Extrinsic and intrinsic apoptosis activate pannexin‐1 to drive NLRP3 inflammasome assembly (original) (raw)
Abstract
Pyroptosis is a form of lytic inflammatory cell death driven by inflammatory caspase‐1, caspase‐4, caspase‐5 and caspase‐11. These caspases cleave and activate the pore‐forming protein gasdermin D (GSDMD) to induce membrane damage. By contrast, apoptosis is driven by apoptotic caspase‐8 or caspase‐9 and has traditionally been classified as an immunologically silent form of cell death. Emerging evidence suggests that therapeutics designed for cancer chemotherapy or inflammatory disorders such as SMAC mimetics, TAK1 inhibitors and BH3 mimetics promote caspase‐8 or caspase‐9‐dependent inflammatory cell death and NLRP3 inflammasome activation. However, the mechanism by which caspase‐8 or caspase‐9 triggers cell lysis and NLRP3 activation is still undefined. Here, we demonstrate that during extrinsic apoptosis, caspase‐1 and caspase‐8 cleave GSDMD to promote lytic cell death. By engineering a novel Gsdmd D88A knock‐in mouse, we further demonstrate that this proinflammatory function of caspase‐8 is counteracted by caspase‐3‐dependent cleavage and inactivation of GSDMD at aspartate 88, and is essential to suppress GSDMD‐dependent cell lysis during caspase‐8‐dependent apoptosis. Lastly, we provide evidence that channel‐forming glycoprotein pannexin‐1, but not GSDMD or GSDME promotes NLRP3 inflammasome activation during caspase‐8 or caspase‐9‐dependent apoptosis.
Keywords: apoptosis, gasdermin, NLRP3, pannexin‐1, pyroptosis
Subject Categories: Autophagy & Cell Death, Immunology
Introduction
Apoptosis and pyroptosis are caspase‐dependent programmed cell death pathways that promote the removal of stressed, damaged, transformed or infected cells. Consequently, abnormalities in these pathways are associated with a range of human diseases including infection, cancer, neurodegeneration and autoinflammatory disease.
Pyroptosis is a form of inflammatory cell death and is best characterized as an innate immune mechanism against intracellular pathogens (Miao et al, 2010; Chen & Schroder, 2013). Pyroptosis is initiated by a multiprotein signalling complex, called the inflammasome, which assembles upon cellular stress or infection (Broz & Dixit, 2016). Inflammasome formation induces the activation of inflammatory caspases, caspase‐1, caspase‐4, caspase‐5 and caspase‐11, which cleave and activate the recently identified pore‐forming protein gasdermin D (GSDMD). In macrophages, cleavage of GSDMD at aspartate 276 (D276) by inflammasome‐activated inflammatory caspases liberates the cytotoxic p30 N‐terminal domain to generate plasma membrane pores and drive pyroptosis (He et al, 2015; Kayagaki et al, 2015; Shi et al, 2015; Aglietti et al, 2016; Ding et al, 2016; Liu et al, 2016; Sborgi et al, 2016), while GSDMD cleavage by caspase‐4, caspase‐11 or neutrophil elastase promotes neutrophil nuclear and plasma membrane damage that culminate in the extrusion of antimicrobial neutrophil extracellular traps (Chen et al, 2018b; Sollberger et al, 2018). A recent study demonstrated that cleavage of human GSDMD at position aspartate 87 (D87) by apoptotic caspase‐3 inactivates the pyroptotic properties of GSDMD (Taabazuing et al, 2017). However, caspase‐1‐driven pyroptosis precedes caspase‐3 activation in inflammasome‐activated cells (He et al, 2015), and a low concentration of active GSDMD is sufficient to trigger pyroptosis (Sborgi et al, 2016). This indicates that caspase‐3 is unlikely to suppress pyroptosis in inflammasome‐activated cells, but instead may suppress GSDMD‐dependent cell lysis during inflammasome‐independent cell death pathways.
In contrast to pyroptosis, apoptosis is traditionally believed to be an immunologically silent form of cell death that is important for the development and the removal of stressed or damaged cells. Apoptosis can be initiated by two major mechanisms, the extrinsic and intrinsic pathways. The extrinsic pathway is activated following the engagement of cell surface death receptors, such as tumour necrosis factor receptor 1 (TNFR1) or Fas (CD95). Engagement of TNFR1 initiates the formation of two distinct signalling complexes (Micheau & Tschopp, 2003; Wang et al, 2008). In healthy macrophages, TNFR1 engagement initiates the assembly of a plasma membrane‐associated signalling complex, termed Complex I, which drives the expression of proinflammatory cytokines and prosurvival genes. However, when core components of Complex I such as the E3 ubiquitin ligases cellular inhibitor of apoptosis 1 (cIAP1) and cIAP2, or the kinases TAK1 or TBK1 are perturbed, TNRF1 ligation promotes the formation of a second, distinct cytosolic death‐inducing complex comprising receptor‐interacting serine/threonine kinase 1 (RIPK1), Fas‐associated protein with death domain (FADD) and the initiator caspase, caspase‐8. This secondary complex is called Complex II or commonly referred as the ripoptosome (Petersen et al, 2007; Varfolomeev et al, 2007; Feoktistova et al, 2011; Tenev et al, 2011; Vince et al, 2012; Dondelinger et al, 2013; Lafont et al, 2018; Malireddi et al, 2018). In contrast to extrinsic apoptosis, the intrinsic pathway is initiated by developmental cues, cytotoxic agents, growth factor withdrawal, infection and chemotherapeutic drugs, which alter the ratio of prosurvival and pro‐apoptotic BCL‐2 family proteins, leading to mitochondrial outer membrane permeabilization and activation of a distinct initiator caspase, caspase‐9 (Czabotar et al, 2014). Both intrinsic and extrinsic pathways converge upon the activation of caspase‐3 and caspase‐7, which execute cellular demise for rapid apoptotic cell clearance and to suppress activation of proinflammatory signalling pathways. For example, caspase‐3 and caspase‐7 cleave the plasma membrane channel pannexin‐1 at its C‐terminus, which triggers membrane permeability and the release of “find‐me” and “eat‐me” signals to promote phagocytic clearance of apoptotic cells (Chekeni et al, 2010; Sandilos et al, 2012).
While these observations suggest that apoptosis is indeed immunologically silent during homeostasis, emerging evidence from us and others indicates that exposure of innate immune cells such as macrophages and neutrophils to various therapeutics designed for cancer chemotherapy or inflammatory disorders such as SMAC mimetics, TAK1 inhibitor and BH3 mimetics promotes inflammation by driving caspase‐8 or caspase‐9‐dependent inflammatory cell death and NLRP3 inflammasome activation (Vince et al, 2012, 2018; Lawlor et al, 2015; Wicki et al, 2016; Chauhan et al, 2018; Chen et al, 2018a; Malireddi et al, 2018). However, the mechanism by which caspase‐8 or caspase‐9 triggers cell lysis and NLRP3 activation is still undefined. Here, we demonstrate that extrinsic apoptosis promotes caspase‐1 and caspase‐8‐dependent GSDMD activation and cell lysis, in parallel with caspase‐3/7‐mediated secondary necrosis. By generating a Gsdmd D88A knock‐in mouse, we further demonstrate that the proinflammatory function of caspase‐8 is counteracted by caspase‐3‐dependent cleavage and inactivation of GSDMD at aspartate 88, and is essential to suppress GSDMD‐dependent cell lysis upon caspase‐8 activation. In contrast, we find no evidence that supports GSDME, a closely related gasdermin family protein, in driving cell lysis during extrinsic or intrinsic apoptosis. Lastly, we demonstrate that channel‐forming glycoprotein pannexin‐1, but not GSDMD or GSDME promotes NLRP3 inflammasome activation during extrinsic and intrinsic apoptosis.
Results
Caspase‐8 activation promotes GSDMD‐dependent cell lysis and caspase‐3/7‐dependent secondary necrosis
Because caspase‐1‐dependent pyroptosis precedes caspase‐3 activation, caspase‐3 is unlikely to suppress pyroptosis in inflammasome‐activated cells (He et al, 2015). Since caspase‐3 activation is a hallmark of apoptosis, we reasoned that caspase‐3‐dependent cleavage and inactivation of GSDMD at position D87 in humans or D88 in mice might counteract the lytic function of GSDMD during apoptosis. First, we investigated whether GSDMD promotes cell lysis during extrinsic apoptosis by stimulating wild‐type (WT) or Gsdmd −/− bone marrow‐derived macrophages (BMDMs) with TNF in combination with the SMAC‐mimetic AZD 5582 (hereafter referred as SM) or the TAK1 inhibitor 5z‐7‐oxozeaenol (hereafter referred as TAK1i), to induce the assembly of the caspase‐8‐activating ripoptosome complex (Vince et al, 2012; Dondelinger et al, 2013; Lawlor et al, 2015; Chen et al, 2018a). Surprisingly, caspase‐8 activation triggered GSDMD‐dependent cell lysis (Fig 1A and B) and induced hallmarks of pyroptosis including membrane ballooning and uptake of the membrane‐impermeable dye propidium iodide (PI) in WT macrophages (Fig 1C; black arrowhead; Movie EV1). By contrast, _Gsdmd_‐deficient cells retained membrane integrity and displayed classical apoptotic morphology such as cell shrinkage and the release of apoptotic bodies (Fig 1C; white arrowhead; Movie EV2). However, Gsdmd deficiency did not completely protect macrophages from caspase‐8‐dependent cell lysis (Fig 1A and B), and _Gsdmd_‐deficient apoptotic bodies incorporate PI over time (Fig 1C; white arrowhead). Therefore, we investigate whether GSDMD‐independent cell lysis is driven by caspase‐3 and/or caspase‐7‐dependent secondary necrosis. To investigate this possibility, we deleted caspase‐3, caspase‐7 or caspase‐3 and caspase‐7 in Gsdmd −/− immortalized BMDMs (iBMDMs) using CRISPR/Cas9 technology and stimulated these cells with TNF and SM or TAK1i. Indeed, Gsdmd −/− Casp3 −/−, Gsdmd −/− Casp7 −/− and Gsdmd −/− Casp3/7 −/− iBMDMs were resistant to cell lysis following caspase‐8 activation (Fig 1D and E). Taken together, our data indicate that extrinsic apoptosis promotes GSDMD‐dependent cell lysis in parallel with caspase‐3/7‐dependent secondary necrosis.
