizumab | Breast cancerb,c Melanomab,c Pancreatic cancerb,c Neuroendocrine cancerb,c | Tumour growth ↓ Animal survival ↑ | 16 |
| Anti-PD-1 mAb (clone RMPI-14; 0.25 mg every other day) | DC101 | Colon cancerc,d | Angiogenesis ↓ T cell infiltration ↑ Cytokine expression ↑ | 181 |
| Anti-PD-1 mAb (clone RMP1-14; 0.25 mg twice per week) | Sunitinib (VEGFR TKI) | Colon cancerc,d | PD-1+CD8+ T cells ↓ Anticancer activity ↑ | 22 |
| Anti-PD-L1 mAb (clone 10F.9G2; 10 mg/kg twice per week) | DC101 | Pancreatic cancerb,c Breast cancerb,c Glioblastomab,c | IFNγ-expressing CD8+ and IFNγ-expressing CD4+ T cells ↑ PD-L1 expression on relapsing tumour cells ↑ Vessel normalization ↑ by PD-L1 blockade and formation of HEVs ↑ via LTβR | 98 |
| Anti-PD-L1 antibody (10F.9G2; 5 mg/kg twice per week) | CVX-060 (ANG2-specific peptide-antibody fusion protein) ± sunitinib or regorafenib (both VEFGR TKIs) or CVX-241 (bi-specific ANG2-VEGF-binding peptide-antibody fusion protein) | Multiple orthotopic human tumour xenograftsb,e and syngeneic mouse tumour modelsb,c | Tumour growth and metastatic progression ↓ with combined inhibition of ANG2 and VEGF signalling (with or without immune-checkpoint blockade) | 90 |
Table 2.
Clinical studies testing combinations of antiangiogenic agents and immunotherapies
| Immunotherapy | Antiangiogenictherapy | Disease | Status and results (end points) | CliniclTrials.gov identifier(publication) |
|---|---|---|---|---|
| Vaccination studies | ||||
| HLA-A24-restricted peptide vaccines derived from VEGFR1, VEGFR2, URLC10, TTK, or CDCA1 | NA | NSCLC | Phase I: completed; 13 of 15 patients had T cell responses to at least one peptide, 7 had SD for ≥2 months, and 2-year OS was 32.8% A strong CTL response and serum CRP level <1.0 were predictive of clinical response | NCT00633724, NCT00874588 (182) |
| OTS11101 (peptide vaccine targeting VEGFR1) | NA | Solid tumours | Phase I: completed; 5 of 9 patients had SD Serum sVEGFR1, sVEGFR2, IL-8, PECAM1, and VEGF levels increased 1 month after vaccination T cell activation and immune response-related gene expression were increased | NA (183) |
| Pox virus vaccine (expressing GM-CSF) | Sorafenib (VEGFR TKI) | HCC | Phase III: recruiting (ORR, PFS, OS, and AEs) | NCT02562755 |
| PF-06755990 (vaccine) | Sunitinib (VEGFR TKI) | Prostate cancer | Phase I: recruiting (DLTs) | NCT02616185 |
| Cell therapy studies | ||||
| CD8+ anti-VEGFR2 CAR T cells | NA | Metastatic cancer (melanoma and RCC) | Phase I/II: completed; 1 of 24 patients had a PR and 1 patient had SD | NCT01218867 |
| NK cell-based immunotherapy | Bevacizumab (anti-VEGF mAb) | Advanced-stage solid tumours | Phase I/II: recruiting (ORR, PFS, and OS) | NCT02857920 |
| Autologous DC immunotherapy | Sunitinib | Advanced-stage RCC | Phase II: completed; clinical benefit in 13 of 21 patients (9 PRs and 4 with SD) Phase III: active, not recruiting | NCT00678119 (65) NCT01582672 |
| Immune-checkpoint blockade studies | ||||
| Ipilimumab (anti-CTLA-4 mAb; 3 or 10 mg/kg) | Bevacizumab (7.5 or 15 mg/kg) | Advanced-stage melanoma | Phase I: 8 of 46 patients had a PR, and 22 had SD Increased numbers of tumour CD8+ T cells and CD163+ dendritic macrophages, and increased numbers of circulating memory T cells with combination versus ipilimumab alone | NA (184) |
| Durvalumab (anti-PD-L1 mAb) | Bevacizumab | Glioblastoma | Phase II: active, not recruiting (PFS, OS, and AEs) | NCT02336165 |
| Pembrolizumab (anti-PD-1 mAb) | Bevacizumab | Glioblastoma | Phase II: active, not recruiting (RP2D and/or MTD of combination, and PFS) | NCT02337491 |
| Nivolumab (anti-PD-1 mAb) | Bevacizumab | NSCLC | Phase I: active, not recruiting (safety and tolerability, ORR, and RFS) | NCT01454102 (185) |
| Ipilimumab or nivolumab | Bevacizumab (combined with ipilimumab only) | Metastatic melanoma | Phase NA: complete High pre-treatment serum ANG2 associated with worse OS Ipilimumab increased serum ANG2 levels and ipilimumab plus bevacizumab decreased serum ANG2 levels | NA (68) |
| MOXR0916 (agonistic anti-TNFRSF4 mAb) + atezolizumab (anti-PD-L1 mAb) | Bevacizumab | Advanced-stage solid tumours | Phase I: active, not recruiting (AEs, DLTs, and ORR) | NCT02410512 |
| Tremelimumab (anti-CTLA-4 mAb) or durvalumab | Bevacizumab | Resectable CRC liver metastases | Phase I: recruiting (feasibility and RFS) | NCT02754856 |
| Ipilimumab | Cabozantinib (VEGFR TKI) | Metastatic genitourinary tumours | Phase I: recruiting (AEs, RP2D, ORR, and PD-L1 and MET expression) | NCT02496208 |
| Atezolizumab (anti-PD-L1 mAb) | Vanucizumab (bi-specific mAb targeting VEGF and ANG2) | Advanced-stage solid tumours | Phase I: active, not recruiting (MTD, AEs, and ORR) | NCT01688206 |
| RO7009789 (agonistic anti-CD40 mAb) | Vanucizumab | Metastatic solid tumours | Phase I: recruiting (safety, pharmacokinetics and pharmacodynamics, and therapeutic activity) | NCT02665416 |
| Atezolizumab | Bevacizumab | Advanced-stage solid tumours (gastric cancer, pancreatic cancer, HCC, and oesophageal cancer) | Phase I: recruiting (safety and tolerability) | NCT02715531 |
| SHR-1210 (anti-PD-1 mAb) | Apatinib (VEGFR TKI) | Gastric cancer and HCC | Phase I/II: recruiting (tumour control rate, disease control rate, OS, and AEs) | NCT02942329 |
| Pembrolizumab | Nintedanib (VEGFR TKI) | Advanced-stage solid tumours | Phase I: recruiting (MTD and DLTs) | NCT02856425 |
| PDR001 (anti-PD-1 mAb) | Regorafenib (VEGFR TKI) | Metastatic CRC | Phase I: recruiting (DLTs and ORR) | NCT03081494 |
| Biomarker study | Sunitinib | Metastatic RCC | Decreased number and function of MDSCs and Treg cells and increased IFNγ production by T cells compared with baseline measurements | NA (186) |
| Nivolumab | Sunitinib or pazopanib (VEGFR TKI) | Metastatic RCC | Phase I: active, not recruiting; 11 out of 33 patients had SD, and grade 3–4 AEs were observed in 20–24% of patients Phase II: recruiting (PFS, OS, and AEs) | NCT01472081 (187) NCT02959554 |
| Immunostimulatory cytokine studies | ||||
| IFNα | Bevacizumab | Metastatic RCC | Phase III (CALGB 90206): completed; among 732 patients, median OS was 18.3 months with IFNα + bevacizumab versus 17.4 months with IFNα alone (P = 0.069) | NA (188) |
| IFNα2A | Bevacizumab | Metastatic RCC | Phase III (AVOREN): completed; in 649 patients, median OS was 23.3 months with IFNα2A + bevacizumab versus 21.3 months with IFNα2A alone (P = 0.1291) | NA (189) |
A major challenge with anti-VEGF therapies is that only transient vascular normalization has been observed62; the window of normalization might be of short duration (days to weeks) and is dependent on the tumour and dose of anti-VEGF agent used27. In our attempts to extend the normalization window in experimental models of cancer, we discovered that ANG1/ANG2–TIE2 signalling (BOX 3) mediates the recruitment of pericytes to the tumour vessels62, and that ectopic expression of ANG2 can abrogate the benefit of anti-VEGF therapy66. Moreover, concomitant blockade of ANG2 and VEGF extends both the window of normalization and the survival benefit compared with inhibition of either pathway alone, in part, by reprogramming the immunosuppressive TME35,36. Interestingly, in patients with glioblastoma, circulating ANG2 levels rebound after a marked initial decrease following treatment with a VEGF pathway inhibitor67. Perhaps most pertinent to immunotherapy, ANG2 levels have been reported to be elevated or increased in patients with melanoma who have an unfavourable response to ICB68. Importantly, ANG2 levels were predictive of treatment response to ICB with or without anti-VEGF therapy68. A number of lessons can be learnt from the findings of these and other preclinical and clinical studies reported to date.
The dose of anti-VEGF agents matters
Preclinical data indicate that anti-VEGF therapy results in vascular normalization via pruning of immature blood vessels, which have poor pericyte coverage, and via active pericyte recruitment62,69. Similar vascular changes have also been observed using advanced MRI techniques in patients with glioblastoma after VEGFR tyrosine kinase inhibitor (TKI) treatment63,67. The onset of vessel normalization conferred by anti-VEGF treatment can occur in as little as 1 day in both mouse62 and human tumours67. The normalization process is transient and reversible, in a tumour-dependent and dose-dependent manner, and lasts for around 1 week in mice and possibly for a few months in humans7,62,63,67. The window of vessel normalization ends when an excessive number of vessels are pruned, or when tumours switch to the use of alternative angiogenic pathways or alternative mechanisms of blood vessel recruitment, such as vessel co-option56,62. Notably, high doses of antiangiogenic agents can result in a short normalization window by causing excessive amounts of vessel pruning, thus exacerbating hypoxia and acidosis in the TME7. High doses of anti-VEGF agents can also lead to increased deposition of extracellular matrix that, together with hypoxia, can promote the infiltration of immunosuppressive and/or pro-tumour immune cells, such as monocytic and granulocytic MDSCs37–39. These results suggest that high-dose anti-VEGF therapy limits the benefits of chemotherapy in mouse models and in patients with metastatic colorectal cancer (CRC) or advanced-stage NSCLC37,70,71. Similarly, blockade of VEGF signalling using high doses of the multi-target, pan-VEGFR TKI sorafenib caused increased hypoxia and the stromal cell-derived factor 1 (SDF1)-mediated recruitment of immunosuppressive cells, such as Treg cells and M2-like macrophages, in a mouse model of advanced-stage hepatocellular carcinoma (HCC); blockade of the SDF1 receptor C-X-C-chemokine receptor 4 (CXCR4) prevented this shift towards an immunosuppressive TME, inhibited tumour growth and metastasis, and improved the survival of sorafenib-treated mice40. In mouse models of CRC, high-dose anti-VEGF treatment can also result in the recruitment of immunosuppressive, nonclassical, lymphocyte antigen 6C (Ly6C)low monocytes that also confer resistance to anti-VEGF therapy, thus setting up a vicious cycle38,39.
