Noradrenergic Modulation of Basolateral Amygdala Neuronal Activity: Opposing Influences of α-2 and β Receptor Activation (original) (raw)
Abstract
Substantial data exists demonstrating the importance of the amygdala and the locus ceruleus (LC) in responding to stress, aversive memory formation, and the development of stress-related disorders; however, little is known about the effects of norepinephrine (NE) on amygdala neuronal activity in vivo. The basolateral nucleus of the amygdala (BLA) receives dense NE projections from the LC, NE increases in the BLA in response to stress, and the BLA can also modulate the LC via reciprocal projections. These experiments examined the effects of noradrenergic agents on spontaneous and evoked responses of BLA neurons. NE iontophoresis inhibited spontaneous firing and decreased the responsiveness of BLA neurons to electrical stimulation of entorhinal cortex and sensory association cortex (Te3). Confirmed BLA projection neurons exhibited exclusively inhibitory responses to NE. Systemic administration of propranolol, a β-receptor antagonist, decreased the spontaneous firing rate and potentiated the NE-evoked inhibition of BLA neurons. In addition, iontophoresis of the α-2 agonist clonidine, footshock administration, and LC stimulation mimicked the effects of NE iontophoresis on spontaneous activity. Furthermore, the effects of LC stimulation were partially blocked by systemic administration of α 2 and β receptor antagonists. This is the first study to demonstrate the actions of directly applied and stimulus-evoked NE in the BLA in vivo, and provides a mechanism by which β receptors can mediate the important behavioral consequences of NE within the BLA. The interaction between these two structures is particularly relevant with regard to their known involvement in stress responses and stress-related disorders.
Keywords: amygdala, norepinephrine, footshock, locus ceruleus, β receptors, α-2 receptors
Introduction
The amygdala is a critical component of the systems mediating many aspects of the stress response, fear-motivated learning, and memory for emotionally evocative events (LeDoux, 2000; McGaugh, 2004; Berretta, 2005). It is also heavily implicated in stress-related disorders such as panic disorder (Shekhar et al., 1999), posttraumatic stress disorder (PTSD) (Rauch et al., 2000), and anxiety (Shekhar et al., 2005). Also heavily researched for its role in stress and stress-related pathologies is the locus ceruleus (LC) (Sved et al., 2002). The basolateral complex of the amygdala (BLA) receives a dense norepinephrine (NE) innervation originating primarily in the LC (Asan 1998), and levels of NE increase in the BLA with presentation of stressful stimuli (Galvez et al., 1996; Hatfield et al., 1999). LC neurons are activated by stressful stimuli (Rasmussen et al., 1986; Abercrombie and Jacobs, 1987; Grant et al., 1988; Aston-Jones et al., 1991), and presumably mediate the stress-induced increase in NE in the BLA. Therefore, the pathway from LC is thought to be critical in providing information about stressful stimuli to the BLA and allowing for appropriate behavioral and neuroendocrine responses to stressors.
The LC provides diffuse projections which target many forebrain nuclei. Major targets relevant to stress include the hippocampus, prefrontal cortex, and amygdala. However, of these targets, the amygdala is unique in three respects. The amygdala is uniquely positioned to provide feedback to the LC via reciprocal projections (Van Bockstaele et al., 2001). Furthermore, although both the prefrontal cortex and hippocampus have been shown to have inhibitory roles over the stress response, leading to dampening of responding to stressful stimuli (Herman and Cullinan, 1997), the amygdala has a facilitatory effect over the stress response, as does the LC. This promotion of the stress response implicates both the amygdala and the LC in the development of maladaptive stress responses and potentially stress-related pathologies and disorders. Finally, the NE input to the amygdala is critical for memory of aversive events (McGaugh, 2002). Thus, NE has been shown to modulate the consolidation of aversive learning within the BLA (Gallagher et al., 1977). Abnormal regulation of emotional memories is thought to be related to the development of PTSD (Debiec and LeDoux, 2006).