Figure 1. Extrinsic apoptosis trigger GSDMD‐dependent and caspase‐3/7‐dependent necrosis.
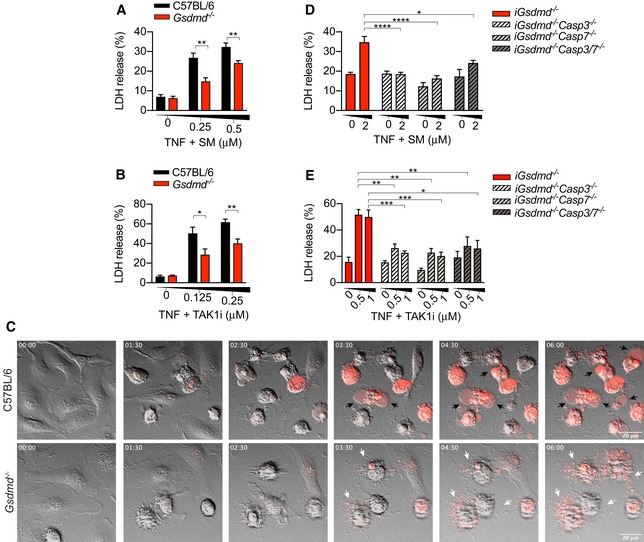
- A–E
Primary (A, B) or immortalized BMDMs (D, E) were stimulated with recombinant murine TNF (100 ng/ml) in combination with (A, D) SM or (B, E) TAK1i for 6 or 4 h, respectively. (C) Time‐lapse confocal images (hour:min) of BMDMs stimulated with recombinant murine TNF (100 ng/ml) and SM (250 nM) stained with propidium iodide (red) for 6 h. Black arrowheads indicate membrane ballooning, while white arrowheads indicate apoptotic bodies.
Data information: Data are means ± SEM of pooled data from (A‐B) five or (D‐E) eight independent experiments. Statistical analyses for normally distributed data sets were analysed using the parametric _t‐_test, whereas non‐normally distributed data sets were analysed using non‐parametric Mann–Whitney _t_‐tests. Data were considered significant when *P < 0.05, **P < 0.01, ***P < 0.001 or ****P < 0.0001. (C) Data are representative of three independent experiments.
GSDMD or GSDME pores do not promote NLRP3 assembly during apoptosis
Since caspase‐8 activation may indirectly promote GSDMD cleavage by activating the NLRP3 inflammasome (Vince et al, 2012; Lawlor et al, 2015), we next investigated cell death and GSDMD processing in WT versus inflammasome‐deficient BMDMs. Indeed, we observed that Nlrp3, Asc, Caspase‐1/11 deficiency similarly reduced TNF and SM‐ or TAK1i‐induced cell death (Figs 2A and EV1A). Unexpectedly, Gsdmd −/− BMDMs were even more protected than Nlrp3 −/− and Caspase‐1/11 −/− BMDMs following TNF and TAK1i stimulation (Fig 2A), suggesting that GSDMD may be activated by caspase‐1‐dependent and caspase‐1‐independent pathways. Consistent with that, we observed robust GSDMD processing into the active p30 and the appearance of an inactive p20 fragment, resulting from the inactivating cleavage by caspase‐3 at position D88, in WT, Nlrp3 and _Caspase‐1/11‐_deficient primary or immortalized BMDM after stimulation with TNF or LPS in combination with SM or TAK1i (Figs 2B and C, and EV1B and C). Caspase‐8 triggers NLRP3 activation through a potassium efflux‐dependent mechanism (Conos et al, 2017), suggesting that caspase‐8 likely triggers NLRP3 assembly by inducing plasma membrane damage. We therefore hypothesize that caspase‐1/11‐independent GSDMD activation promotes plasma membrane pore formation, potassium efflux and NLRP3 activation, analogous to NLRP3 activation by the non‐canonical inflammasome (Kayagaki et al, 2011; Ruhl & Broz, 2015). However, TNF or LPS costimulated with SM or TAK1i induced comparable levels of caspase‐1 p20 autoprocessing, a hallmark of inflammasome activation, between WT and Gsdmd −/− macrophages (Figs 2B, D, E and G, and EV1D and E), and extracellular potassium similarly reduced caspase‐1 processing in WT and Gsdmd −/− BMDMs (Fig 2D and E). Gasdermin E (GSDME), another pyroptotic effector from the gasdermin protein family, is activated by caspase‐3 during apoptosis and proposed to mediate secondary necrosis in BMDMs (Rogers et al, 2017; Wang et al, 2017). Consistent with previous reports, we observed robust GSDME processing into the active p30 fragment during apoptosis; however, Gsdme deficiency did not reduce cell death or caspase‐1 processing in WT or Gsdmd −/− macrophages (Figs 2F and G, and EV1F), indicating that caspase‐8 triggers potassium efflux and NLRP3 activation through GSDMD‐ or GSDME‐independent pores.
Figure 2. Extrinsic apoptosis triggers caspase‐1/11‐independent GSDMD processing and GSDMD/E‐independent NLRP3 activation.
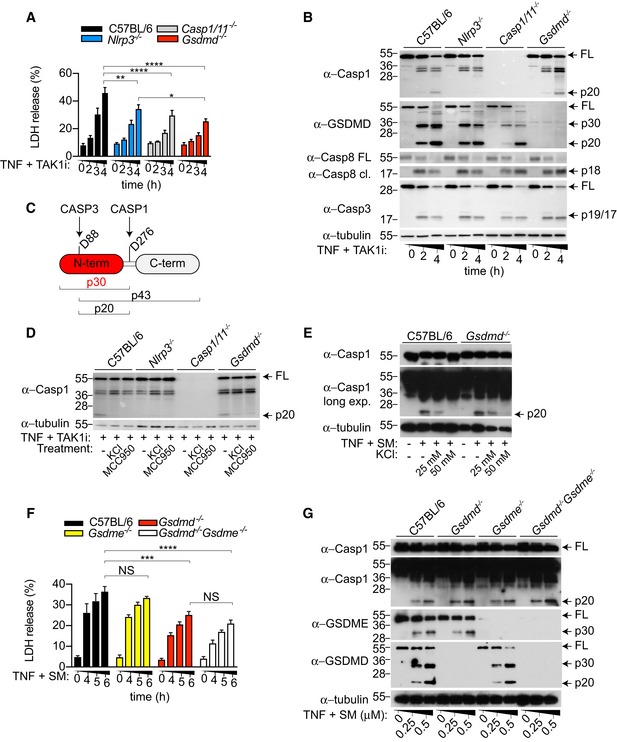
- A, B
BMDMs were stimulated with TNF (100 ng/ml) in combination with TAK1i (125 nM) for the indicated time points. (A) LDH release and (B) mixed supernatant and cell extracts were analysed. - C
Representation of known caspase cleavage site and molecular weight of corresponding cleavage fragment in mouse GSDMD. - D
BMDMs were costimulated with TNF (100 ng/ml) and TAK1i (125 nM) for 4 h in the presence or absence of KCl (50 mM). Where indicated, cells were pre‐incubated with MCC950 (10 μM) 20–30 min prior to TNF/TAK1i stimulation. - E–G
BMDMs were costimulated with TNF (100 ng/ml) and SM (E) (250 nM; 6 h), mixed supernatant and cell extracts were analysed by immunoblot, or (F) LDH release in the cell culture supernatant was quantified at the indicated time points.
Data information: Data are means ± SEM of pooled data from (A) four or (F) five independent experiments. Statistical analyses were performed using a two‐way ANOVA. Data were considered significant when *P < 0.05, **P < 0.01, ***P < 0.001 or ****P < 0.0001. All immunoblots are representative of three independent experiments.Source data are available online for this figure.
Figure EV1. Caspase‐8 triggers NLRP3/caspase‐1‐independent GSDMD processing.
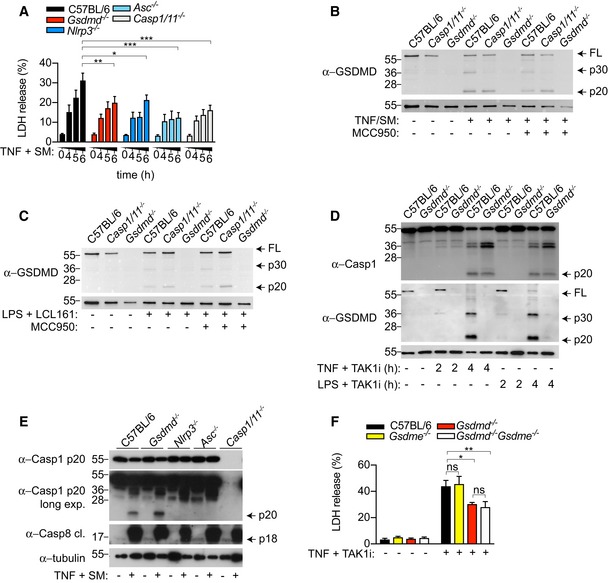
- A
BMDMs were costimulated with TNF (100 ng/ml) and SM (500 nM) for the indicated time points, and LDH release was quantified. - B, C
Immortalized BMDMs were (B) costimulated with TNF (100 ng/ml) and SM (500 nM) for 6 h or (C) primed with ultrapure E. coli K12 LPS (100 ng/ml) for 3 h prior to stimulation with the SMAC‐mimetic LCL161 (1 μM) for a further 16 h. Mixed supernatant and extracts were analysed by immunoblot. - D
BMDM were costimulated with ultrapure E. coli K12 LPS (100 ng/ml) or TNF (100 ng/ml) and TAK1i (125 nM) for 2 or 4 h, and mixed supernatant and extracts were analysed by immunoblot. - E
BMDMs were costimulated with TNF (100 ng/ml) and SM (500 nM) for 6 h, and mixed supernatant and extracts were analysed by immunoblot. - F
BMDMs were costimulated with TNF (100 ng/ml) and TAK1i (125 nM), and LDH release was quantified at 4 h.