By contrast, use of lower doses of antiangiogenic agents — for instance, as low as one-quarter of the doses that induce antivascular effects and/or vessel pruning in animal studies — has the potential to induce prolonged vessel normalization7,27. Achieving vascular normalization, and thereby reducing tumour hypoxia and acidosis, is crucial because these stressors confer cancer cells with an invasive metastatic phenotype72. Moreover, a normalized vasculature provides a conduit for the efficient infiltration of immune cells and delivery of anticancer therapeutics into tumours6,27. Importantly, restoring tumour oxygenation and a physiological pH might improve the anticancer activity of infiltrating immune cells27, as well as that of radiation therapy and many drugs7 (FIGS 2,4). Treatment-resistant cancer stem cells are hypothesized to reside in the hypoxic niche, and excessive antiangiogenic therapy can cause hypoxia and thereby support these cells73. Therefore, low doses of antiangiogenic agents might also reduce the number of these stem cells, hence avoiding or delaying therapy resistance. In fact, the results of two retrospective studies have shown that low-dose bevacizumab (<3.6 mg/kg per week) in combination with chemoradiotherapy confers a greater survival benefit than high-dose bevacizumab (5 mg/kg per week) in patients with glioblastoma74–77. Hence, to realize the full potential of antiangiogenic therapy, the dose and schedule of anti-VEGF agents might need to be tailored to baseline levels of tumour vascularity78 and/or VEGF. Notably, pre-treatment levels of circulating VEGF are prognostic for treatment outcome in different solid tumour settings, but are not predictive of anti-VEGF therapy outcome79. Whether pre-treatment VEGF levels are predictive in the context of personalized bevacizumab dosing regimens is unknown. This hypothesis needs to be tested in prospective studies using low-dose antiangiogenic therapy.
Figure 4. Vascular-normalizing therapies can reprogramme the immunosuppressive tumour microenvironment to an immunosupportive one.
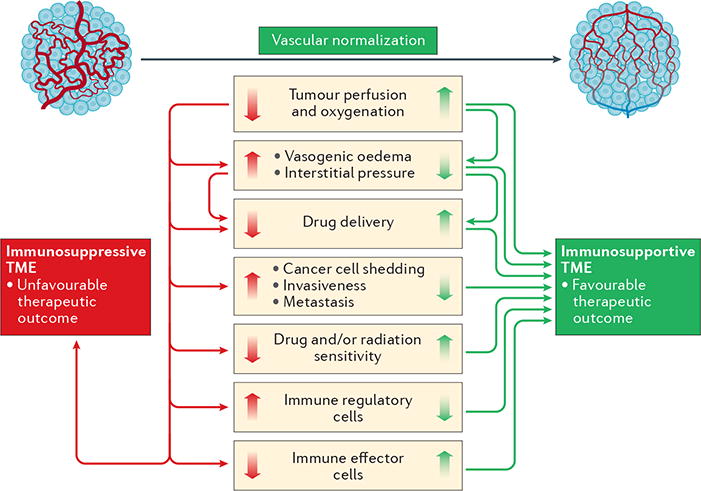
The structural and functional abnormalities of tumour blood vessels lead to impaired blood flow and perfusion, thus resulting in a hypoxic tumour microenvironment (TME). Hypoxic conditions in tumours limit the effectiveness of cytotoxic therapies, such as radiation therapy and certain chemotherapies. In addition, drug delivery to the tumour via these abnormal vessels is impaired, and this problem is further exacerbated by the high interstitial fluid pressure — a consequence of the leaky blood vessels and irregular lymphatic system in the tumour. Moreover, abnormal and leaky tumour blood vessels facilitate the intravasation of cancer cells into the systemic circulation, promoting metastasis. Furthermore, the abnormal TME is associated with increased infiltration of immunosuppressive regulatory T cells and polarizes tumour-associated macrophages from an anticancer M1-like phenotype towards an immunosuppressive M2 phenotype. The dysfunctional vasculature also restricts the accumulation and homogeneous intratumoural distribution of effector T cells. Consequently, anticancer immune responses are severely impaired. Vascular normalization could potentially prevent or reverse many of these adverse effects in patients, enhance the delivery and effectiveness of chemotherapy and immunotherapy, and improve the anticancer effects of radiotherapy.
ANG2 might confer resistance to anti-VEGF therapy
Similar to other anticancer drugs (such as molecularly targeted agents), tumours inevitably develop resistance to anti-VEGF agents, and one key mediator of resistance is the hypoxia induced by exposure to these drugs7. Several cellular and molecular mechanisms of escape from antiangiogenic therapies have been postulated and investigated, including activation of alternative angiogenic pathways, recruitment of proangiogenic myeloid cells, increased pericyte cell coverage of tumour blood vessels, increased tumour cell invasion, and alternative modes of vessel recruitment7,9,80.
Herein, we focus on activation of the ANG2–TIE2 axis as an escape mechanism from anti-VEGF treatment for the following reasons. First, ANG1 mediates vascular normalization in preclinical models62, whereas overexpression of ANG2 can compromise the benefit of VEGF pathway inhibition by abrogating vessel normalization66. Second, ANG2 is co-expressed with VEGF in many human solid tumours, such as glioblastoma35, HCC81, and CRC82. Third, circulating levels of ANG2 transiently decrease during the window of normalization and return back to baseline levels as the tumours progress after anti-VEGF therapy in patients with glioblastoma67. Fourth, tumour-derived VEGF can increase the expression of ANG2 in ECs, which results in activation of the endothelium and thus promotes lung metastasis in mouse models, suggesting that an interaction between these angiogenic pathways promotes tumour progression44. Finally, ANG2 is a candidate predictive and/or prognostic marker of therapeutic responsiveness to anticancer treatments, including immunotherapy using immune-checkpoint inhibitors, as discussed below (BOX 4; Supplementary Table S6).
Box 4. VEGF and ANG2 as prognostic and/or predictive biomarkers.
No validated predictive biomarkers of a response to therapy targeting the VEGF signalling pathway have been identified in patients with cancer7,77. Angiopoietin 2 (ANG2) is one potential biomarker of resistance to VEGF-targeted therapy. The results of some clinical studies using the VEGFA-neutralizing antibody bevacizumab have shown increased levels of circulating ANG2 after treatment165, whereas others have shown that serum ANG2 levels remain stable throughout the course of bevacizumab treatment166. In patients with ovarian cancer, however, a decrease in circulating ANG2 levels in response to VEGF pathway inhibition with sunitinib is correlated with a trend towards longer progression-free survival167. Moreover, we demonstrated in patients with glioblastoma that treatment with the pan-VEGF receptor tyrosine kinase inhibitor cediranib transiently decreases levels of plasma ANG2 (REF. 67). Nevertheless, a paucity of data exists on the kinetics of circulating ANG2 levels and tumour ANG2 expression in response to anti-VEGF therapy.
ANG2 needs to be explored as a potential predictive and/or prognostic biomarker because high levels of ANG2 in the tumour or in the circulation are correlated with unfavourable patient survival across multiple cancer types, including glioblastoma166, neuroendocrine tumours159, ovarian cancer160, metastatic melanoma161, breast cancer45, hepatocellular carcinoma168, pancreatic ductal carcinoma169, and gastric carcinoma170 (Supplementary Table S6). Moreover, high levels of circulating ANG2 are associated with unfavourable outcomes of bevacizumab-based therapy in colorectal carcinoma171. Notably, a study in patients with metastatic melanoma treated with immune-checkpoint inhibitors revealed that high levels of circulating ANG2 correlate with an unfavourable treatment response and prognosis, possibly reflecting hypoxia and immunosuppression in the tumour microenvironment68.
Blocking both ANG1 and ANG2 might not be of benefit to patients
Therapeutic agents that target the ANG2–TIE2 axis can lead to tumour regression in a number of subcutaneous and orthotopic mouse models (Supplementary Table S7); many of these agents have been advanced to testing in clinical trials (Supplementary Table S8). Notably, however, the results of clinical studies targeting both ANG1 and ANG2 using the bi-specific peptibody trebananib have been disappointing. In a phase II trial involving patients with CRC83, trebananib combined with 5-fluorouracil, leucovorin, and irinotecan (FOLFIRI) chemotherapy did not significantly improve the estimated median progression-free survival (PFS; HR 1.23, 95% CI 0.81–1.86; P = 0.33) or overall survival durations (HR 0.90, 95% CI 0.53–1.54; P = 0.70) compared with FOLFIRI plus placebo. Similarly, the addition of trebananib to cisplatin and capecitabine chemotherapy failed to prolong the estimated median PFS duration in patients with metastatic gastroesophageal cancer (HR 0.98, 95% CI 0.67–1.43; P = 0.92) in a randomized, double-blind, placebo-controlled phase II study84. In a different phase II trial85, trebananib in combination with sorafenib failed to improve the PFS of patients with metastatic clear cell RCC when compared with sorafenib plus placebo (HR 0.88, 95% CI 0.60–1.30; P = 0.52). Interestingly, in the phase III TRINOVA-1 trial of trebananib plus paclitaxel in women with recurrent ovarian cancer86, only patients with ascites at baseline had a statistically significant improvement in overall survival (14.5 months versus 12.3 months with paclitaxel plus placebo; HR 0.72, 95% CI 0.55–0.93; P = 0.011). PFS was also significantly improved in the overall intent-to-treat population with the addition of trebananib to chemotherapy (12.5 months versus 10.9 months; HR 0.85, 95% CI 0.74–0.98; P = 0.024), but overall survival was not improved (19.3 months versus 18.3 months; HR 0.95, 95% CI 0.81–1.11; P = 0.52). The benefits observed in the context of ascites point to possible vessel-normalizing effects of trebananib that might improve chemotherapy. On the other hand, the failure of trebananib to improve the PFS of patients with ovarian cancer when combined with pegylated liposomal doxorubicin in the phase III TRINOVA 2 trial87, as well as the results of the aforementioned phase II studies of this agent83–85, suggests that the vessel-normalizing effects of ANG1–ANG2 blockade are generally insufficient to improve patient outcomes. One possible reason for the clinical failure of dual ANG1–ANG2 blockade is that inhibition of ANG1, which is an agonistic ligand of TIE2 (BOX 3), might compromise the benefits of vascular normalization conferred by the blockade of the antagonistic TIE2 ligand ANG2 (REF. 88). Importantly, clinical trials of ANG2-specific inhibitors are currently ongoing (Supplementary Table S8).
Targeting both VEGF and ANG2 might increase vascular normalization and promote anticancer immunity
The complementary roles of VEGF and ANG2 in tumour angiogenesis have motivated a plethora of preclinical studies attempting to investigate concurrent blockade of both receptors for these cytokines in multiple tumour models (Supplementary Table S9). This dual blockade approach can provide tumour control in some preclinical models, although the degree of benefit and the underlying mechanism of action seem to be context-dependent31,32,80,89. For example, dual VEGF–ANG2 blockade prolonged survival compared with anti-VEGF or anti-ANG2 monotherapy in mouse models of glioblastoma35,36, melanoma16, pancreatic neuroendocrine tumours (PNETs)16, and metastatic16,90 or early stage (resected) breast cancer90. By contrast, a survival benefit was not observed in a model of metastatic RCC (RENCA) with VEGF–ANG2-neutralizing therapy90. Treatment with the pan-VEGFR TKI sunitinib in combination with an ANG2-neutralizing antibody, however, led to a decreased frequency of lung metastases from orthotopic RENCA tumours, possibly owing to inhibitory effects on multiple kinases with diverse activities90. Dual VEGF–ANG2 inhibition has also been shown to be more efficacious than anti-VEGF monotherapy in breast cancer models, as indicated by reduced metastatic dissemination and micrometastatic growth54. Taken together, these findings suggest that the benefit of dual antiangiogenic therapy is dependent on the tumour type (or model used), the stage of disease progression, and the specific characteristics of the TME. With regard to the nature of the TME, the therapeutic benefits of dual VEGF–ANG2 blockade seem to be conferred through the lymphoid (for example, in breast and colon tumour models16) and/or myeloid cell populations (in both central nervous system (CNS) and non-CNS tumour models16,35,36,90).