Despite evidence confirming the amygdala and the LC as regions involved in stress-related behaviors and disorders, and much behavioral data validating the importance of BLA NE in aversive learning, there is a paucity of information about how NE affects the BLA in vivo. The present studies examine the effects of microiontophoresis of NE on BLA neuron spontaneous activity and activity evoked by stimulation of entorhinal cortex (EC) or sensory association cortex (Te3). Furthermore, we examined the contribution of β receptors and α 2 receptors to these effects of NE. Finally, we investigated similarities in the responses of BLA neurons to footshock or LC stimulation and NE iontophoresis to gain insight into the role that NE plays in BLA neuronal response to acute stressors, as well as the role of the LC-BLA pathway in mediating these responses.
Materials and Methods
Surgical preparation.
All studies were conducted in accordance with the Guide for the Care and Use of Laboratory Animals published by the United States Public Health Service, and all experimental procedures were approved by the University of Pittsburgh Institutional Animal Care and Use Committee. Male Sprague Dawley rats (250–400 g), housed in pairs and kept on a 12 h light/dark cycle, were maintained at constant temperature and given food and water ad libitum. After 5 d of pair housing, one cage mate was removed from the cage and control housing room for use in other studies. The remaining rats were housed singly until removal from the cage for experimental use. Rats were anesthetized with 8% chloral hydrate (400 mg/kg, i.p.) and then implanted with jugular catheters for anesthetic or drug administration. Rats were then placed in a stereotaxic apparatus, with supplemental doses of chloral hydrate given intravenously to maintain a constant level of anesthesia, and temperature was monitored with a rectal thermometer probe and maintained at 37°C. An incision was made along the scalp and the underlying skull was exposed. Burr holes were drilled in the skull and the dura under the burr holes was removed. The locations of the BLA [−5.0 mm lateral (L), −3.0 mm caudal (C) from bregma], Te3 [−6.1 mm L, −6.8 mm C, −4.7 mm ventral (V) from bregma], LC (−1.1 mm L, −3.6 mm C, −5.8 mm V from λ), and the EC (−5.0 mm L, −6.8 mm C, −8.2 mm V from bregma) were calculated using a stereotaxic atlas (Paxinos and Watson, 1997). Because of the small size of the LC nucleus and its close apposition to many surrounding nuclei, the location of the LC was verified by lowering a recording electrode, confirming the presence of neurons that displayed characteristic LC action potential waveforms and exhibited an excitatory response to foot pinch and replacing the recording electrode with a stimulating electrode in the identified location. Stimulating electrodes were lowered into the brain, and recordings did not begin until at least 45 min after placement of the stimulating electrodes.
Electrophysiology.
The electrophysiological activity of individual neurons within the BLA (single-unit extracellular recordings) was isolated using five-barrel microiontophoretic recording electrodes, and only those neurons with a signal-to-noise ratio >3:1 were used for data analysis. In all experiments, spontaneous activity was recorded for a minimum of 5 min and expressed as spikes per second. After recording of spontaneous activity, the neuron was examined for responsiveness to stimulation of EC, Te3, LC, or footshock stimulation, depending on the experiment. If responsive, the nature of the response was characterized and recorded. EC and Te3 stimulation protocols were also run concomitant with NE application. If unresponsive, NE (200 μm) or clonidine (50 μm) was applied via iontophoresis in successively increasing doses (5–40 nA), and the nature of the response recorded. In a subset of neurons, NE was applied via iontophoresis before and after systemic propranolol administration (0.5 mg/kg).
Electrical stimulation.