Data information: Data are means ± SEM of pooled data from (A) three to six or (F) four individual experiments. (A) Statistical analyses were performed using a two‐way ANOVA (F) or a non‐parametric Mann–Whitney t_‐tests. Data were considered significant when *P < 0.05, **P < 0.01 and ***P_ < 0.001. Immunoblots are representative of two (E) or three (B–D) individual experiments.Source data are available online for this figure.
Caspase‐3 counteracts caspase‐8‐dependent GSDMD activation during extrinsic apoptosis
Since we observed that GSDMD processing is ablated in Ripk3 −/− Casp8 −/− during extrinsic apoptosis (Fig EV2A), and caspase‐1 and caspase‐8 recognize and cleave overlapping sequences in substrates such as pro‐IL‐1β (Maelfait et al, 2008; Vince et al, 2012), we hypothesized that caspase‐8 could directly trigger GSDMD activation. For this, we engineered a doxycycline‐inducible caspase‐8 construct, in which we replaced the DED domains of caspase‐8 with a DmrB domain to control caspase‐8 dimerization upon addition of the B/B homodimerizer drug. Next, we ectopically expressed DmrB‐caspase‐8 with WT full‐length GSDMD, a caspase‐1/11‐uncleavable D276A mutant, a caspase‐3‐uncleavable D88A mutant and a D88A D276A double mutant in HEK293T cells which are naturally deficient in caspase‐1 (Fig EV2B). Indeed, addition of doxycycline and B/B homodimerizer to induce caspase‐8 expression and dimerization triggered cleavage of WT GSDMD into the active p30 fragment and the appearance of the inactive p43 and p20 fragments, corresponding to cleavage of full‐length GSDMD or further processing of active p30 at position D88 by caspase‐3 (Figs 3A and 2C). Consistent with that, we observed an accumulation of active p30 and complete disappearance of the inactive p43 and p20 fragment in the D88A mutant. Conversely, caspase‐8 dimerization triggered an accumulation of the inactive p43 but not the appearance of active p30 and inactive p20 when D276 was mutated, indicating that caspase‐8 and caspase‐1 can process GSDMD at the same residue (Fig 3A); consistent with a recent report that caspase‐8 triggers direct GSDMD during Yersinia infection (Orning et al, 2018). To verify in vitro that human caspase‐1 and caspase‐8 cleave GSDMD at the same residue, we designed a GSDMD‐based substrate derived from the caspase‐1 cleavage site in GSDMD and monitored its cleavage using a fluorometric assay (Fig EV2C). Indeed, human caspase‐8 processed the GSDMD‐based substrate, though 30‐fold less efficiently than caspase‐1 (Fig EV2D and E). Since caspase‐3 is activated downstream of caspase‐8 during apoptosis, we next investigated whether the direct caspase‐8‐dependent GSDMD activation is counteracted by caspase‐3‐dependent GSDMD inactivation at position D88. For this, we reconstituted immortalized Gsdmd −/− BMDMs with either WT GSDMD (GSDMDWT) or a caspase‐3‐uncleavable D88A mutant (GSDMDD88A) by lentiviral transduction and monitored cell death after TNF and SM or LPS and LCL161 (SMAC mimetic)‐induced caspase‐8 activation. Remarkably, GSDMDD88A‐expressing iBMDMs were significantly more susceptible than GSDMDWT‐expressing controls to both TNF‐ and LPS‐induced apoptosis (Fig 3B and C). To investigate whether GSDMD inactivation at D88 is observed in primary macrophages, we generated a Gsdmd D88A knock‐in mouse. Gsdmd D88A/D88A animals were born at expected Mendelian ratio, healthy and developed a normal immune system (Appendix Fig S1A–F). Indeed, we observed an accumulation of the cytotoxic GSDMD p30 fragment and the disappearance of the inactive p20 fragment upon TNF and TAK1i or SM stimulation of Gsdmd D88A/D88A cells, which correlated with a 1.5‐fold enhancement in cell death (Figs 3D and E, and EV3A and B). Although the cytotoxic GSDMD p30 fragment was significantly enriched in Gsdmd D88A/D88A cells compared to WT littermates, caspase‐1 processing was unchanged between Gsdmd D88A/D88A and WT littermate controls (Figs 3D and EV3B), reiterating that GSDMD pores do not promote caspase‐8‐dependent NLRP3 activation.
Figure EV2. Caspase‐8 cleaves GSDMD at a lower efficiency than caspase‐1.

- BMDMs were stimulated with TNF (100 ng/ml) and SM (500 nM) for 6 h, and mixed supernatant and extracts were analysed by immunoblot, representative of three independent experiments.
- Caspase‐1 expression in HEK293T versus HeLa cells.
- Amino acid sequence of human gasdermin D. The fluorescence lifetime substrate Ac‐Cys(Pt14)‐FLTD^GVPY‐NH2 was designed around D276 as highlighted (red); ^ indicates the Casp1/8 cleavage site.
- The kinetic constants of the proteolysis of the FLT‐substrate Ac‐Cys(Pt14)‐FLTD^GVPY‐NH2 by Casp1/8 were determined from the time courses of product formation under initial velocity conditions. The K M value was obtained from measurements conducted at constant enzyme concentration (Casp1 = 30 nM; Casp8 = 833 nM) and different substrate concentrations as indicated.
- Comparison of kinetic constants determined for caspase‐1/8 cleavage of (Pt14)‐LETD^Y‐NH2 and Ac‐Cys(Pt14)‐FLTD^GVPY‐NH2.
Source data are available online for this figure.
Figure 3. Caspase‐3 suppresses caspase‐8‐dependent GSDMD activation and cell lysis during extrinsic apoptosis.
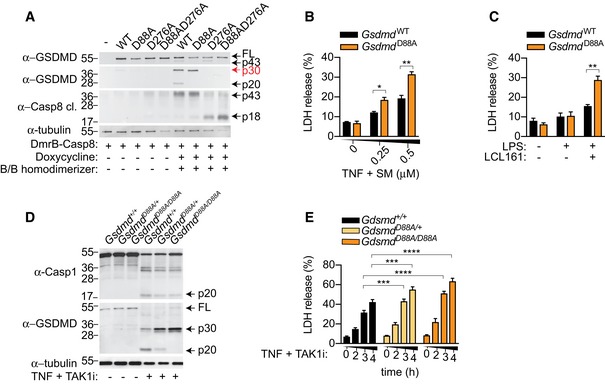
- A
HEK293T cells were transfected with doxycycline‐inducible DmrB‐caspase‐8 and the indicated GSDMD constructs. Cells were stimulated with doxycycline (10 μg/ml) for 18 h to induce DmrB‐caspase‐8 expression and exposed to B/B homodimerizer (12.5 nM) for another 2 h to activate caspase‐8. Mixed supernatant and extracts were analysed by immunoblot. - B, C
Immortalized Gsdmd −/− BMDM expressing GSDMDWT and GSDMDD88A were (B) costimulated with TNF (100 ng/ml) and SM for 6 h or (C) primed for 3 h with ultrapure E. coli K12 LPS (100 ng/ml) and stimulated with LCL161 (1 μM) for 24 h, and LDH release was quantified. - D, E
BMDMs were costimulated with TNF (100 ng/ml) and TAK1i for 4 h, (D) mixed supernatant and extracts were analysed by immunoblot, or (E) LDH release was quantified at the indicated time points.
Data information: All immunoblots are representative of three independent experiments. Data are means ± SEM of pooled data from (B‐C) three or (E) seven individual experiments. (B–C) Statistical analyses for normally distributed data sets were analysed using the parametric t‐_test, whereas non‐normally distributed data sets were analysed using non‐parametric Mann–Whitney t_‐tests. (E) Statistical analyses were performed using a two‐way ANOVA. Data were considered significant when *P < 0.05, **P < 0.01, ***P < 0.001 or ****P < 0.0001.Source data are available online for this figure.
Figure EV3. Gsdmd D88A/D88A BMDMs are more susceptible to extrinsic apoptosis.
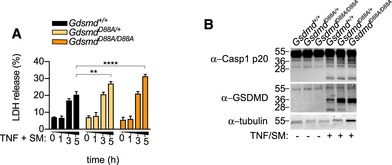
- A, B
BMDMs were costimulated with TNF (100 ng/ml) and SM (500 nM), (A) LDH release was quantified at the indicated time points, or (B) mixed supernatant and extracts were analysed at 5 h. (A) Data are means ± SEM of pooled data from three independent experiments. Statistical analyses were performed using a two‐way ANOVA Data were considered significant when **P < 0.01 or ****P < 0.0001. (B) Immunoblots are representative of three independent experiments.
Source data are available online for this figure.