In the lymphoid compartment, the extravasation and perivascular accumulation of both CD4+ and CD8+ T cells increased in response to treatment with an anti-VEGF–ANG2 bispecific antibody (A2V) in genetically engineered and xenograft mouse models of metastatic breast cancer, PNETs, or melanoma16. As expected, T cell accumulation was confined to tumour areas with normalized vessels16. The overall number of T cells in the tumour mass, however, was unchanged16,35,36. Nevertheless, the anticancer activity — an important determinant of durable tumour control — of the T cells was improved, with increased production of IFNγ and increased expression of the activation marker CD69 on CTLs with this treatment compared with the results of monotherapy with neutralizing antibodies to either ANG2 or VEGF alone16.
In the myeloid compartment, reprogramming of pro-tumour, M2-like TAMs towards an anticancer M1 phenotype has been shown to be a major contributor to the therapeutic efficacy of dual antiangiogenic therapy targeting VEGF and ANG2, independent of the tumour model used16,35,36. Reprogramming of TAMs to an anticancer phenotype might be critical in patients with glioblastoma, in which macrophages constitute up to 40% of the tumour mass91. In preclinical glioblastoma models, macrophage depletion with an antibody against macrophage colony-stimulating factor 1 (CSF1) abrogated the benefits of dual VEGF–ANG2 inhibition, thus demonstrating the importance of TAMs in mediating the treatment effects36. Of note, CSF1 pathway inhibition is currently being pursued in clinical trials (NCT02526017, NCT02777710, and NCT02471716); caution should be taken if these agents should ever be combined with dual VEGF–ANG2 inhibition.
Currently, multiple clinical trials are being conducted to investigate dual inhibition of VEGF and ANG2 in patients with cancer (Supplementary Table S10). We anticipate that an extended window of vessel normalization and reprogramming of the TME towards an immunostimulatory phenotype will be achieved using this approach compared with standard-dose anti-VEGF therapy. We speculate that the effects of dual VEGF–ANG2 targeting could even be further improved through the judicious use of low doses of anti-VEGF agents75,92,93. Notably, a knowledge gap exists with respect to the dosing and scheduling (for example, simultaneous or sequential) of agents for dual targeting of VEGF and ANG2; careful dose selection for dual antiangiogenic treatments in clinical trials will be crucial for avoiding excessive pruning of vessels and for increasing the delivery of drugs to the tumour.
On the basis of evidence from multiple preclinical studies16,35,36, we anticipate that dual VEGF–ANG2 blockade alone will not enable durable clinical responses similar to those conferred in some patients by immune-checkpoint inhibitors. For example, in the phase II McCAVE study involving patients with untreated metastatic CRC (NCT02141295), addition of the bispecific anti-VEGF–ANG2 antibody vanucizumab to 5-fluorouracil, folinic acid, and oxaliplatin (FOLFOX) chemotherapy was not sufficient to prolong PFS (the primary end point of the trial) compared with FOLFOX plus bevacizumab. The failure to meet the primary end point of this clinical trial might be explained by the high doses of vanucizumab used (2,000 mg every 2 weeks); the use of lower doses might have provided a better therapeutic outcome over an extended period of time through the aforementioned mechanisms7. Moreover, emerging preclinical evidence suggests an upregulation of counter-regulatory pathways in response to vanucizumab treatment. Specifically, dual antiangiogenic therapy with the anti-VEGF–ANG2 antibody A2V led to upregulation of PD-L1 on ECs and tumour cells in multiple mouse tumour models, resulting in suppression of anticancer immunity16. Of note, this immune-evasive mechanism could be counteracted by concomitant treatment with an anti-PD-1 antibody, resulting in improved tumour control16.
The benefits of dual VEGF–ANG2 inhibition are likely to be tumour dependent. We hypothesize that dual VEGF–ANG2 blockade might be particularly effective against tumours of the CNS. Glioblastomas are particularly hypoxic, which poses a considerable challenge to therapy7,77, and normalizing brain tumour blood vessels for an extended period of time might help overcome this obstacle. In addition, the high abundance of macrophages in glioblastomas91 could potentially render these tumours particularly sensitive to dual VEGF–ANG2 inhibition owing to the ability of these treatments to reprogramme macrophages towards an anticancer phenotype, as observed in preclinical models35,36. Targeting nonredundant vessel normalization pathways by inhibiting the renin–angiotensin system is another way to potentially further extend the window of normalization94,95. In line with this hypothesis, the results of a retrospective study demonstrated that patients with recurrent glioblastoma who were treated with bevacizumab and used angiotensin receptor blockers (ARBs) had prolonged overall survival compared with those who did not use ARBs (HR 0.65, 95% CI 0.46–0.92; P = 0.016), including the subgroup treated with low-dose bevacizumab (area under the curve for bevacizumab <3.6 mg/kg per week)75. In summary, the available clinical data underscore that the successful translation of dual anti-VEGF–ANG2 therapy will require careful mechanistic studies and biomarker-driven clinical trial designs.
Normalization improves immunotherapy
Immunotherapy has rapidly transformed the anticancer treatment and drug development landscape; however, many patients do not benefit from this approach. The success of immunotherapy relies on the recruitment, expansion, and effective anticancer activity of immune cells, especially CTLs, in the TME.
The accumulation of T cells occurs via a multistep process. First, T cells must interact with adhesion molecules, such as E-selectin, ICAM1, and VCAM1, expressed on the luminal surface of ECs lining blood vessels and must then cross the endothelial barriers. Dual anti-VEGF–ANG2 therapy with A2V has been shown to upregulate the expression of adhesion molecules during the window of vascular normalization and can thereby facilitate the accumulation of anticancer T cells within multiple types of tumours in mice16. This finding is in line with the results of earlier preclinical studies that demonstrated increased tumour infiltration by T cells in response to low doses of an anti-VEGFR2 antibody — during the window of normalization27. In these models, antiangiogenic treatment several days before immunotherapy, by either vaccination or adoptive cell transfer approaches, increased the accumulation of T cells within the tumour and thus increased anticancer efficacy compared with vaccination therapy alone27,96. Anti-VEGF therapy is also known to induce DC maturation and thus inhibit the priming of T cells. Additionally, loss of VEGF in T cells results in vessel normalization and an improved chemotherapeutic response in mice97. Importantly, although loss of VEGF expression by T cells might potentiate anticancer responses through vascular normalization, this effect might be compromised by the decreased capacity of VEGF-deficient T cells to infiltrate tumours and thus control tumour growth97. Indeed, the aforementioned upregulation of PD-L1 expression on ECs, which renders PD-1-expressing CTLs dysfunctional, has been highlighted as a possible mechanism of resistance to dual VEGF–ANG2 blockade16. Similarly, upregulation of PD-L1 has been demonstrated on both ECs and tumour cells following treatment with anti-VEGFR2 therapy98. Thus, a strong rationale supports the combination of antiangiogenic agents and therapies that improve the functions of T cells in tumours. In fact, the findings of multiple preclinical studies provide evidence of the efficacy of combining vascular-normalizing agents with interventions that alleviate functional blockade of T cells, such as immune-checkpoint inhibitors (TABLE 1). For example, ICB with an anti-PD-1 antibody improved the degree of tumour control achieved with the anti-ANG2–VEGF antibody A2V in various cancer models16.
The combination of anti-VEGFR2 and anti-PD-L1 antibodies, however, did not improve survival in a glioblastoma model98. This lack of efficacy was attributed to a low incidence of high endothelial venules (HEVs) within these tumours, contrary to findings in models of breast cancer and PNETs, in which this combination was efficacious. HEVs are specialized vascular units, usually organized as tertiary lymphoid structures, which facilitate the recruitment and extravasation of naive T cells, and then promote the differentiation of these cells into CTLs. The ECs lining HEVs are known to express ICAM1 and VCAM1, and thus facilitate homing and migration of immune cells to tumours98. These findings are of particular importance because anti-PD-1 treatment alone conferred a survival benefit for only a small fraction of patients with glioblastoma99 and failed to improve the outcomes of patients with this disease in a randomized phase III trial100. Anti-PD-1 antibodies have also been shown to provide limited therapeutic benefit in a less immunogenic breast tumour model (MMTV-PyMT)16. Thus, treatments that can induce HEV formation and/or increase antigen load, which include radiation, chemotherapy, and cancer vaccines, might enhance the therapeutic benefits of ICB101–104. Notably, HEV formation in the aforementioned breast cancer and PNET models was found to be mediated by lymphotoxin-β receptor (LTβR) signalling, and treatment with an agonistic anti-LTβR antibody induced HEVs in the previously recalcitrant glioblastoma model, increased CTL activity, and thereby increased the efficacy of combined anti-VEGFR2 and anti-PD-L1 therapy98.
Interestingly, ICB of cytotoxic T lymphocyte protein 4 (CTLA-4) and PD-1 can normalize blood vessels in multiple models of transplanted breast and other tumours105. The vascular normalization effect observed in this study was attributed to increased CD4+ type 1 T helper (TH1) cell accumulation and anticancer activity105. However, this evidence was derived using CD4+ T cell-deficient animals or anti-CD4 antibody treatment before breast tumour inoculation; thus, the roles of Treg cells versus those of other populations of CD4+ T cells could not be separated. Hence, the effects of TH1 cells on vascular normalization in the context of ICB need to be further validated in other preclinical models and through comparisons of patient biopsy specimens obtained before and after ICB. Importantly, although the generalizability of the reciprocal regulation of tumour vessel normalization and immunostimulatory reprogramming is supported by analyses of large transcriptomic databases in human tumours, the experimental studies were limited to breast cancer models105. Whether ICB-mediated vessel normalization is induced across a range of tumour types remains to be determined. If found to be broadly applicable, this mechanism provides another rationale for the pursuit of combinatorial approaches using antiangiogenic agents and immune-checkpoint inhibitors, as these therapies target nonredundant pathways for vessel normalization and abrogation of immunosuppression in the TME.
Perspectives
Compelling preclinical and clinical evidence indicates that anti-VEGF therapy creates a transient window of vessel normalization during which the delivery of oxygen — a radiosensitizer and immunostimulator — and various therapeutic agents to tumours is improved7,62. The extent of normalization varies with tumour type, as well as with the duration of treatment and the dosage of drug, and closure of the normalization window is marked by the development of adaptive tumour resistance and immunosuppression7. Tumours might escape anti-VEGF therapies mainly through alternative modes of vascularization (such as vessel co-option) and/or upregulation of alternative angiogenic pathways (such as ANG2–TIE2 signalling). Thus, targeting both the VEGF and ANG2 pathways concomitantly increases the efficacy of antiangiogenic treatments beyond that of monotherapy targeting either of these cytokines in preclinical studies35,36. The treatment benefits observed with antiangiogenic therapy in animal models stem, in part, from restoration of anticancer immunity in the TME16, and this mechanism has formed the basis for combining antiangiogenic therapies with various immunotherapies, including tumour vaccines, chimeric antigen receptor (CAR) T cells, and immune-checkpoint inhibitors (TABLE 1). On the basis of positive preclinical data, these combinations are now being tested in multiple clinical trials, with promising outcomes reported for patients with RCC in a phase II trial65 (TABLE 2). However, these therapies have complex biological effects, and combining them is likely to further increase this complexity; an increased risk of toxicities, particularly immune-related adverse events such as autoimmune colitis5, might ensue. Here, we outline some of the challenges and opportunities that lie ahead, highlighting future research priorities.