The channel‐forming membrane protein pannexin‐1 promotes NLRP3 activation during extrinsic apoptosis
Since the RIPK1 kinase inhibitor Nec‐1s suppressed caspase‐8‐mediated cell death (Wang et al, 2008; Feoktistova et al, 2011; Tenev et al, 2011) (Fig 4A), and RIPK1 signalling is often associated with RIPK3, we next investigated the role of RIPK3 in promoting cell death and NLRP3 activation during TNF‐induced apoptosis. RIPK3 did not contribute to TNF and TAK1i‐induced cell lysis and GSDMD activation (Fig 4B and C); however, caspase‐1 processing was remarkably reduced in Ripk3 −/− compared to WT macrophages. Extracellular potassium and the NLRP3‐specific inhibitor MCC950 reduced GSDMD and processing in WT but not Ripk3 −/− cells, confirming that RIPK3 is indeed upstream of NLRP3 during TNF‐induced apoptosis (Fig 4C). RIPK3 kinase activity can drive NLRP3 activation by promoting MLKL pore formation and potassium efflux (Conos et al, 2017; Gutierrez et al, 2017). However, TNF and TAK1i did not trigger RIPK3‐dependent MLKL phosphorylation (Fig 4C) and MLKL did not contribute to cell death or caspase‐1 processing (Appendix Fig S2A and B). In agreement with a RIPK3 kinase‐independent role for driving NLRP3 activation, the RIPK3 kinase inhibitor GSK'872 did not suppress cell lysis (Appendix Fig S2C) but paradoxically promoted caspase‐1 processing (Fig 4C), most probably through altering the ripoptosome conformation (Mandal et al, 2014; Newton et al, 2014; Moriwaki et al, 2015). Consistent with previous reports, our data provide support for the hypothesis that it is the RIPK3 scaffolding function that promotes ripoptosome‐mediated caspase‐3 activation (Fig 4D) (Vince et al, 2012; Dondelinger et al, 2013). Since both caspase‐3 activation and caspase‐1 activation were reduced in Ripk3 −/− cells, we hypothesized that ripoptosome‐induced caspase‐3 activity drives potassium efflux and NLRP3 activation. Pannexin‐1, a channel‐forming glycoprotein, is activated by caspase‐3/7‐dependent cleavage at its C‐terminus during apoptosis (Chekeni et al, 2010; Sandilos et al, 2012). This cleavage event promotes the removal of its inhibitory C‐terminal domain, resulting in the opening of the pannexin‐1 channel, membrane permeability, ATP release and potassium efflux (Chekeni et al, 2010; Yang et al, 2015). We therefore investigated whether the ripoptosome promotes pannexin‐1 activity for NLRP3 assembly by using two well‐established pannexin‐1 inhibitors, probenecid (Silverman et al, 2008) and the antibiotic trovafloxacin (Poon et al, 2014). Remarkably, probenecid and trovafloxacin strongly reduced caspase‐1 activation during TNF‐induced apoptosis (Fig 4E; Appendix Fig S3A). By contrast, both inhibitors had no effect on caspase‐1 processing following nigericin or poly(dAdT) stimulation to activate NLRP3 or AIM2 inflammasome, respectively (Fig 4E, Appendix Fig S3B and C). Consistent with a potential role for pannexin‐1 in driving caspase‐8‐dependent NLRP3 activation following TNFR1 engagement, probenecid similarly reduced caspase‐1 processing in LPS and SM‐stimulated cells (Fig 4F). In summary, this indicated that the ripoptosome promotes pannexin‐1 activity to drive NLRP3 assembly upon TNFR1 and TLR4 stimulation. To validate genetically that pannexin‐1 drives NLRP3 activation upon ripoptosome activation, we stimulated WT and Panx1 −/− BMDMs with TNF and SM or TAK1i. Consistent with our inhibitor data (Fig 4E and F, Appendix Fig S3A), Panx1 deficiency dramatically reduced caspase‐1 processing compared to WT cells upon TNF and SM treatment (Figs 4G and EV4A). In line with the requirement for NLRP3 to amplify inflammatory cell death during caspase‐8 activation (Figs 2A and EV1A), Panx1 deficiency significantly reduced cell lysis and GSDMD processing following TNF and SM treatment (Fig 4G and H). Interestingly, while caspase‐1 processing was reduced in Panx1 −/− compared to WT BMDMs, it was not sufficient to reduce TNF‐ or LPS‐ and TAK1i‐induced cell lysis (Fig EV4A–C), possibly due to compensatory mechanisms from caspase‐8‐dependent GSDMD cleavage (Fig 2B) and caspase‐3/7‐dependent secondary necrosis during TAK1 inhibition (Fig 1E).
Figure 4. RIPK3 promotes caspase‐3 activation and pannexin‐1 activity to drive NLRP3 assembly during extrinsic apoptosis.
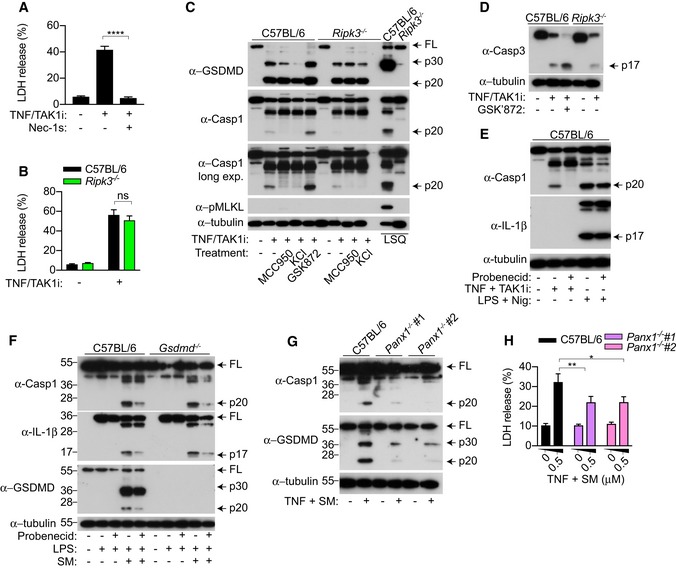
- A–E
BMDMs were costimulated with TNF (100 ng/ml) and TAK1i (125 nM) for 4 h, (A, B) LDH release was quantified, or (C, D, E) mixed supernatant and extracts were analysed by immunoblot. Where indicated, cells were treated with the inhibitors Nec‐1s (50 μM), MCC950 (10 μM), GSK'872 (1 μM), probenecid (1 mM) 20–30 min prior to cell stimulation. KCl (50 mM) was added together with TNF and TAK1i. (C) To induce necroptosis, BMDMs were primed for 3 h with ultrapure E. coli K12 LPS (100 ng/ml) and Q‐VD‐OPh (10 μM) was added at the last 20–30 min of priming and stimulated with SM (500 nM) for 4 h. (E) To activate the NLRP3 inflammasome, BMDMs were primed with ultrapure E. coli K12 LPS (100 ng/ml) for 4 h and stimulated with nigericin (10 μM) for 1 h. - F
BMDMs were primed for 3 h with ultrapure E. coli K12 LPS (100 ng/ml) and stimulated with SM (0.5 μM) for a further 4 h. Probenecid (1 mM) was added 20–30 min prior to cell stimulation, and mixed supernatant and extracts were analysed by immunoblot. - G, H
BMDMs were stimulated with TNF (100 ng/ml) and SM (0.5 μM) for 6 h, (G) mixed supernatant and extracts were analysed by immunoblot, or (H) LDH release was quantified.
Data information: All immunoblots are representative of three independent experiments. (A, B, H) Data are means ± SEM of pooled data from (A, H) four or (B) three independent experiments. Statistical analyses for normally distributed data sets were analysed using the parametric t‐_test, whereas non‐normally distributed data sets were analysed using non‐parametric Mann–Whitney t_‐tests. Data were considered significant when *P < 0.05, **P < 0.01 or ****P < 0.0001.Source data are available online for this figure.
Figure EV4. Extrinsic and intrinsic apoptosis promote NLRP3 assembly through pannexin‐1.
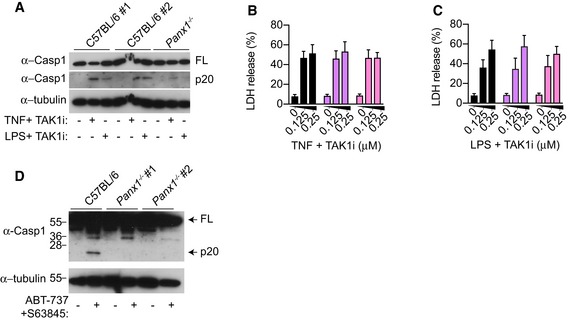
- A–C
BMDMs were stimulated with TNF (100 ng/ml) or E. coli K12 LPS (100 ng/ml) and TAK1i (125 nM) for 4 h, mixed supernatant and extracts were analysed by immunoblot (A), or (B‐C) LDH release was quantified. - D
Unprimed BMDMs were stimulated with ABT‐737 (500 nM) and S63845 (500 nM) for 6 h, and mixed supernatant and extracts were analysed by immunoblot.
Data information: Immunoblots are representative of three independent experiments. Data are mean ± SEM of pooled data from (B, C) four independent experiments.Source data are available online for this figure.
Pannexin‐1 but not GSDMD or GSDME promotes NLRP3 activation during intrinsic apoptosis
Having established that extrinsic apoptosis triggers GSDMD‐dependent cell lysis and pannexin‐1‐dependent NLRP3 activation, we next investigate whether the same pathway occurs during intrinsic apoptosis. For this, we stimulated WT or Gsdmd −/− BMDM with a combination of the BH3 mimetic ABT‐737 and the MCL‐1 inhibitor S63845 to activate mitochondrial apoptosis (van Delft et al, 2006; Kotschy et al, 2016). ABT‐737 and S63845 cotreatment triggered cell lysis over time; however, in contrast to extrinsic apoptosis (Fig 1A and B), ABT‐737/S63845‐induced cell lysis was GSDMD‐independent (Fig 5A and C). In agreement with that, BMDMs derived from Gsdmd D88A/D88A mice in which GSDMD cannot be inactivated by caspase‐3 did not display an enhancement in cell lysis upon ABT‐737/S63845 treatment (Fig 5B). Caspase‐3 is activated during mitochondrial apoptosis, and a recent study reported that caspase‐3‐dependent GSDME activation drives secondary necrosis in BMDMs (Rogers et al, 2017). In agreement with our earlier data (Fig 2G), ABT‐737/S63845 also triggered robust GSDME processing into the active p30 fragment (Fig 5D); however, GSDME p30 did not contribute to mitochondrial apoptosis‐induced cell lysis, since intracellular LDH release was GSDME‐independent (Fig 5E). Gasdermin pores may promote potassium efflux to activate NLRP3 during intrinsic apoptosis; however, consistent with earlier observations (Figs 2B, D, E and G, and EV1D and E), caspase‐1 processing was comparable between WT, Gsdmd −/− , Gsmde −/− or Gsdmd −/− Gsmde −/− BMDMs, indicating that NLRP3 assembly is driven by GSDMD‐ or GSDME‐independent mechanisms (Fig 5F). Since caspase‐8‐dependent NLRP3 activation is pannexin‐1‐dependent (Fig 4G), we examined whether pannexin‐1 similarly promotes NLRP3 activation during mitochondrial apoptosis. Caspase‐1 processing was remarkably reduced in LPS‐primed and LPS‐unprimed Panx1 −/− BMDMs compared to WT cells (Figs 5G and EV4D). Given that caspase‐1 and caspase‐8 both promote pro‐IL‐1β cleavage during apoptosis (Chauhan et al, 2018; Vince et al, 2018), Panx1 deficiency only partially reduced IL‐1β secretion into the cell culture supernatant compared to WT cells (Fig 5H).
Figure 5. Intrinsic apoptosis drives gasdermin‐independent cell lysis but promotes NLPR3 assembly through pannexin‐1 activity.
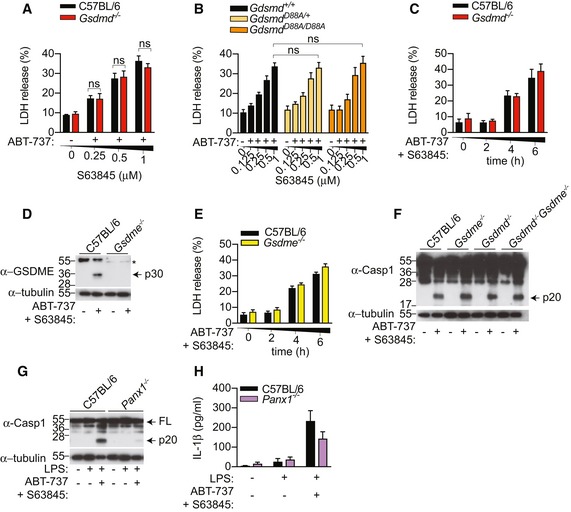
- A, B
BMDMs were stimulated with an increasing dose of S63845 in the presence of ABT‐737 (0.5 μM), and LDH release was quantified at 6 h. - C–F
BMDMs were stimulated with ABT‐737 (0.5 μM) and S63845 (0.5 μM), LDH release was quantified (C, E), or mixed supernatant and extracts were analysed by immunoblot at 6 h (D, F). - G, H
BMDMs were primed with ultrapure E. coli K12 LPS (100 ng/ml) for 3 h and further stimulated with ABT‐737 (1 μM) and S63845 (1 μM) for 24 h, mixed supernatant and extracts were analysed by immunoblot (G), and IL‐1β in cell‐free supernatant was quantified by ELISA (H).
Data information: All immunoblots are representative of three independent experiments. Data are means ± SEM of pooled data from (A) four, (B) five or (C, E, H) three independent experiments.Source data are available online for this figure.
Discussion
Here, we demonstrate that during extrinsic apoptosis caspase‐8 directly cleaves GSDMD to initiate inflammatory cell death that is further amplified by the NLRP3 inflammasome. By contrast, the closely related gasdermin family member GSDME is dispensable for macrophage cell lysis downstream of the ripoptosome (Fig EV5). A recent study proposed that caspase‐8‐dependent GSDMD activation triggers plasma membrane pores and potassium efflux to activate the NLRP3 inflammasome (Orning et al, 2018). However, we observed that Gsdmd deficiency did not impact caspase‐1 processing upon caspase‐8 activation, and extracellular potassium similarly reduced caspase‐1 processing in both WT and _Gsdmd‐_deficient cells. In addition, by generating a novel Gsdmd D88A/D88A mouse, we provide genetic evidence that accumulation of GSDMD p30 pores upon caspase‐8 activation does not enhance caspase‐1 cleavage. Instead, we propose that the RIPK3 scaffolding function promotes apoptotic caspase activation, pannexin‐1 channel activity and potassium efflux downstream of the ripoptosome to trigger NLRP3 inflammasome activation (Fig EV5). In line with this, we also demonstrate that gasdermins do not promote macrophage cell lysis or NLRP3 activation during mitochondrial apoptosis. Instead, our data indicate that pannexin‐1 is a universal requirement for both intrinsic and extrinsic apoptosis to drive NLRP3 activation (Fig EV5). During apoptosis, caspase‐3 and caspase‐7 cleave the channel‐forming glycoprotein pannexin‐1 at its C‐terminus, which activates its channel activity to promote membrane permeability, ATP release and potassium efflux (Chekeni et al, 2010; Yang et al, 2015). We propose that pannexin‐1 likely drives NLRP3 assembly by promoting potassium efflux, but not autocrine or paracrine ATP signalling, since caspase‐1 activation is unaffected in P2x7 −/− macrophages during apoptosis (Vince et al, 2012).
Figure EV5. Schematic for ripoptosome‐ and apoptosome‐mediated cell death and NLRP3 activation.
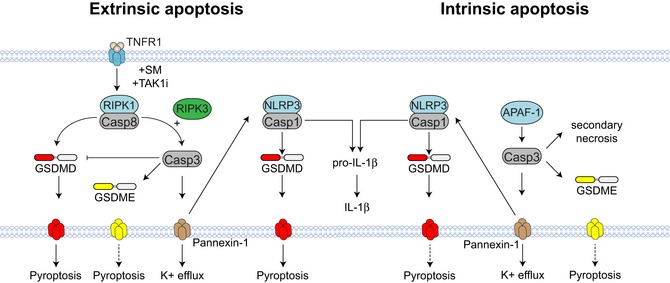
TNFR1 signalling in the presence of SM or TAK1i promotes the assembly of the ripoptosome, a caspase‐8 activating platform. Caspase‐8 triggers direct GSDMD activation to induce cell lysis, which is amplified by the NLRP3 inflammasome. The cytotoxic function of caspase‐8 is suppressed by caspase‐3‐mediated inactivation of GSDMD. Caspase‐3 cleaves GSDME, but GSDME does not contribute to cell death in BMDM. Finally, RIPK3 promotes caspase‐3 activation downstream of the ripoptosome to promote pannexin‐1 activity, potassium efflux and NLRP3 activation. By contrast, GSDMD does not promote cell lysis during mitochondrial apoptosis. The related gasdermin family member, GSDME, is processed into its active form during mitochondrial apoptosis, but does not promote cell lysis. Consistent with TNF‐induced apoptosis, pannexin‐1 but not gasdermins promote NLRP3 activation during mitochondrial apoptosis.
Our finding that certain cancer chemotherapeutics such as SMAC mimetics, BH3 mimetics and MCL1 inhibitors drive inflammation by promoting GSDMD‐dependent cell lysis and activation of the pannexin‐1‐NLRP3 signalling axis has major clinical implications. While some studies documented that _Nlrp3_‐ and _Il1b‐_deficient animals are susceptible to tumorigenesis and are poorly able to clear tumours during chemotherapy, emerging literature now indicates that excessive IL‐1β levels correlate with poor prognosis in a variety of cancers (Lewis et al, 2006; Ghiringhelli et al, 2009). This indicates that optimal IL‐1β signalling is required for antitumour immunity, while prolonged IL‐1β signalling may be detrimental. Whether chemotherapy‐induced NLRP3 activation promotes or suppresses tumour clearance is unclear, and is likely to be tumour‐specific. Therefore, future studies should determine whether known pannexin‐1 inhibitors, such as probenecid (Silverman et al, 2008) and spironolactone (Good et al, 2018), can suppress NLRP3 and IL‐1β activation during chemotherapy to promote tumour clearance. Further, it is unclear whether side effects associated with chemotherapeutics (e.g. SMAC mimetics) and clinical inhibitors (e.g. TAK1i) are a result of cell death, cytokine processing or both. Therefore, future studies should investigate whether GSDMD (Sollberger et al, 2018), NLRP3 (Coll et al, 2015) or pannexin‐1 inhibition can reduce side effects of such clinical inhibitors. Lastly, our finding that trovafloxacin, an antibiotic with known safety concerns (Ahmad, 2001), blocks NLRP3 inflammasome activation during apoptosis suggests that additional studies should be performed to ensure the safe usage of these drugs in the clinic.