The results of both preclinical and retrospective clinical studies have demonstrated that the dosage of agents targeting the VEGF pathway is a key consideration6,7. Historically, the clinical development of these agents has been predicated on the use of maximum tolerated doses until disease progression. High doses and/or a prolonged duration of anti-VEGF treatment are correlated with lower levels of tumour perfusion and elevated hypoxia27. The addition of agents that target the ANG2–TIE2 pathway to VEGF pathway blockade seems to extend the window of vascular normalization and convert an immunosuppressive micro-environment into an immunostimulatory milieu35,36; however, over time, tumours again become hypoxic and escape from this combined treatment approach via upregulation of PD-L1 on tumour cells and stromal cells of the TME, including ECs and immune cells98. Thus, a critical need exists to optimize the dosage, duration, and sequence of administration of antiangiogenic agents for use in combination with ICB.
ICB can have vessel-normalizing effects in preclinical models of breast tumours105, although the kinetics of vascular normalization are unknown. Obtaining this knowledge would facilitate efforts to optimally sequence the administration of immune-checkpoint inhibitors and antiangiogenic agents, in order to extend the window of normalization and, thus, the survival benefits106.
Of note, ICB is known to increase the risk of oedema in patients with brain tumours, sometimes resulting in death107,108. This observation contrasts with the vascular normalization induced by ICB in breast cancer models105 and suggests that the vascular effects are dependent on the tumour location and/or type — a possibility that needs to be further investigated. Nevertheless, anti-VEGF agents can reduce glioblastoma-associated brain oedema in mice and patients63,109, providing a strong rationale for combining low-dose anti-VEGF therapy with ICB and other forms of immunotherapy for this disease, and potentially also brain metastases.
ICBs are known to cause sometimes severe immune-related adverse events. These toxicities can often be resolved by discontinuing ICB therapy or reducing the doses of the immune-checkpoint inhibitors used5. Considering that vascular normalization can improve the delivery of therapeutic agents to tumours7, the proposed combination strategy might require lower doses of immune-checkpoint inhibitors to confer immunostimulation but also to reduce the risk of toxicity.
Neither anti-VEGF therapy nor ICB seem to be effective in highly desmoplastic tumours, such as pancreatic ductal adenocarcinoma, breast cancer, and cholangiocarcinoma7,110 — with the notable exception of desmoplastic melanomas, which have been shown to respond to ICB111. The reasons for this lack of responsiveness are currently unknown, although results from our laboratory have shown that the elevated levels of extracellular matrix molecules, including collagen I and hyaluronan, in such tumours can compress blood vessels, impairing their function and resulting in hypoxia94. We have also shown that widely prescribed ARBs can normalize the stroma and decompress blood vessels, thereby improving the outcome of chemotherapy94. Moreover, RNA sequencing analyses of tumour specimens from patients with pancreatic ductal adenocarcinoma have revealed that ARBs can activate both the innate and adaptive immune system112. Thus, the testing of combinations of ARBs with antiangiogenic agents113 and immune-checkpoint inhibitors95 is warranted.
Obesity is associated with an unfavourable prognosis across multiple types of cancer114. Notably, an increased severity of adverse effects has been observed in obese versus nonobese mice following immunotherapy115; however, the mechanisms underlying this increase are not understood. Given that obesity is an emerging public health problem worldwide, this relationship demands urgent investigation with regard to combined antiangiogenesis and immunotherapies.
We have focused on VEGF and angiopoietins as key targets for achieving vascular normalization; however, the duration and extent of normalization that can be achieved by specifically targeting these pathways might be limited. Certain molecularly targeted agents, such as the anti-HER2 antibody trastuzumab, and metronomic chemotherapy can also induce vascular normalization via upregulation of thrombospondin expression28,116,117. Moreover, endocrine therapy for hormone-dependent tumours can also reduce VEGF levels and normalize tumour vessels118. Similarly, novel approved drugs, including poly [ADP-ribose] polymerase inhibitors and cyclin-dependent kinase 4 (CDK4) and CDK6 inhibitors, might potentiate the effectiveness of immunotherapies, in part, by vascular normalization119. Novel vascular normalization strategies targeting these additional pathways should be further explored.
In addition to ICB, other immunotherapy approaches are also showing promise for the treatment of solid tumours120. Thus, the concepts discussed herein might also be applicable to CAR T cells and immunostimulatory or bi-functional antibodies in terms of improving their delivery and effectiveness.
A number of preclinical studies have demonstrated that cytotoxic therapies (radiation and chemotherapy) can be used to increase the neoantigen load of tumours that have a low baseline neoantigen load, which is a barrier to effective immunotherapy121. Again, by increasing the functionality of tumour vessels and fostering an immunosupportive TME (FIG. 4), vascular normalization approaches might not only improve the development of anticancer immunity, but also decrease the required doses of cytotoxic therapies and thus reduce the adverse effects of combinations incorporating immunotherapies.
The microbiota is emerging as a determinant of responses to various anticancer therapies, including immunotherapy122–125. Virtually nothing is known, however, about the role of the microbiome in antiangiogenic therapy and vice versa. Thus, exploring the interactions among the microbiota, immunotherapy, and antiangiogenic therapy might offer exciting opportunities to improve the survival of patients with cancer.
Finally, no validated biomarkers are available to guide the use of antiangiogenic drugs7,126. Similarly, biomarkers to evaluate responses to ICB, beyond MSI-H, dMMR, and PD-L1 positivity (which have suboptimal predictive utility), are urgently needed5. Optimally combining antiangiogenic therapy with ICB would require a concerted effort in this area owing to the costs and adverse effects of these treatments, which necessitate efforts to limit their use to patients who will derive clinical benefit.
Conclusions
In summary, immune-checkpoint inhibitors and antiangiogenic drugs are widely and increasingly prescribed, and are under continued developed in clinical trials, for the treatment of a range of solid tumours (Supplementary Table S1 and Supplementary Table S2). ICB can dramatically prolong the survival of patients who have durable responses, but has generally failed to improve the outcomes of the remaining majority of patients, at the cost of substantial toxicities in many recipients. Antiangiogenic drugs offer only a modest survival benefit of a few weeks to months, with rare durable responses. Herein, we have discussed the potential to rationally combine these two approaches in order to increase patient survival beyond that currently conferred by each approach individually. Despite the outstanding challenges, including the currently high costs and considerable risks of adverse effects associated with these treatment modalities, compelling evidence supports the combination of antiangiogenic and ICB therapies. The evidence indicates that the potential benefit of such combinations will be manifested though modulation of both the tumour vasculature and the tumour immune microenvironment. We await, with interest, the outcomes of ongoing clinical trials that are investigating whether the combination of antiangiogenic therapy with ICB will realize the full potential of this approach to improve the survival of patients with cancer. We also anticipate that the development of predictive biomarkers and orally available inhibitors of immune checkpoints, as well as competition among the companies manufacturing these agents, will help bring down the costs associated with these treatments in the future.
Supplementary Material
Supplementary Information
Acknowledgments
The work of the authors is supported in part by US National Institutes of Health (NIH) National Cancer Institute grants P01CA080124 (D.F, D.G.D., and R.K.J.), R01-CA096915 (D.F.), R01-CA159258 and R41-CA213678 (D.G.D.), and R01-CA129371, R01-CA208205, U01-CA 224348, and Outstanding Investigator Award R35-CA197743 and P50-CA165962 (R.K.J.). J.K. has received fellowships from the German Research Foundation (DFG) and the Solidar-Immun Foundation. Z.A. has received a fellowship from the Aid for Cancer Research Foundation and Tosteson Fund for Medical Discovery (FMD). R.K.J. has also received research funding from the Gates Foundation, the Ludwig Center at Harvard, the Lustgarten Foundation, and the National Foundation for Cancer Research. The authors thank M. Badeaux, M. Datta, Y. Huang, K. Jung, J. Martin, N. Wang, and C. Wong (who are all past or current fellows of the Edwin L. Steele Laboratories) for their critical reading of, and input into, this manuscript.
Footnotes
Competing interests
D.G.D. has received consultancy fees from Bayer and research funding from Bayer, Bristol-Myers Squibb, Exelixis, Leap, and Merrimack. R.K.J. has received consultancy fees from Merck, Ophthotech, Pfizer, SPARC, and SynDevRx; owns equity in Enlight, Ophthotech, SynDevRx, and XTuit; and serves on the board of directors of XTuit and the boards of trustees of Tekla Healthcare Investors, Tekla Life Sciences Investors, Tekla Healthcare Opportunities Fund, and Tekla World Healthcare Fund. D.F., Z.A., and J.K. do not have any conflicts of interest to declare.
Author contributions
J.K. and Z.A. contributed equally to the preparation of the manuscript. All authors made substantial contributions to researching data for the article, discussions of content, and the writing, reviewing, and editing of the manuscript.
SUPPLEMENTARY INFORMATION
See online article: S1 (table) | S2 (table) | S3 (table) | S4 (table) | S5 (table) | S6 (table) | S7 (table) | S8 (table) | S9 (table) | S10 (table)
ALL LINKS ARE ACTIVE IN THE ONLINE PDF
References
- 1.Mellman I, Coukos G, Dranoff G. Cancer immunotherapy comes of age. Nature. 2011;480:480–489. doi: 10.1038/nature10673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Topalian SL, Taube JM, Anders RA, Pardoll DM. Mechanism-driven biomarkers to guide immune checkpoint blockade in cancer therapy. Nat Rev Cancer. 2016;16:275–287. doi: 10.1038/nrc.2016.36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Broderick JM. FDA approves pembrolizumab for microsatellite instability-high and mismatch repair deficient cancers. OncLive. 2017 http://www.onclive.com/web-exclusives/fda-approves-pembrolizumab-for-microsatellite-instabilityhigh-and-mismatch-repair-deficient-cancers.