Materials and Methods
Mice
All experiments were performed with approval from the veterinary office of the Canton de Vaud and according to the guidelines from the Swiss animal protection law (licence VD3257). C57BL/6J mice were purchased from Janvier Labs (France) and housed at specific‐pathogen‐free facility at the University of Lausanne. Nlrp3 −/− , Asc −/− , Caspase‐1/11 −/− , Gsdmd −/− , Ripk3 −/−, Ripk3 −/− Casp8 −/−, Mlkl −/− and Panx1 −/− mice have been previously described. Gsdme −/− and Gsdmd D88A/D88A mice were generated at Center for Transgenic model of the University of Basel using CRISPR/Cas9 genome targeting as follows: Gsdme −/−: Guide RNAs targeting exon 2 of the mouse Gsdme gene were designed using gRNA sequence (including PAM) ACTCTTCGTTTGGAACCCTGAGG. Injection of the gRNAs and Cas9 protein into C57BL/6 embryos was done according to standard methods. Biopsies for genotyping were taken at an age of 10–12 days. DNA extraction was performed using the KAPA HotStart Mouse Genotyping Kit according to the manufacturer's protocol. Genotyping PCR was done using Q5 Polymerase (NEB) using primers GSDME_ex2_fw2 (CTGCCCATGACAACTGAGGT) and GSDME_ex_rv2 (AGGGCAGTTACAGGAGCCTA), which were designed using Primer3 v.0.4.0 giving a fragment of 529 bp. The mutation was identified as a 1‐bp insertion in exon 2 of Gsdme resulting in a premature stop codon. GSDMD D88A/D88A: Guide RNAs targeting exon 2 of the Gsdmd gene were designed using gRNA sequence (including PAM) AAGTCTCTGATGTCGTCGATGGG. gRNAs and Cas9 protein were coinjected with a 200nt HDR oligo inducing the following mutations: an AT>CC mutation to affect the D>A change and a silent G>C mutation to destroy the PAM sequence. This also created a GCCGGC restriction site (NaeI) to genotype the KI animals. Genotyping PCR was done using primers GSDMD_fw2 (TACAGACGGTTGTGAGCCA) and GSDMD_rv2 (GCTTCCCTCATTCAGTGCT) giving a fragment of 597 bp. Gsdmd −/− Gsdme −/− mice were created by crossing Gsdmd −/− and Gsdme −/− mice.
Cell culture
Bone marrow‐derived macrophages were differentiated in DMEM (Gibco) supplemented with 20% MCSF (3T3 supernatant), 10% heat‐inactivated FCS (Bioconcept), 10 mM HEPES (Bioconcept), penicillin/streptomycin (Bioconcept) and non‐essential amino acids (Gibco), and stimulated on day 7–9 of differentiation. Immortalized BMDMs were maintained in DMEM (Gibco) supplemented with 10% MCSF (3T3 supernatant), 10% heat‐inactivated FCS (Bioconcept), 10 mM HEPES (Bioconcept) and non‐essential amino acids (Gibco). HEK293T and HeLa cells were cultured in DMEM (Gibco) supplemented with 10 mM HEPES (Bioconcept), non‐essential amino acids (Gibco) and 5 or 10% heat‐inactivated FCS (Bioconcept), respectively.
Lentiviral transduction of mouse immortalized Gsdmd −/− BMDMs
To produce lentiviral particles, 1 × 106 HEK293T cells were seeded in a six‐well plate and transfected with 2 μg lentiviral plasmid, 2 μg psPax2 and 0.4 μg VSV‐G using polyethylenimine (Polysciences, Inc) for 6 h. The cell culture media were replaced with fresh DMEM (Gibco) supplemented with 10% MCSF (3T3 supernatant), 10% heat‐inactivated FCS (Bioconcept), 10 mM HEPES (Bioconcept), non‐essential amino acids (Gibco) and 1× penicillin/streptomycin (Bioconcept), and lentiviral particles were collected and filtered (0.45 μm) 24 h later. Polybrene (6.25 μg/ml; Merck) was added to the filtered solution and added dropwise to the immortalized Gsdmd −/− BMDMs (seeded 1 day before at 5 × 105 cells per 6 wells), and cells were spin‐infected (1,800 g, 90 min, 37°C) with these particles and incubated at 37°C with 5% CO2. 3 days later, cells were split into medium containing puromycin (10 μg/ml, InvivoGen) for selection and used 4 days after selection.
Generation of CRISPR knockouts in immortalized BMDMs
_Caspase‐3, Caspase‐7 and Caspase‐3/7‐_deficient immortalized BMDM (iBMDM) were generated using the genome editing system Alt‐R‐CRISPR/Cas (IDT) according to the manufacturer's protocol. Briefly, the gene‐specific targeting crRNA (Caspase‐7: GATAAGTGGGCACTCGGTCC TGG, Caspase‐3: AATGTCATCTCGCTCTGGTA CGG or TGGGCCTGAAATACCAAGTC AGG) was mixed with the universal RNA oligo tracrRNA to form a gRNA complex (crRNA:tracrRNA). The addition of the recombinant Cas9 nuclease V3 allowed the formation of an RNP complex specific for targeting the Caspase‐3 or Caspase‐7 genes. The tracrRNA only or RNP complexes were subsequently reverse‐transfected into immortalized Gsdmd −/− iBMDM using RNAiMax (Invitrogen). The bulk population was tested for successful gene mutation using the T7 endonuclease digestion assay as follows: cells were lysed by the KAPA Biosystems Kit according to the manufacturer's protocol, and genomic DNA flanking the guide RNA (crRNA) binding site was amplified by PCR using gene‐specific primers (Caspase‐7: fw: TTGCCTGACCCAAGGTTTGT, rv: CCCAGCAACAGGAAAGCAAC; Caspase‐3: fw: GTGGGGGATATCGCTGTCAT, rv: TGTGTAAGGATGCGGACTGC). The amplified genomic DNA was used to perform the heteroduplex analysis according to the manufacturer's protocol (IDT). Single clones were derived from the bulk population by limiting dilution, and the absence of protein expression in single clones was verified by immunoblotting.
HEK293T DmrB‐caspase‐8 dimerization system
HEK293T cells were seeded in 96‐well plate at 2.5 × 104 cells per well the day prior to transfection. Cells were transfected with 300 ng of plasmid using linear polyethylenimine (900 ng; Polysciences, Inc) per well, according to the manufacturer's protocol, in plain DMEM (Gibco) and centrifuged at 300 g for 5 min at 37°C. Media were replaced with fresh DMEM 6 h later and supplemented with 10 μg/ml doxycycline (Sigma) for 18 h to induce DmrB‐caspase‐8 expression. Cells were exposed with 12.5 nM B/B homodimerizer (Clontech) for 2 h in plain DMEM to induce DmrB‐caspase‐8 homodimerization, and cell extracts were lysed in boiling lysis buffer and analysed by immunoblotting.
Apoptosis and necroptosis assay
Primary and immortalized BMDMs were seeded in 96‐well plates at 5 × 104 cells per well a day prior to stimulation. For all macrophage cell stimulation, the cell culture medium was replaced with Opti‐MEM (Gibco). Macrophages were treated with ABT‐737 (500 nM; Selleckchem) and S63845 (125–1,000 nM; Selleckchem) in Opti‐MEM (Gibco) to induce mitochondrial apoptosis. Alternatively, cells were simultaneously stimulated with recombinant murine TNF (100 ng/ml; Peprotech) and the SMAC‐mimetic AZD 5582 (250–500 nM; Selleckchem) or the TAK1 inhibitor 5z 7‐oxozeaenol (125–250 nM; Sigma) in Opti‐MEM (Gibco) to trigger TNF‐dependent apoptosis. To trigger TLR4‐apoptosis, cells were primed with ultrapure E. coli K12 LPS (InvivoGen) for 3 h and stimulated with AZD 5582 (500 nM) for a further 4 h. To induce LPS‐induced necroptosis, cells were treated with Q‐VD‐OPh (10 μM; Selleckchem) during the last 20 min of LPS priming to block caspase‐8 activity prior to AZD 5582 (500 nM) treatment for a further 4 h. In some experiments, BMDMs were treated with MCC950 (10 μM; Sigma), GSK872 (1 μM; Selleckchem), Nec‐1s (50 μM; Abcam), Probenecid (1 mM; Sigma) and trovafloxacin (5–10 μM; Sigma) 20–30 min prior to cell stimulation.
Inflammasome assay
Primary BMDMs were plated in 96‐well plates at 5 × 104 cells per well a day prior to stimulation. Cell were primed with ultrapure E. coli K12 LPS (InvivoGen) for 4 h in Opti‐MEM and stimulated with nigericin (10 μM; Sigma) for 1 h or transfected with 0.2 μg/well of poly(dA:dT) (InvivoGen) using Polyethylenimine (Polysciences, Inc) and centrifuged at 300 g for 5 min at room temperature and incubated for 1 h. Where indicated, cells were treated with Probenecid (1 mM; Sigma) and trovafloxacin (5–10 μM; Sigma) at the last 20–30 min of priming.
LDH release assay
LDH release into the cell culture supernatant was quantified using CytoTox 96 non‐radioactive cytotoxicity assay (Promega) and expressed as a percentage of total cellular LDH (100% lysis).
Western blotting
Cell‐free methanol/chloroform‐precipitated supernatant was resuspended with cell extracts lysed in boiling lysis buffer (66 mM Tris–Cl pH 7.4, 2% SDS, 10 mM DTT, NuPage LDS sample buffer; Thermo Fisher) and separated on 14% polyacrylamide gels. Proteins were transferred onto nitrocellulose membrane using Trans‐Blot Turbo (Bio‐Rad). Antibodies for immunoblot were against GSDMD (EPR19828; Abcam; 1:1,000), GSDME (EPR19859; Abcam; 1:1,000), caspase‐1 p20 (casper‐1; Adipogen; 1:1,000), full‐length caspase‐8 (4927; Cell Signaling; 1:1,000), cleaved caspase‐8 (9429; Cell Signalling; 1:1,000), caspase‐3 (9662; Cell Signalling; 1:1,000), phosphorylated MLKL (EPR9515(2); Abcam; 1:1,000), pro‐IL‐1β (AF‐401‐NA, R&D; 1:1,000) and alpha‐tubulin (DM1A; Abcam; 1:2,000).
Live cell imaging
BMDMs were seeded at 2.5 × 104 per well in eight‐well tissue culture plates (Ibidi) a day before imaging. Cells were stimulated with TNF (100 ng/ml) and SM (250 nM) in Opti‐MEM, and cells were stained with propidium iodide (0.5 μg/ml; Sigma). Images were acquired every 5 min over 6 h using a Zeiss LSM800 point scanning confocal microscope equipped with 63× Plan‐Apochromat NA 1.4 oil objective, Zeiss ESID detector module, LabTek heating/CO2 chamber and motorized scanning stage.