- 4.Chen DS, Mellman I. Elements of cancer immunity and the cancer-immune set point. Nature. 2017;541:321–330. doi: 10.1038/nature21349. [DOI] [PubMed] [Google Scholar]
- 5.Nishino M, Ramaiya NH, Hatabu H, Hodi FS. Monitoring immune-checkpoint blockade: response evaluation and biomarker development. Nat Rev Clin Oncol. 2017;14:655–668. doi: 10.1038/nrclinonc.2017.88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Jain RK. Normalizing tumor microenvironment to treat cancer: bench to bedside to biomarkers. J Clin Oncol. 2013;31:2205–2218. doi: 10.1200/JCO.2012.46.3653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Jain RK. Antiangiogenesis strategies revisited: from starving tumors to alleviating hypoxia. Cancer Cell. 2014;26:605–622. doi: 10.1016/j.ccell.2014.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Palucka AK, Coussens LM. The basis of oncoimmunology. Cell. 2016;164:1233–1247. doi: 10.1016/j.cell.2016.01.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Carmeliet P, Jain RK. Molecular mechanisms and clinical applications of angiogenesis. Nature. 2011;473:298–307. doi: 10.1038/nature10144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Jain RK, et al. Leukocyte-endothelial adhesion and angiogenesis in tumors. Cancer Metastasis Rev. 1996;15:195–204. doi: 10.1007/BF00437472. [DOI] [PubMed] [Google Scholar]
- 11.Hoshida T, et al. Imaging steps of lymphatic metastasis reveals that vascular endothelial growth factor-C increases metastasis by increasing delivery of cancer cells to lymph nodes: therapeutic implications. Cancer Res. 2006;66:8065–8075. doi: 10.1158/0008-5472.CAN-06-1392. [DOI] [PubMed] [Google Scholar]
- 12.Hiratsuka S, et al. Endothelial focal adhesion kinase mediates cancer cell homing to discrete regions of the lungs via E-selectin up-regulation. Proc Natl Acad Sci USA. 2011;108:3725–3730. doi: 10.1073/pnas.1100446108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chen DS, Mellman I. Oncology meets immunology: the cancer-immunity cycle. Immunity. 2013;39:1–10. doi: 10.1016/j.immuni.2013.07.012. [DOI] [PubMed] [Google Scholar]
- 14.Melder RJ, et al. During angiogenesis, vascular endothelial growth factor and basic fibroblast growth factor regulate natural killer cell adhesion to tumor endothelium. Nat Med. 1996;2:992–997. doi: 10.1038/nm0996-992. [DOI] [PubMed] [Google Scholar]
- 15.Hendry SA, et al. The role of the tumor vasculature in the host immune response: implications for therapeutic strategies targeting the tumor microenvironment. Front Immunol. 2016;7:621. doi: 10.3389/fimmu.2016.00621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Schmittnaegel M, et al. Dual angiopoietin-2 and VEGFA inhibition elicits antitumor immunity that is enhanced by PD-1 checkpoint blockade. Sci Transl Med. 2017;9:eaak9670. doi: 10.1126/scitranslmed.aak9670. [DOI] [PubMed] [Google Scholar]
- 17.Facciabene A, et al. Tumour hypoxia promotes tolerance and angiogenesis via CCL28 and Treg cells. Nature. 2011;475:226–230. doi: 10.1038/nature10169. [DOI] [PubMed] [Google Scholar]
- 18.Rolny C, et al. HRG inhibits tumor growth and metastasis by inducing macrophage polarization and vessel normalization through downregulation of PlGF. Cancer Cell. 2011;19:31–44. doi: 10.1016/j.ccr.2010.11.009. [DOI] [PubMed] [Google Scholar]
- 19.Chang AL, et al. CCL2 produced by the glioma microenvironment is essential for the recruitment of regulatory T cells and myeloid-derived suppressor cells. Cancer Res. 2016;76:5671–5682. doi: 10.1158/0008-5472.CAN-16-0144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Raman D, Baugher PJ, Thu YM, Richmond A. Role of chemokines in tumor growth. Cancer Lett. 2007;256:137–165. doi: 10.1016/j.canlet.2007.05.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Huang YH, Goel S, Duda DG, Fukumura D, Jain RK. Vascular normalization as an emerging strategy to enhance cancer immunotherapy. Cancer Res. 2013;73:2943–2948. doi: 10.1158/0008-5472.CAN-12-4354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Voron T, et al. VEGF-A modulates expression of inhibitory checkpoints on CD8+ T cells in tumors. J Exp Med. 2015;212:139–148. doi: 10.1084/jem.20140559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gabrilovich D, et al. Vascular endothelial growth factor inhibits the development of dendritic cells and dramatically affects the differentiation of multiple hematopoietic lineages in vivo. Blood. 1998;92:4150–4166. [PubMed] [Google Scholar]
- 24.Gabrilovich DI, et al. Production of vascular endothelial growth factor by human tumors inhibits the functional maturation of dendritic cells. Nat Med. 1996;2:1267. doi: 10.1038/nm1096-1096. [DOI] [PubMed] [Google Scholar]
- 25.Maenhout SK, Thielemans K, Aerts JL. Location, location, location: functional and phenotypic heterogeneity between tumor-infiltrating and non-infiltrating myeloid-derived suppressor cells. Oncoimmunology. 2014;3:e956579. doi: 10.4161/21624011.2014.956579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chaudhary B, Khaled YS, Ammori BJ, Elkord E. Neuropilin 1: function and therapeutic potential in cancer. Cancer Immunol Immunother. 2014;63:81–99. doi: 10.1007/s00262-013-1500-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Huang Y, et al. Vascular normalizing doses of antiangiogenic treatment reprogram the immunosuppressive tumor microenvironment and enhance immunotherapy. Proc Natl Acad Sci USA. 2012;109:17561–17566. doi: 10.1073/pnas.1215397109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Jain RK. Normalization of tumor vasculature: an emerging concept in antiangiogenic therapy. Science. 2005;307:58–62. doi: 10.1126/science.1104819. [DOI] [PubMed] [Google Scholar]
- 29.Scholz A, et al. Angiopoietin-2 promotes myeloid cell infiltration in a β2-integrin-dependent manner. Blood. 2011;118:5050–5059. doi: 10.1182/blood-2011-03-343293. [DOI] [PubMed] [Google Scholar]
- 30.Coffelt SB, et al. Angiopoietin 2 stimulates TIE2-expressing monocytes to suppress T cell activation and to promote regulatory T cell expansion. J Immunol. 2011;186:4183–4190. doi: 10.4049/jimmunol.1002802. [DOI] [PubMed] [Google Scholar]
- 31.De Palma M, et al. Tie2 identifies a hematopoietic lineage of proangiogenic monocytes required for tumor vessel formation and a mesenchymal population of pericyte progenitors. Cancer Cell. 2005;8:211–226. doi: 10.1016/j.ccr.2005.08.002. [DOI] [PubMed] [Google Scholar]
- 32.Holopainen T, et al. Effects of angiopoietin-2-blocking antibody on endothelial cell-cell junctions and lung metastasis. J Natl Cancer Inst. 2012;104:461–475. doi: 10.1093/jnci/djs009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Saharinen P, Eklund L, Alitalo K. Therapeutic targeting of the angiopoietin-TIE pathway. Nat Rev Drug Discov. 2017;16:635–661. doi: 10.1038/nrd.2016.278. [DOI] [PubMed] [Google Scholar]
- 34.Murdoch C, Tazzyman S, Webster S, Lewis CE. Expression of Tie-2 by human monocytes and their responses to angiopoietin-2. J Immunol. 2007;178:7405–7411. doi: 10.4049/jimmunol.178.11.7405. [DOI] [PubMed] [Google Scholar]
- 35.Kloepper J, et al. Ang-2/VEGF bispecific antibody reprograms macrophages and resident microglia to anti-tumor phenotype and prolongs glioblastoma survival. Proc Natl Acad Sci USA. 2016;113:4476–4481. doi: 10.1073/pnas.1525360113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Peterson TE, et al. Dual inhibition of Ang-2 and VEGF receptors normalizes tumor vasculature and prolongs survival in glioblastoma by altering macrophages. Proc Natl Acad Sci USA. 2016;113:4470–4475. doi: 10.1073/pnas.1525349113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Rahbari NN, et al. Anti-VEGF therapy induces ECM remodeling and mechanical barriers to therapy in colorectal cancer liver metastases. Sci Transl Med. 2016;8:360ra135. doi: 10.1126/scitranslmed.aaf5219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Jung K, et al. Targeting CXCR4-dependent immunosuppressive Ly6Clow monocytes improves antiangiogenic therapy in colorectal cancer. Proc Natl Acad Sci USA. 2017;114:10455–10460. doi: 10.1073/pnas.1710754114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Jung K, et al. Ly6Clo monocytes drive immunosuppression and confer resistance to anti-VEGFR2 cancer therapy. J Clin Invest. 2017;127:3039–3051. doi: 10.1172/JCI93182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Chen Y, et al. CXCR4 inhibition in tumor microenvironment facilitates anti-programmed death receptor-1 immunotherapy in sorafenib-treated hepatocellular carcinoma in mice. Hepatology. 2015;61:1591–1602. doi: 10.1002/hep.27665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Shojaei F, et al. Tumor refractoriness to anti-VEGF treatment is mediated by CD11b+Gr1+ myeloid cells. Nat Biotechnol. 2007;25:911–920. doi: 10.1038/nbt1323. [DOI] [PubMed] [Google Scholar]
- 42.Ilhan-Mutlu A, et al. Bevacizumab prevents brain metastases formation in lung adenocarcinoma. Mol Cancer Ther. 2016;15:702–710. doi: 10.1158/1535-7163.MCT-15-0582. [DOI] [PubMed] [Google Scholar]
- 43.Kodack DP, et al. Combined targeting of HER2 and VEGFR2 for effective treatment of HER2-amplified breast cancer brain metastases. Proc Natl Acad Sci USA. 2012;109:E3119–E3127. doi: 10.1073/pnas.1216078109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Minami T, et al. The calcineurin–NFAT–angiopoietin-2 signaling axis in lung endothelium is critical for the establishment of lung metastases. Cell Rep. 2013;4:709–723. doi: 10.1016/j.celrep.2013.07.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Keskin D, et al. Targeting vascular pericytes in hypoxic tumors increases lung metastasis via angiopoietin-2. Cell Rep. 2015;10:1066–1081. doi: 10.1016/j.celrep.2015.01.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Engblom C, Pfirschke C, Pittet MJ. The role of myeloid cells in cancer therapies. Nat Rev Cancer. 2016;16:447–462. doi: 10.1038/nrc.2016.54. [DOI] [PubMed] [Google Scholar]
- 47.Mazzieri R, et al. Targeting the ANG2/TIE2 axis inhibits tumor growth and metastasis by impairing angiogenesis and disabling rebounds of proangiogenic myeloid cells. Cancer Cell. 2011;19:512–526. doi: 10.1016/j.ccr.2011.02.005. [DOI] [PubMed] [Google Scholar]
- 48.Park JS, et al. Normalization of tumor vessels by Tie2 activation and Ang2 inhibition enhances drug delivery and produces a favorable tumor microenvironment. Cancer Cell. 2016;30:953–967. doi: 10.1016/j.ccell.2016.10.018. [DOI] [PubMed] [Google Scholar]
- 49.Goel S, et al. Effects of vascular-endothelial protein tyrosine phosphatase inhibition on breast cancer vasculature and metastatic progression. J Natl Cancer Inst. 2013;105:1188–1201. doi: 10.1093/jnci/djt164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Rigamonti N, et al. Role of angiopoietin-2 in adaptive tumor resistance to VEGF signaling blockade. Cell Rep. 2014;8:696–706. doi: 10.1016/j.celrep.2014.06.059. [DOI] [PubMed] [Google Scholar]
- 51.Imanishi Y, et al. Angiopoietin-2 stimulates breast cancer metastasis through the α5β1 integrin-mediated pathway. Cancer Res. 2007;67:4254–4263. doi: 10.1158/0008-5472.CAN-06-4100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Sfiligoi C, et al. Angiopoietin-2 expression in breast cancer correlates with lymph node invasion and short survival. Int J Cancer. 2003;103:466–474. doi: 10.1002/ijc.10851. [DOI] [PubMed] [Google Scholar]
- 53.Srivastava K, et al. Postsurgical adjuvant tumor therapy by combining anti-angiopoietin-2 and metronomic chemotherapy limits metastatic growth. Cancer Cell. 2014;26:880–895. doi: 10.1016/j.ccell.2014.11.005. [DOI] [PubMed] [Google Scholar]
- 54.Kienast Y, et al. Ang-2–VEGF-A CrossMab, a novel bispecific human IgG1 antibody blocking VEGF-A and Ang-2 functions simultaneously, mediates potent antitumor, antiangiogenic, and antimetastatic efficacy. Clin Cancer Res. 2013;19:6730–6740. doi: 10.1158/1078-0432.CCR-13-0081. [DOI] [PubMed] [Google Scholar]
- 55.Pezzella F, Gatter K. Non-angiogenic tumours unveil a new chapter in cancer biology. J Pathol. 2015;235:381–383. doi: 10.1002/path.4474. [DOI] [PubMed] [Google Scholar]
- 56.Griveau A, et al. A glial signature and Wnt7 signaling regulate glioma-vascular interactions and tumor microenvironment. Cancer Cell. doi: 10.1016/j.ccell.2018.03.020. (in press) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Eggermont AM, et al. Adjuvant ipilimumab versus placebo after complete resection of high-risk stage III melanoma (EORTC 18071): a randomised, double-blind, phase 3 trial. Lancet Oncol. 2015;16:522–530. doi: 10.1016/S1470-2045(15)70122-1. [DOI] [PubMed] [Google Scholar]
- 58.Antonia SJ, et al. Durvalumab after chemoradiotherapy in stage III non-small-cell lung cancer. N Engl J Med. 2017;377:1919–1929. doi: 10.1056/NEJMoa1709937. [DOI] [PubMed] [Google Scholar]
- 59.Wolchok JD, et al. Overall survival with combined nivolumab and ipilimumab in advanced melanoma. N Engl J Med. 2017;377:1345–1356. doi: 10.1056/NEJMoa1709684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Jain RK, Martin JD, Stylianopoulos T. The role of mechanical forces in tumor growth and therapy. Annu Rev Biomed Eng. 2014;16:321–346. doi: 10.1146/annurev-bioeng-071813-105259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Jain RK. Normalizing tumor vasculature with anti-angiogenic therapy: a new paradigm for combination therapy. Nat Med. 2001;7:987–989. doi: 10.1038/nm0901-987. [DOI] [PubMed] [Google Scholar]
- 62.Winkler F, et al. Kinetics of vascular normalization by VEGFR2 blockade governs brain tumor response to radiation: role of oxygenation, angiopoietin-1, and matrix metalloproteinases. Cancer Cell. 2004;6:553–563. doi: 10.1016/j.ccr.2004.10.011. [DOI] [PubMed] [Google Scholar]
- 63.Batchelor TT, et al. AZD2171, a pan-VEGF receptor tyrosine kinase inhibitor, normalizes tumor vasculature and alleviates edema in glioblastoma patients. Cancer Cell. 2007;11:83–95. doi: 10.1016/j.ccr.2006.11.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Willett CG, et al. Direct evidence that the VEGF-specific antibody bevacizumab has antivascular effects in human rectal cancer. Nat Med. 2004;10:145–147. doi: 10.1038/nm988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Amin A, et al. Survival with AGS-003, an autologous dendritic cell-based immunotherapy, in combination with sunitinib in unfavorable risk patients with advanced renal cell carcinoma (RCC): phase 2 study results. J Immunother Cancer. 2015;3:14. doi: 10.1186/s40425-015-0055-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Chae SS, et al. Angiopoietin-2 interferes with anti-VEGFR2-induced vessel normalization and survival benefit in mice bearing gliomas. Clin Cancer Res. 2010;16:3618–3627. doi: 10.1158/1078-0432.CCR-09-3073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Batchelor TT, et al. Phase II study of cediranib, an oral pan-vascular endothelial growth factor receptor tyrosine kinase inhibitor, in patients with recurrent glioblastoma. J Clin Oncol. 2010;28:2817–2823. doi: 10.1200/JCO.2009.26.3988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Wu X, et al. Angiopoietin-2 as a biomarker and target for immune checkpoint therapy. Cancer Immunol Res. 2017;5:17–28. doi: 10.1158/2326-6066.CIR-16-0206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Tong RT, et al. Vascular normalization by vascular endothelial growth factor receptor 2 blockade induces a pressure gradient across the vasculature and improves drug penetration in tumors. Cancer Res. 2004;64:3731–3736. doi: 10.1158/0008-5472.CAN-04-0074. [DOI] [PubMed] [Google Scholar]
- 70.Van der Veldt AA, et al. Rapid decrease in delivery of chemotherapy to tumors after anti-VEGF therapy: implications for scheduling of anti-angiogenic drugs. Cancer Cell. 2012;21:82–91. doi: 10.1016/j.ccr.2011.11.023. [DOI] [PubMed] [Google Scholar]
- 71.Heist RS, et al. Improved tumor vascularization after anti-VEGF therapy with carboplatin and nab-paclitaxel associates with survival in lung cancer. Proc Natl Acad Sci USA. 2015;112:1547–1552. doi: 10.1073/pnas.1424024112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Yang MH, et al. Direct regulation of TWIST by HIF-1α promotes metastasis. Nat Cell Biol. 2008;10:295–305. doi: 10.1038/ncb1691. [DOI] [PubMed] [Google Scholar]
- 73.Conley SJ, et al. Antiangiogenic agents increase breast cancer stem cells via the generation of tumor hypoxia. Proc Natl Acad Sci USA. 2012;109:2784–2789. doi: 10.1073/pnas.1018866109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Levin VA, et al. Impact of bevacizumab administered dose on overall survival of patients with progressive glioblastoma. J Neurooncol. 2015;122:145–150. doi: 10.1007/s11060-014-1693-x. [DOI] [PubMed] [Google Scholar]
- 75.Levin VA, Chan J, Datta M, Yee JL, Jain RK. Effect of angiotensin system inhibitors on survival in newly diagnosed glioma patients and recurrent glioblastoma patients receiving chemotherapy and/or bevacizumab. J Neurooncol. 2017;134:325–330. doi: 10.1007/s11060-017-2528-3. [DOI] [PubMed] [Google Scholar]
- 76.Lorgis V, et al. Relation between bevacizumab dose intensity and high-grade glioma survival: a retrospective study in two large cohorts. J Neurooncol. 2012;107:351–358. doi: 10.1007/s11060-011-0748-5. [DOI] [PubMed] [Google Scholar]
- 77.Lu-Emerson C, et al. Lessons from anti-vascular endothelial growth factor and anti-vascular endothelial growth factor receptor trials in patients with glioblastoma. J Clin Oncol. 2015;33:1197–1213. doi: 10.1200/JCO.2014.55.9575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Tolaney SM, et al. Role of vascular density and normalization in response to neoadjuvant bevacizumab and chemotherapy in breast cancer patients. Proc Natl Acad Sci USA. 2015;112:14325–14330. doi: 10.1073/pnas.1518808112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Hegde PS, et al. Predictive impact of circulating vascular endothelial growth factor in four phase III trials evaluating bevacizumab. Clin Cancer Res. 2013;19:929–937. doi: 10.1158/1078-0432.CCR-12-2535. [DOI] [PubMed] [Google Scholar]
- 80.Bergers G, Hanahan D. Modes of resistance to anti-angiogenic therapy. Nat Rev Cancer. 2008;8:592–603. doi: 10.1038/nrc2442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Moon WS, et al. Overexpression of VEGF and angiopoietin 2: a key to high vascularity of hepatocellular carcinoma? Mod Pathol. 2003;16:552–557. doi: 10.1097/01.MP.0000071841.17900.69. [DOI] [PubMed] [Google Scholar]
- 82.Wang HL, et al. Expression of angiopoietin-2 is correlated with vascularization and tumor size in human colorectal adenocarcinoma. Tohoku J Exp Med. 2007;213:33–40. doi: 10.1620/tjem.213.33. [DOI] [PubMed] [Google Scholar]
- 83.Peeters M, et al. A randomised, double-blind, placebo-controlled phase 2 study of trebananib (AMG 386) in combination with FOLFIRI in patients with previously treated metastatic colorectal carcinoma. Br J Cancer. 2013;108:503–511. doi: 10.1038/bjc.2012.594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Eatock MM, et al. Phase II randomized, double-blind, placebo-controlled study of AMG 386 (trebananib) in combination with cisplatin and capecitabine in patients with metastatic gastro-oesophageal cancer. Ann Oncol. 2013;24:710–718. doi: 10.1093/annonc/mds502. [DOI] [PubMed] [Google Scholar]
- 85.Rini B, et al. AMG 386 in combination with sorafenib in patients with metastatic clear cell carcinoma of the kidney: a randomized, double-blind, placebo-controlled, phase 2 study. Cancer. 2012;118:6152–6161. doi: 10.1002/cncr.27632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Monk BJ, et al. Final results of a phase 3 study of trebananib plus weekly paclitaxel in recurrent ovarian cancer (TRINOVA-1): long-term survival, impact of ascites, and progression-free survival-2. Gynecol Oncol. 2016;143:27–34. doi: 10.1016/j.ygyno.2016.07.112. [DOI] [PubMed] [Google Scholar]
- 87.Marth C, et al. ENGOT-ov-6/TRINOVA-2: Randomised, double-blind, phase 3 study of pegylated liposomal doxorubicin plus trebananib or placebo in women with recurrent partially platinum-sensitive or resistant ovarian cancer. Eur J Cancer. 2017;70:111–121. doi: 10.1016/j.ejca.2016.09.004. [DOI] [PubMed] [Google Scholar]
- 88.Reardon DA, et al. Phase 2 and biomarker study of trebananib, an angiopoietin-blocking peptibody, with and without bevacizumab for patients with recurrent glioblastoma. Cancer. 2017 doi: 10.1002/cncr.31172. https://doi.org/10.1002/cncr.31172. [DOI] [PubMed]
- 89.Liu C, Workman CJ, Vignali DA. Targeting regulatory T cells in tumors. FEBS J. 2016;283:2731–2748. doi: 10.1111/febs.13656. [DOI] [PubMed] [Google Scholar]
- 90.Wu FT, et al. Efficacy of cotargeting angiopoietin-2 and the VEGF pathway in the adjuvant postsurgical setting for early breast, colorectal, and renal cancers. Cancer Res. 2016;76:6988–7000. doi: 10.1158/0008-5472.CAN-16-0888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Charles NA, Holland EC, Gilbertson R, Glass R, Kettenmann H. The brain tumor microenvironment. Glia. 2011;59:1169–1180. doi: 10.1002/glia.21136. [DOI] [PubMed] [Google Scholar]
- 92.Kabbinavar F, et al. Phase II, randomized trial comparing bevacizumab plus fluorouracil (FU)/leucovorin (LV) with FU/LV alone in patients with metastatic colorectal cancer. J Clin Oncol. 2003;21:60–65. doi: 10.1200/JCO.2003.10.066. [DOI] [PubMed] [Google Scholar]
- 93.Willett CG, et al. Surrogate markers for antiangiogenic therapy and dose-limiting toxicities for bevacizumab with radiation and chemotherapy: continued experience of a phase I trial in rectal cancer patients. J Clin Oncol. 2005;23:8136–8139. doi: 10.1200/JCO.2005.02.5635. [DOI] [PubMed] [Google Scholar]