Statistical analyses
Statistical analyses were performed using GraphPad Prism 7 software. All data sets were analysed for normality using Shapiro–Wilk normality test. Normally distributed data sets were analysed using the parametric _t‐_test, whereas non‐normally distributed data sets were analysed using non‐parametric Mann–Whitney _t_‐tests. A two‐way ANOVA was used to analyse repeated measures over time. Data were considered significant when *P ≤ 0.05, **P ≤ 0.01, ***P ≤ 0.001 or ****P ≤ 0.0001.
Author contributions
KWC and BD designed, performed and analysed all experiments with the exception of confocal microscopy and GSDMD substrate cleavage assay that were performed by KS, and AB and CJF, respectively. RH generated CRISPR knockouts. PP generated Gsdme −/− and Gsdmd D88A/D88A mice. KWC and PB designed and supervised the study and wrote the paper. PB oversaw the study.
Conflict of interest
A.B and C.J.F are employees of Novartis, Inc.
Supporting information
Appendix
Expanded View Figures PDF
Movie EV1
Movie EV2
Source Data for Expanded View and Appendix
Review Process File
Source Data for Figure 2
Source Data for Figure 3
Source Data for Figure 4
Source Data for Figure 5
Acknowledgements
This work was supported by grants from the European Research Council (ERC‐2017‐CoG—770988—InflamCellDeath) to P.B. and a Swiss Government Excellence (ESKAS) postdoctoral fellowship (2018.0618) to K.W.C. Microscopy and FACS analyses were performed at the UNIL Core Facilities. We thank Prof. Andy Wullaert, Prof. Wei‐Lynn Wong and Prof. Nathalie Rouach for sharing Ripk3 −/−, Mlkl −/− and Panx1 −/− bone marrow respectively; Prof. Thomas Henry and Aubry Tardivel for sharing Asc −/− bone marrow; Cristina Ramon‐Barros and Heide Oller for technical assistance; and Dr. James Vince and Dr. Ivan Poon for advice and discussion.
The EMBO Journal (2019) 38: e101638
References
- Aglietti RA, Estevez A, Gupta A, Ramirez MG, Liu PS, Kayagaki N, Ciferri C, Dixit VM, Dueber EC (2016) GsdmD p30 elicited by caspase‐11 during pyroptosis forms pores in membranes. Proc Natl Acad Sci USA 113: 7858–7863 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahmad K (2001) Drug company sued over research trial in Nigeria. Lancet 358: 815 [DOI] [PubMed] [Google Scholar]
- Broz P, Dixit VM (2016) Inflammasomes: mechanism of assembly, regulation and signalling. Nat Rev Immunol 16: 407–420 [DOI] [PubMed] [Google Scholar]
- Chauhan D, Bartok E, Gaidt MM, Bock FJ, Herrmann J, Seeger JM, Broz P, Beckmann R, Kashkar H, Tait SWG, Muller R, Hornung V (2018) BAX/BAK‐induced apoptosis results in caspase‐8‐dependent IL‐1beta maturation in macrophages. Cell Rep 25: 2354–2368.e5 [DOI] [PubMed] [Google Scholar]
- Chekeni FB, Elliott MR, Sandilos JK, Walk SF, Kinchen JM, Lazarowski ER, Armstrong AJ, Penuela S, Laird DW, Salvesen GS, Isakson BE, Bayliss DA, Ravichandran KS (2010) Pannexin 1 channels mediate “find‐me” signal release and membrane permeability during apoptosis. Nature 467: 863–867 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen KW, Schroder K (2013) Antimicrobial functions of inflammasomes. Curr Opin Microbiol 16: 311–318 [DOI] [PubMed] [Google Scholar]
- Chen KW, Lawlor KE, von Pein JB, Boucher D, Gerlic M, Croker BA, Bezbradica JS, Vince JE, Schroder K (2018a) Cutting edge: blockade of inhibitor of apoptosis proteins sensitizes neutrophils to TNF‐ but not lipopolysaccharide‐mediated cell death and IL‐1beta secretion. J Immunol 200: 3341–3346 [DOI] [PubMed] [Google Scholar]
- Chen KW, Monteleone M, Boucher D, Sollberger G, Ramnath D, Condon ND, von Pein JB, Broz P, Sweet MJ, Schroder K (2018b) Noncanonical inflammasome signaling elicits gasdermin D‐dependent neutrophil extracellular traps. Sci Immunol 3: pii: eaar6676 [DOI] [PubMed] [Google Scholar]
- Coll RC, Robertson AA, Chae JJ, Higgins SC, Munoz‐Planillo R, Inserra MC, Vetter I, Dungan LS, Monks BG, Stutz A, Croker DE, Butler MS, Haneklaus M, Sutton CE, Nunez G, Latz E, Kastner DL, Mills KH, Masters SL, Schroder K_et al_ (2015) A small‐molecule inhibitor of the NLRP3 inflammasome for the treatment of inflammatory diseases. Nat Med 21: 248–255 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conos SA, Chen KW, De Nardo D, Hara H, Whitehead L, Nunez G, Masters SL, Murphy JM, Schroder K, Vaux DL, Lawlor KE, Lindqvist LM, Vince JE (2017) Active MLKL triggers the NLRP3 inflammasome in a cell‐intrinsic manner. Proc Natl Acad Sci USA 114: E961–E969 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Czabotar PE, Lessene G, Strasser A, Adams JM (2014) Control of apoptosis by the BCL‐2 protein family: implications for physiology and therapy. Nat Rev Mol Cell Biol 15: 49–63 [DOI] [PubMed] [Google Scholar]
- van Delft MF, Wei AH, Mason KD, Vandenberg CJ, Chen L, Czabotar PE, Willis SN, Scott CL, Day CL, Cory S, Adams JM, Roberts AW, Huang DC (2006) The BH3 mimetic ABT‐737 targets selective Bcl‐2 proteins and efficiently induces apoptosis via Bak/Bax if Mcl‐1 is neutralized. Cancer Cell 10: 389–399 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding J, Wang K, Liu W, She Y, Sun Q, Shi J, Sun H, Wang DC, Shao F (2016) Pore‐forming activity and structural autoinhibition of the gasdermin family. Nature 535: 111–116 [DOI] [PubMed] [Google Scholar]
- Dondelinger Y, Aguileta MA, Goossens V, Dubuisson C, Grootjans S, Dejardin E, Vandenabeele P, Bertrand MJ (2013) RIPK3 contributes to TNFR1‐mediated RIPK1 kinase‐dependent apoptosis in conditions of cIAP1/2 depletion or TAK1 kinase inhibition. Cell Death Differ 20: 1381–1392 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feoktistova M, Geserick P, Kellert B, Dimitrova DP, Langlais C, Hupe M, Cain K, MacFarlane M, Hacker G, Leverkus M (2011) cIAPs block ripoptosome formation, a RIP1/caspase‐8 containing intracellular cell death complex differentially regulated by cFLIP isoforms. Mol Cell 43: 449–463 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghiringhelli F, Apetoh L, Tesniere A, Aymeric L, Ma Y, Ortiz C, Vermaelen K, Panaretakis T, Mignot G, Ullrich E, Perfettini JL, Schlemmer F, Tasdemir E, Uhl M, Genin P, Civas A, Ryffel B, Kanellopoulos J, Tschopp J, Andre F_et al_ (2009) Activation of the NLRP3 inflammasome in dendritic cells induces IL‐1beta‐dependent adaptive immunity against tumors. Nat Med 15: 1170–1178 [DOI] [PubMed] [Google Scholar]
- Good ME, Chiu YH, Poon IKH, Medina CB, Butcher JT, Mendu SK, DeLalio LJ, Lohman AW, Leitinger N, Barrett E, Lorenz UM, Desai BN, Jaffe IZ, Bayliss DA, Isakson BE, Ravichandran KS (2018) Pannexin 1 channels as an unexpected new target of the anti‐hypertensive drug spironolactone. Circ Res 122: 606–615 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gutierrez KD, Davis MA, Daniels BP, Olsen TM, Ralli‐Jain P, Tait SW, Gale M Jr, Oberst A (2017) MLKL activation triggers NLRP3‐mediated processing and release of IL‐1beta independently of gasdermin‐D. J Immunol 198: 2156–2164 [DOI] [PMC free article] [PubMed] [Google Scholar]
- He WT, Wan H, Hu L, Chen P, Wang X, Huang Z, Yang ZH, Zhong CQ, Han J (2015) Gasdermin D is an executor of pyroptosis and required for interleukin‐1beta secretion. Cell Res 25: 1285–1298 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kayagaki N, Warming S, Lamkanfi M, Vande Walle L, Louie S, Dong J, Newton K, Qu Y, Liu J, Heldens S, Zhang J, Lee WP, Roose‐Girma M, Dixit VM (2011) Non‐canonical inflammasome activation targets caspase‐11. Nature 479: 117–121 [DOI] [PubMed] [Google Scholar]
- Kayagaki N, Stowe IB, Lee BL, O'Rourke K, Anderson K, Warming S, Cuellar T, Haley B, Roose‐Girma M, Phung QT, Liu PS, Lill JR, Li H, Wu J, Kummerfeld S, Zhang J, Lee WP, Snipas SJ, Salvesen GS, Morris LX_et al_ (2015) Caspase‐11 cleaves gasdermin D for non‐canonical inflammasome signalling. Nature 526: 666–671 [DOI] [PubMed] [Google Scholar]
- Kotschy A, Szlavik Z, Murray J, Davidson J, Maragno AL, Le Toumelin‐Braizat G, Chanrion M, Kelly GL, Gong JN, Moujalled DM, Bruno A, Csekei M, Paczal A, Szabo ZB, Sipos S, Radics G, Proszenyak A, Balint B, Ondi L, Blasko G_et al_ (2016) The MCL1 inhibitor S63845 is tolerable and effective in diverse cancer models. Nature 538: 477–482 [DOI] [PubMed] [Google Scholar]