- 94.Chauhan VP, et al. Angiotensin inhibition enhances drug delivery and potentiates chemotherapy by decompressing tumour blood vessels. Nat Commun. 2013;4:2516. doi: 10.1038/ncomms3516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Pinter M, Jain RK. Targeting the renin-angiotensin system to improve cancer treatment: Implications for immunotherapy. Sci Transl Med. 2017;9:eaan5616. doi: 10.1126/scitranslmed.aan5616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Shrimali RK, et al. Antiangiogenic agents can increase lymphocyte infiltration into tumor and enhance the effectiveness of adoptive immunotherapy of cancer. Cancer Res. 2010;70:6171–6180. doi: 10.1158/0008-5472.CAN-10-0153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Palazon A, et al. An HIF-1α/VEGF-A axis in cytotoxic T cells regulates tumor progression. Cancer Cell. 2017;32:669–683.e5. doi: 10.1016/j.ccell.2017.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Allen E, et al. Combined antiangiogenic and anti-PD-L1 therapy stimulates tumor immunity through HEV formation. Sci Transl Med. 2017;9:eaak9679. doi: 10.1126/scitranslmed.aak9679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Reardon DA, et al. OS10. 3 Randomized phase 3 study evaluating the efficacy and safety of nivolumab vs bevacizumab in patients with recurrent glioblastoma: CheckMate 143 [abstract] Neuro-Oncol. 2017;19(Suppl 3):iii21. [Google Scholar]
- 100.Martins I. Opdivo fails to demonstrate survival benefit in phase 3 brain cancer trial. Immuno-Oncology News. 2017 https://immuno-oncologynews.com/2017/04/18/opdivo-fails-demonstrate-increased-survival-glioblastoma-multiforme-phase-3-study/
- 101.D’Souza NM, et al. Combining radiation therapy with immune checkpoint blockade for central nervous system malignancies. Front Oncol. 2016;6:212. doi: 10.3389/fonc.2016.00212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Zeng J, et al. Anti-PD-1 blockade and stereotactic radiation produce long-term survival in mice with intracranial gliomas. Int J Radiat Oncol Biol Phys. 2013;86:343–349. doi: 10.1016/j.ijrobp.2012.12.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Sampson JH, et al. A randomized, phase 3, open-label study of nivolumab versus temozolomide (TMZ) in combination with radiotherapy (RT) in adult patients (pts) with newly diagnosed, O-6-methylguanine DNA methyltransferase (MGMT)-unmethylated glioblastoma (GBM): CheckMate-498 [abstract] J Clin Oncol. 2016;34(Suppl):TPS2079. [Google Scholar]
- 104.Weller M, et al. A randomized phase 2, single-blind study of temozolomide (TMZ) and radiotherapy (RT) combined with nivolumab or placebo (PBO) in newly diagnosed adult patients (pts) with tumor O6-methylguanine DNA methyltransferase (MGMT)-methylated glioblastoma (GBM) — CheckMate-548 [abstract] Ann Oncol. 2016;27(Suppl 6):356TiP. [Google Scholar]
- 105.Tian L, et al. Mutual regulation of tumour vessel normalization and immunostimulatory reprogramming. Nature. 2017;544:250–254. doi: 10.1038/nature21724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.De Palma M, Jain RK. CD4+ T cell activation and vascular normalization: two sides of the same coin? Immunity. 2017;46:773–775. doi: 10.1016/j.immuni.2017.04.015. [DOI] [PubMed] [Google Scholar]
- 107.Zhu X, et al. Severe cerebral edema following nivolumab treatment for pediatric glioblastoma: case report. J Neurosurg Pediatr. 2017;19:249–253. doi: 10.3171/2016.8.PEDS16326. [DOI] [PubMed] [Google Scholar]
- 108.Qin L, et al. Advanced MRI assessment to predict benefit of anti-programmed cell death 1 protein immunotherapy response in patients with recurrent glioblastoma. Neuroradiology. 2017;59:135–145. doi: 10.1007/s00234-016-1769-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Kamoun WS, et al. Edema control by cediranib, a vascular endothelial growth factor receptor-targeted kinase inhibitor, prolongs survival despite persistent brain tumor growth in mice. J Clin Oncol. 2009;27:2542–2552. doi: 10.1200/JCO.2008.19.9356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Salmon H, et al. Matrix architecture defines the preferential localization and migration of T cells into the stroma of human lung tumors. J Clin Invest. 2012;122:899–910. doi: 10.1172/JCI45817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Eroglu Z, et al. High response rate to PD-1 blockade in desmoplastic melanomas. Nature. 2018;553:347–350. doi: 10.1038/nature25187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Liu H, et al. Use of angiotensin system inhibitors is associated with immune activation and longer survival in nonmetastatic pancreatic ductal adenocarcinoma. Clin Cancer Res. 2017;23:5959–5969. doi: 10.1158/1078-0432.CCR-17-0256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Stylianopoulos T, Jain RK. Combining two strategies to improve perfusion and drug delivery in solid tumors. Proc Natl Acad Sci USA. 2013;110:18632–18637. doi: 10.1073/pnas.1318415110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Fukumura D, Incio J, Shankaraiah RC, Jain RK. Obesity and cancer: an angiogenic and inflammatory link. Microcirculation. 2016;23:191–206. doi: 10.1111/micc.12270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Mirsoian A, Murphy WJ. Obesity and cancer immunotherapy toxicity. Immunotherapy. 2015;7:319–322. doi: 10.2217/imt.15.12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Izumi Y, Xu L, di Tomaso E, Fukumura D, Jain RK. Tumour biology: herceptin acts as an anti-angiogenic cocktail. Nature. 2002;416:279–280. doi: 10.1038/416279b. [DOI] [PubMed] [Google Scholar]
- 117.Mpekris F, Baish JW, Stylianopoulos T, Jain RK. Role of vascular normalization in benefit from metronomic chemotherapy. Proc Natl Acad Sci USA. 2017;114:1994–1999. doi: 10.1073/pnas.1700340114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Jain RK, et al. Endothelial cell death, angiogenesis, and microvascular function after castration in an androgen-dependent tumor: role of vascular endothelial growth factor. Proc Natl Acad Sci USA. 1998;95:10820–10825. doi: 10.1073/pnas.95.18.10820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Goel S, et al. CDK4/6 inhibition triggers anti-tumour immunity. Nature. 2017;548:471–475. doi: 10.1038/nature23465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Fesnak AD, June CH, Levine BL. Engineered T cells: the promise and challenges of cancer immunotherapy. Nat Rev Cancer. 2016;16:566–581. doi: 10.1038/nrc.2016.97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Garg AD, et al. Dendritic cell vaccines based on immunogenic cell death elicit danger signals and T cell-driven rejection of high-grade glioma. Sci Transl Med. 2016;8:328ra27. doi: 10.1126/scitranslmed.aae0105. [DOI] [PubMed] [Google Scholar]
- 122.Roy S, Trinchieri G. Microbiota: a key orchestrator of cancer therapy. Nat Rev Cancer. 2017;17:271–285. doi: 10.1038/nrc.2017.13. [DOI] [PubMed] [Google Scholar]
- 123.Gopalakrishnan V, et al. Gut microbiome modulates response to anti-PD-1 immunotherapy in melanoma patients. Science. 2018;359:97–103. doi: 10.1126/science.aan4236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Routy B, et al. Gut microbiome influences efficacy of PD-1-based immunotherapy against epithelial tumors. Science. 2018;359:91–97. doi: 10.1126/science.aan3706. [DOI] [PubMed] [Google Scholar]
- 125.Matson V, et al. The commensal microbiome is associated with anti-PD-1 efficacy in metastatic melanoma patients. Science. 2018;359:104–108. doi: 10.1126/science.aao3290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Jain RK, et al. Biomarkers of response and resistance to antiangiogenic therapy. Nat Rev Clin Oncol. 2009;6:327–338. doi: 10.1038/nrclinonc.2009.63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Goel S, et al. Normalization of the vasculature for treatment of cancer and other diseases. Physiol Rev. 2011;91:1071–1121. doi: 10.1152/physrev.00038.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Batchelor TT, et al. Phase III randomized trial comparing the efficacy of cediranib as monotherapy, and in combination with lomustine, versus lomustine alone in patients with recurrent glioblastoma. J Clin Oncol. 2013;31:3212–3218. doi: 10.1200/JCO.2012.47.2464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Chinot OL, et al. Bevacizumab plus radiotherapy-temozolomide for newly diagnosed glioblastoma. N Engl J Med. 2014;370:709–722. doi: 10.1056/NEJMoa1308345. [DOI] [PubMed] [Google Scholar]
- 130.Gilbert MR, et al. A randomized trial of bevacizumab for newly diagnosed glioblastoma. N Engl J Med. 2014;370:699–708. doi: 10.1056/NEJMoa1308573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Hurwitz H, et al. Bevacizumab plus irinotecan, fluorouracil, and leucovorin for metastatic colorectal cancer. N Engl J Med. 2004;350:2335–2342. doi: 10.1056/NEJMoa032691. [DOI] [PubMed] [Google Scholar]
- 132.Sandler A, et al. Paclitaxel-carboplatin alone or with bevacizumab for non-small-cell lung cancer. N Engl J Med. 2006;355:2542–2550. doi: 10.1056/NEJMoa061884. [DOI] [PubMed] [Google Scholar]
- 133.Tewari KS, et al. Improved survival with bevacizumab in advanced cervical cancer. N Engl J Med. 2014;370:734–743. doi: 10.1056/NEJMoa1309748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Zalcman G, et al. Bevacizumab for newly diagnosed pleural mesothelioma in the Mesothelioma Avastin Cisplatin Pemetrexed Study (MAPS): a randomised, controlled, open-label, phase 3 trial. Lancet. 2016;387:1405–1414. doi: 10.1016/S0140-6736(15)01238-6. [DOI] [PubMed] [Google Scholar]
- 135.Goel HL, Mercurio AM. VEGF targets the tumour cell. Nat Rev Cancer. 2013;13:871–882. doi: 10.1038/nrc3627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Senger DR, et al. Tumor cells secrete a vascular permeability factor that promotes accumulation of ascites fluid. Science. 1983;219:983–985. doi: 10.1126/science.6823562. [DOI] [PubMed] [Google Scholar]
- 137.Leung DW, Cachianes G, Kuang WJ, Goeddel DV, Ferrara N. Vascular endothelial growth factor is a secreted angiogenic mitogen. Science. 1989;246:1306–1309. doi: 10.1126/science.2479986. [DOI] [PubMed] [Google Scholar]
- 138.Carmeliet P, et al. Abnormal blood vessel development and lethality in embryos lacking a single VEGF allele. Nature. 1996;380:435–439. doi: 10.1038/380435a0. [DOI] [PubMed] [Google Scholar]
- 139.Ferrara N, et al. Heterozygous embryonic lethality induced by targeted inactivation of the VEGF gene. Nature. 1996;380:439–442. doi: 10.1038/380439a0. [DOI] [PubMed] [Google Scholar]
- 140.Fukumura D, et al. Tumor induction of VEGF promoter activity in stromal cells. Cell. 1998;94:715–725. doi: 10.1016/s0092-8674(00)81731-6. [DOI] [PubMed] [Google Scholar]
- 141.Simon T, Gagliano T, Giamas G. Direct effects of anti-angiogenic therapies on tumor cells: VEGF signaling. Trends Mol Med. 2017;23:282–292. doi: 10.1016/j.molmed.2017.01.002. [DOI] [PubMed] [Google Scholar]
- 142.Hamrah P, Zhang Q, Dana MR. Expression of vascular endothelial growth factor receptor-3 (VEGFR-3) in the conjunctiva — a potential link between lymphangiogenesis and leukocyte trafficking on the ocular surface. Adv Exp Med Biol. 2002;506:851–860. doi: 10.1007/978-1-4615-0717-8_120. [DOI] [PubMed] [Google Scholar]
- 143.Sawano A, et al. Flt-1, vascular endothelial growth factor receptor 1, is a novel cell surface marker for the lineage of monocyte-macrophages in humans. Blood. 2001;97:785–791. doi: 10.1182/blood.v97.3.785. [DOI] [PubMed] [Google Scholar]
- 144.Basu A, et al. Cutting edge: Vascular endothelial growth factor-mediated signaling in human CD45RO+ CD4+ T cells promotes Akt and ERK activation and costimulates IFN-γ production. J Immunol. 2010;184:545–549. doi: 10.4049/jimmunol.0900397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Gavalas NG, et al. VEGF directly suppresses activation of T cells from ascites secondary to ovarian cancer via VEGF receptor type 2. Br J Cancer. 2012;107:1869–1875. doi: 10.1038/bjc.2012.468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Wada J, et al. The contribution of vascular endothelial growth factor to the induction of regulatory T-cells in malignant effusions. Anticancer Res. 2009;29:881–888. [PubMed] [Google Scholar]
- 147.Lichtenberger BM, et al. Autocrine VEGF signaling synergizes with EGFR in tumor cells to promote epithelial cancer development. Cell. 2010;140:268–279. doi: 10.1016/j.cell.2009.12.046. [DOI] [PubMed] [Google Scholar]
- 148.Sundberg C, Kowanetz M, Brown LF, Detmar M, Dvorak HF. Stable expression of angiopoietin-1 and other markers by cultured pericytes: phenotypic similarities to a subpopulation of cells in maturing vessels during later stages of angiogenesis in vivo. Lab Invest. 2002;82:387–401. doi: 10.1038/labinvest.3780433. [DOI] [PubMed] [Google Scholar]
- 149.Gamble JR, et al. Angiopoietin-1 is an antipermeability and anti-inflammatory agent in vitro and targets cell junctions. Circ Res. 2000;87:603–607. doi: 10.1161/01.res.87.7.603. [DOI] [PubMed] [Google Scholar]
- 150.McGuire PG, Rangasamy S, Maestas J, Das A. Pericyte-derived sphingosine 1-phosphate induces the expression of adhesion proteins and modulates the retinal endothelial cell barrier. Arterioscler Thromb Vasc Biol. 2011;31:e107–e115. doi: 10.1161/ATVBAHA.111.235408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Metheny-Barlow LJ, Li LY. The enigmatic role of angiopoietin-1 in tumor angiogenesis. Cell Res. 2003;13:309–317. doi: 10.1038/sj.cr.7290176. [DOI] [PubMed] [Google Scholar]
- 152.Franco M, Roswall P, Cortez E, Hanahan D, Pietras K. Pericytes promote endothelial cell survival through induction of autocrine VEGF-A signaling and Bcl-w expression. Blood. 2011;118:2906–2917. doi: 10.1182/blood-2011-01-331694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Daly C, et al. Angiopoietin-1 modulates endothelial cell function and gene expression via the transcription factor FKHR (FOXO1) Genes Dev. 2004;18:1060–1071. doi: 10.1101/gad.1189704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Hori S, Ohtsuki S, Hosoya K, Nakashima E, Terasaki T. A pericyte-derived angiopoietin-1 multimeric complex induces occludin gene expression in brain capillary endothelial cells through Tie-2 activation in vitro. J Neurochem. 2004;89:503–513. doi: 10.1111/j.1471-4159.2004.02343.x. [DOI] [PubMed] [Google Scholar]
- 155.Thurston G, et al. Angiopoietin-1 protects the adult vasculature against plasma leakage. Nat Med. 2000;6:460–463. doi: 10.1038/74725. [DOI] [PubMed] [Google Scholar]
- 156.Fiedler U, et al. The Tie-2 ligand angiopoietin-2 is stored in and rapidly released upon stimulation from endothelial cell Weibel-Palade bodies. Blood. 2004;103:4150–4156. doi: 10.1182/blood-2003-10-3685. [DOI] [PubMed] [Google Scholar]
- 157.Ziegler T, et al. Angiopoietin 2 mediates microvascular and hemodynamic alterations in sepsis. J Clin Invest. 2013;123:3436–3445. doi: 10.1172/JCI66549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Pichiule P, Chavez JC, LaManna JC. Hypoxic regulation of angiopoietin-2 expression in endothelial cells. J Biol Chem. 2004;279:12171–12180. doi: 10.1074/jbc.M305146200. [DOI] [PubMed] [Google Scholar]
- 159.Detjen KM, et al. Angiopoietin-2 promotes disease progression of neuroendocrine tumors. Clin Cancer Res. 2010;16:420–429. doi: 10.1158/1078-0432.CCR-09-1924. [DOI] [PubMed] [Google Scholar]
- 160.Sallinen H, et al. Preoperative angiopoietin-2 serum levels: a marker of malignant potential in ovarian neoplasms and poor prognosis in epithelial ovarian cancer. Int J Gynecol Cancer. 2010;20:1498–1505. doi: 10.1111/IGC.0b013e3181f936e3. [DOI] [PubMed] [Google Scholar]
- 161.Helfrich I, et al. Angiopoietin-2 levels are associated with disease progression in metastatic malignant melanoma. Clin Cancer Res. 2009;15:1384–1392. doi: 10.1158/1078-0432.CCR-08-1615. [DOI] [PubMed] [Google Scholar]
- 162.Gale NW, et al. Angiopoietin-2 is required for postnatal angiogenesis and lymphatic patterning, and only the latter role is rescued by angiopoietin-1. Dev Cell. 2002;3:411–423. doi: 10.1016/s1534-5807(02)00217-4. [DOI] [PubMed] [Google Scholar]
- 163.Maisonpierre PC, et al. Angiopoietin-2, a natural antagonist for Tie2 that disrupts in vivo angiogenesis. Science. 1997;277:55–60. doi: 10.1126/science.277.5322.55. [DOI] [PubMed] [Google Scholar]
- 164.Daly C, et al. Angiopoietin-2 functions as a Tie2 agonist in tumor models, where it limits the effects of VEGF inhibition. Cancer Res. 2013;73:108–118. doi: 10.1158/0008-5472.CAN-12-2064. [DOI] [PubMed] [Google Scholar]
- 165.Labussiere M, et al. Angiopoietin-2 may be involved in the resistance to bevacizumab in recurrent glioblastoma. Cancer Invest. 2016;34:39–44. doi: 10.3109/07357907.2015.1088948. [DOI] [PubMed] [Google Scholar]
- 166.Scholz A, et al. Endothelial cell-derived angiopoietin-2 is a therapeutic target in treatment-naive and bevacizumab-resistant glioblastoma. EMBO Mol Med. 2016;8:39–57. doi: 10.15252/emmm.201505505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Bauerschlag DO, et al. Evaluation of potentially predictive markers for anti-angiogenic therapy with sunitinib in recurrent ovarian cancer patients. Transl Oncol. 2013;6:305–310. doi: 10.1593/tlo.13205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Wada H, et al. Expression pattern of angiogenic factors and prognosis after hepatic resection in hepatocellular carcinoma: importance of angiopoietin-2 and hypoxia-induced factor-1α. Liver Int. 2006;26:414–423. doi: 10.1111/j.1478-3231.2006.01243.x. [DOI] [PubMed] [Google Scholar]
- 169.Schulz P, et al. Angiopoietin-2 drives lymphatic metastasis of pancreatic cancer. FASEB J. 2011;25:3325–3335. doi: 10.1096/fj.11-182287. [DOI] [PubMed] [Google Scholar]
- 170.Hacker UT, et al. Evaluation of angiopoietin-2 as a biomarker in gastric cancer: results from the randomised phase III AVAGAST trial. Br J Cancer. 2016;114:855–862. doi: 10.1038/bjc.2016.30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Goede V, et al. Identification of serum angiopoietin-2 as a biomarker for clinical outcome of colorectal cancer patients treated with bevacizumab-containing therapy. Br J Cancer. 2010;103:1407–1414. doi: 10.1038/sj.bjc.6605925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Nagorsen D, et al. Tumor-infiltrating macrophages and dendritic cells in human colorectal cancer: relation to local regulatory T cells, systemic T-cell response against tumor-associated antigens and survival. J Transl Med. 2007;5:62. doi: 10.1186/1479-5876-5-62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Renner DN, et al. Improved treatment efficacy of antiangiogenic therapy when combined with picornavirus vaccination in the GL261 glioma model. Neurotherapeutics. 2016;13:226–236. doi: 10.1007/s13311-015-0407-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Manning EA, et al. A vascular endothelial growth factor receptor-2 inhibitor enhances antitumor immunity through an immune-based mechanism. Clin Cancer Res. 2007;13:3951–3959. doi: 10.1158/1078-0432.CCR-07-0374. [DOI] [PubMed] [Google Scholar]
- 175.Li B, et al. Vascular endothelial growth factor blockade reduces intratumoral regulatory T cells and enhances the efficacy of a GM-CSF-secreting cancer immunotherapy. Clin Cancer Res. 2006;12:6808–6816. doi: 10.1158/1078-0432.CCR-06-1558. [DOI] [PubMed] [Google Scholar]
- 176.Farsaci B, Higgins JP, Hodge JW. Consequence of dose scheduling of sunitinib on host immune response elements and vaccine combination therapy. Int J Cancer. 2012;130:1948–1959. doi: 10.1002/ijc.26219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Foy KC, et al. Combined vaccination with HER-2 peptide followed by therapy with VEGF peptide mimics exerts effective anti-tumor and anti-angiogenic effects in vitro and in vivo. Oncoimmunology. 2012;1:1048–1060. doi: 10.4161/onci.20708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Huang XJ, et al. Combined therapy of local and metastatic 4T1 breast tumor in mice using SU6668, an inhibitor of angiogenic receptor tyrosine kinases, and the immunostimulator B7.2–IgG fusion protein. Cancer Res. 2002;62:5727–5735. [PubMed] [Google Scholar]
- 179.Wang W, et al. Specificity redirection by CAR with human VEGFR-1 affinity endows T lymphocytes with tumor-killing ability and anti-angiogenic potency. Gene Ther. 2013;20:970–978. doi: 10.1038/gt.2013.19. [DOI] [PubMed] [Google Scholar]
- 180.Gabrilovich DI, Ishida T, Nadaf S, Ohm JE, Carbone DP. Antibodies to vascular endothelial growth factor enhance the efficacy of cancer immunotherapy by improving endogenous dendritic cell function. Clin Cancer Res. 1999;5:2963–2970. [PubMed] [Google Scholar]
- 181.Yasuda S, et al. Simultaneous blockade of programmed death 1 and vascular endothelial growth factor receptor 2 (VEGFR2) induces synergistic anti-tumour effect in vivo. Clin Exp Immunol. 2013;172:500–506. doi: 10.1111/cei.12069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Suzuki H, et al. Multiple therapeutic peptide vaccines consisting of combined novel cancer testis antigens and anti-angiogenic peptides for patients with non-small cell lung cancer. J Transl Med. 2013;11:97. doi: 10.1186/1479-5876-11-97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Hayashi H, et al. Phase I trial of OTS11101, an anti-angiogenic vaccine targeting vascular endothelial growth factor receptor 1 in solid tumor. Cancer Sci. 2013;104:98–104. doi: 10.1111/cas.12034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Hodi FS, et al. Bevacizumab plus ipilimumab in patients with metastatic melanoma. Cancer Immunol Res. 2014;2:632–642. doi: 10.1158/2326-6066.CIR-14-0053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Ott PA, Hodi FS, Buchbinder EI. Inhibition of immune checkpoints and vascular endothelial growth factor as combination therapy for metastatic melanoma: an overview of rationale, preclinical evidence, and initial clinical data. Front Oncol. 2015;5:202. doi: 10.3389/fonc.2015.00202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Ko JS, et al. Sunitinib mediates reversal of myeloid-derived suppressor cell accumulation in renal cell carcinoma patients. Clin Cancer Res. 2009;15:2148–2157. doi: 10.1158/1078-0432.CCR-08-1332. [DOI] [PubMed] [Google Scholar]
- 187.Amin A, et al. Nivolumab (anti-PD-1; BMS-936558, ONO-4538) in combination with sunitinib or pazopanib in patients (pts) with metastatic renal cell carcinoma (mRCC) [abstract] J Clin Oncol. 2014;32(Suppl):5010. [Google Scholar]
- 188.Rini BI, et al. Phase III trial of bevacizumab plus interferon alfa versus interferon alfa monotherapy in patients with metastatic renal cell carcinoma: final results of CALGB 90206. J Clin Oncol. 2010;28:2137–2143. doi: 10.1200/JCO.2009.26.5561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Escudier B, et al. Phase III trial of bevacizumab plus interferon alfa-2a in patients with metastatic renal cell carcinoma (AVOREN): final analysis of overall survival. J Clin Oncol. 2010;28:2144–2150. doi: 10.1200/JCO.2009.26.7849. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Information