- Lafont E, Draber P, Rieser E, Reichert M, Kupka S, de Miguel D, Draberova H, von Massenhausen A, Bhamra A, Henderson S, Wojdyla K, Chalk A, Surinova S, Linkermann A, Walczak H (2018) TBK1 and IKKepsilon prevent TNF‐induced cell death by RIPK1 phosphorylation. Nat Cell Biol 20: 1389–1399 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lawlor KE, Khan N, Mildenhall A, Gerlic M, Croker BA, D'Cruz AA, Hall C, Kaur Spall S, Anderton H, Masters SL, Rashidi M, Wicks IP, Alexander WS, Mitsuuchi Y, Benetatos CA, Condon SM, Wong WW, Silke J, Vaux DL, Vince JE (2015) RIPK3 promotes cell death and NLRP3 inflammasome activation in the absence of MLKL. Nat Commun 6: 6282 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis AM, Varghese S, Xu H, Alexander HR (2006) Interleukin‐1 and cancer progression: the emerging role of interleukin‐1 receptor antagonist as a novel therapeutic agent in cancer treatment. J Transl Med 4: 48 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu X, Zhang Z, Ruan J, Pan Y, Magupalli VG, Wu H, Lieberman J (2016) Inflammasome‐activated gasdermin D causes pyroptosis by forming membrane pores. Nature 535: 153–158 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maelfait J, Vercammen E, Janssens S, Schotte P, Haegman M, Magez S, Beyaert R (2008) Stimulation of Toll‐like receptor 3 and 4 induces interleukin‐1beta maturation by caspase‐8. J Exp Med 205: 1967–1973 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malireddi RKS, Gurung P, Mavuluri J, Dasari TK, Klco JM, Chi H, Kanneganti TD (2018) TAK1 restricts spontaneous NLRP3 activation and cell death to control myeloid proliferation. J Exp Med 215: 1023–1034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mandal P, Berger SB, Pillay S, Moriwaki K, Huang C, Guo H, Lich JD, Finger J, Kasparcova V, Votta B, Ouellette M, King BW, Wisnoski D, Lakdawala AS, DeMartino MP, Casillas LN, Haile PA, Sehon CA, Marquis RW, Upton J_et al_ (2014) RIP3 induces apoptosis independent of pronecrotic kinase activity. Mol Cell 56: 481–495 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miao EA, Leaf IA, Treuting PM, Mao DP, Dors M, Sarkar A, Warren SE, Wewers MD, Aderem A (2010) Caspase‐1‐induced pyroptosis is an innate immune effector mechanism against intracellular bacteria. Nat Immunol 11: 1136–1142 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Micheau O, Tschopp J (2003) Induction of TNF receptor I‐mediated apoptosis via two sequential signaling complexes. Cell 114: 181–190 [DOI] [PubMed] [Google Scholar]
- Moriwaki K, Bertin J, Gough PJ, Chan FK (2015) A RIPK3‐caspase 8 complex mediates atypical pro‐IL‐1beta processing. J Immunol 194: 1938–1944 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newton K, Dugger DL, Wickliffe KE, Kapoor N, de Almagro MC, Vucic D, Komuves L, Ferrando RE, French DM, Webster J, Roose‐Girma M, Warming S, Dixit VM (2014) Activity of protein kinase RIPK3 determines whether cells die by necroptosis or apoptosis. Science 343: 1357–1360 [DOI] [PubMed] [Google Scholar]
- Orning P, Weng D, Starheim K, Ratner D, Best Z, Lee B, Brooks A, Xia S, Wu H, Kelliher MA, Berger SB, Gough PJ, Bertin J, Proulx MM, Goguen JD, Kayagaki N, Fitzgerald KA, Lien E (2018) Pathogen blockade of TAK1 triggers caspase‐8‐dependent cleavage of gasdermin D and cell death. Science 362: 1064–1069 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petersen SL, Wang L, Yalcin‐Chin A, Li L, Peyton M, Minna J, Harran P, Wang X (2007) Autocrine TNFalpha signaling renders human cancer cells susceptible to Smac‐mimetic‐induced apoptosis. Cancer Cell 12: 445–456 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poon IK, Chiu YH, Armstrong AJ, Kinchen JM, Juncadella IJ, Bayliss DA, Ravichandran KS (2014) Unexpected link between an antibiotic, pannexin channels and apoptosis. Nature 507: 329–334 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rogers C, Fernandes‐Alnemri T, Mayes L, Alnemri D, Cingolani G, Alnemri ES (2017) Cleavage of DFNA5 by caspase‐3 during apoptosis mediates progression to secondary necrotic/pyroptotic cell death. Nat Commun 8: 14128 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruhl S, Broz P (2015) Caspase‐11 activates a canonical NLRP3 inflammasome by promoting K(+) efflux. Eur J Immunol 45: 2927–2936 [DOI] [PubMed] [Google Scholar]
- Sandilos JK, Chiu YH, Chekeni FB, Armstrong AJ, Walk SF, Ravichandran KS, Bayliss DA (2012) Pannexin 1, an ATP release channel, is activated by caspase cleavage of its pore‐associated C‐terminal autoinhibitory region. J Biol Chem 287: 11303–11311 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sborgi L, Ruhl S, Mulvihill E, Pipercevic J, Heilig R, Stahlberg H, Farady CJ, Muller DJ, Broz P, Hiller S (2016) GSDMD membrane pore formation constitutes the mechanism of pyroptotic cell death. EMBO J 35: 1766–1778 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi J, Zhao Y, Wang K, Shi X, Wang Y, Huang H, Zhuang Y, Cai T, Wang F, Shao F (2015) Cleavage of GSDMD by inflammatory caspases determines pyroptotic cell death. Nature 526: 660–665 [DOI] [PubMed] [Google Scholar]
- Silverman W, Locovei S, Dahl G (2008) Probenecid, a gout remedy, inhibits pannexin 1 channels. Am J Physiol Cell Physiol 295: C761–C767 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sollberger G, Choidas A, Burn GL, Habenberger P, Di Lucrezia R, Kordes S, Menninger S, Eickhoff J, Nussbaumer P, Klebl B, Kruger R, Herzig A, Zychlinsky A (2018) Gasdermin D plays a vital role in the generation of neutrophil extracellular traps. Sci Immunol 3: pii: eaar6689 [DOI] [PubMed] [Google Scholar]
- Taabazuing CY, Okondo MC, Bachovchin DA (2017) Pyroptosis and apoptosis pathways engage in bidirectional crosstalk in monocytes and macrophages. Cell Chem Biol 24: 507–514.e4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tenev T, Bianchi K, Darding M, Broemer M, Langlais C, Wallberg F, Zachariou A, Lopez J, MacFarlane M, Cain K, Meier P (2011) The ripoptosome, a signaling platform that assembles in response to genotoxic stress and loss of IAPs. Mol Cell 43: 432–448 [DOI] [PubMed] [Google Scholar]
- Varfolomeev E, Blankenship JW, Wayson SM, Fedorova AV, Kayagaki N, Garg P, Zobel K, Dynek JN, Elliott LO, Wallweber HJ, Flygare JA, Fairbrother WJ, Deshayes K, Dixit VM, Vucic D (2007) IAP antagonists induce autoubiquitination of c‐IAPs, NF‐kappaB activation, and TNFalpha‐dependent apoptosis. Cell 131: 669–681 [DOI] [PubMed] [Google Scholar]
- Vince JE, Wong WW, Gentle I, Lawlor KE, Allam R, O'Reilly L, Mason K, Gross O, Ma S, Guarda G, Anderton H, Castillo R, Hacker G, Silke J, Tschopp J (2012) Inhibitor of apoptosis proteins limit RIP3 kinase‐dependent interleukin‐1 activation. Immunity 36: 215–227 [DOI] [PubMed] [Google Scholar]
- Vince JE, De Nardo D, Gao W, Vince AJ, Hall C, McArthur K, Simpson D, Vijayaraj S, Lindqvist LM, Bouillet P, Rizzacasa MA, Man SM, Silke J, Masters SL, Lessene G, Huang DCS, Gray DHD, Kile BT, Shao F, Lawlor KE (2018) The mitochondrial apoptotic effectors BAX/BAK activate caspase‐3 and ‐7 to trigger NLRP3 inflammasome and caspase‐8 driven IL‐1beta activation. Cell Rep 25: 2339–2353 e4 [DOI] [PubMed] [Google Scholar]
- Wang L, Du F, Wang X (2008) TNF‐alpha induces two distinct caspase‐8 activation pathways. Cell 133: 693–703 [DOI] [PubMed] [Google Scholar]
- Wang Y, Gao W, Shi X, Ding J, Liu W, He H, Wang K, Shao F (2017) Chemotherapy drugs induce pyroptosis through caspase‐3 cleavage of a gasdermin. Nature 547: 99–103 [DOI] [PubMed] [Google Scholar]
- Wicki S, Gurzeler U, Wei‐Lynn Wong W, Jost PJ, Bachmann D, Kaufmann T (2016) Loss of XIAP facilitates switch to TNFalpha‐induced necroptosis in mouse neutrophils. Cell Death Dis 7: e2422 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang D, He Y, Munoz‐Planillo R, Liu Q, Nunez G (2015) Caspase‐11 requires the pannexin‐1 channel and the purinergic P2X7 pore to mediate pyroptosis and endotoxic shock. Immunity 43: 923–932 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix
Expanded View Figures PDF
Movie EV1
Movie EV2
Source Data for Expanded View and Appendix
Review Process File
Source Data for Figure 2
Source Data for Figure 3
Source Data for Figure 4
Source Data for Figure 5