Bipolar, concentric stimulation electrodes were lowered into either the LC, Te3, or the EC at the conclusion of surgical preparation. Neurons responsive to LC stimulation were isolated based on spontaneous activity (not during tonic stimulation of LC) and then tested for responsivity to LC stimulation (50–300 μA, 0.25 ms, 0.2 Hz). This was done to avoid massive release of NE with repeated LC stimulation during a search-stimulation protocol. Furthermore, in experiments in which an attempt was made to block LC responses with systemic administration of propranolol or yohimbine, LC stimulation current levels were limited to 50 μA to further ensure specific stimulation of the LC alone and not surrounding brainstem nuclei. Neurons responsive to either Te3 or EC stimulation were found using a search-stimulate protocol. Single-pulse stimuli (300–900 μA, 0.25 ms, 0.5 Hz) were delivered to the Te3 or EC while the recording electrode was lowered through the BLA, and responsive neurons were identified and isolated. Neurons were characterized as having presumed orthodromic monosynaptic, orthodromic polysynaptic, or antidromic responses to EC stimulation by the following criteria: responses were operationally defined as orthodromic if they had an onset latency of <20 ms, showed a failure to substantially change latency in response to increases in current intensity, exhibited a consistent onset latency with ∼1–5 ms of variability in evoked spike latency, and followed paired pulse stimulation at 50 Hz but not 400 Hz. Antidromic responses were characterized by a constant onset latency with absence of variability in evoked spike latency, and their ability to followed paired pulse stimulation at 400 Hz. Monosynaptic responses were differentiated from polysynaptic spikes based on onset latency and variability of evoked spike latencies, as well as the failure of presumed polysynaptic spikes to follow paired-pulse stimulation at 50 Hz.
Iontophoresis.
Five-barrel microiontophoretic electrodes (Activational Systems, Warren, MI) were constructed using a vertical microelectrode puller, broken back under microscopic control to yield electrodes with a central recording barrel impedance of 4–8 MΩ for electrophysiological recordings. These electrodes were lowered through the BLA in successive vertical tracks. The central barrel of the microelectrode was filled with 2% Pontamine sky blue in 2 m NaCl. One of the outer barrels was filled with 3 m NaCl for automatic current balancing, and the remaining barrels were filled with 200 μm NE, pH 4.0, or 50 μm clonidine, pH 4.0. All drugs were dissolved in 0.1 m NaCl. Drugs were retained using currents that ranged between −12 and −18 nA, and were ejected at currents that ranged between 5 and 40 nA. The ejection was done using repeated, increasing doses from 5 to 40 nA with at least 2 min of spontaneous activity recorded between each period of drug ejection.
Neurons were tested for dose responsivity to NE (5–40 nA). Those neurons that responded to lower doses of NE (5–10 nA) did so in a moderate, variable, and often nonsignificant manner, and these effects were often difficult to evaluate reliably because of the very low firing rates of many neurons. More clear, significant, and consistent responses emerged in BLA neurons to higher doses of NE (40 nA). Therefore, this dose was used for analysis of NE effects on spontaneous activity. However, because of the greater reliability in measuring changes in evoked activity, which does not rely on evaluating small changes in neurons with low levels of spontaneous spike activity, we found that evoked activity was significantly altered by lower doses of NE (10 nA). Therefore, the effects of NE on BLA evoked activity focused on the 10 nA dose, although effects are also reported for higher doses where tested.
Drug administration.
Propranolol (0.5 mg/kg) and yohimbine (0.5 mg/kg) were given intravenously via a jugular vein catheter.
Footshock.
A single footshock (5 mA, 0.5 ms) was administered via two 28-gauge pins inserted into the lateral side of the footpad, and single unit responses were recorded. The footshock was repeated up to five times per neuron.
Histology.
At the conclusion of electrophysiological experiments, Pontamine sky blue dye was ejected by passing constant current through the recording electrode (10 nA, 15 min) to mark the location of the recording site. Anodal current was passed through the stimulating electrodes to create small lesions to localize placement of the stimulating electrodes. Rats were killed by an overdose of anesthetic, followed by decapitation and removal of the brain. Brains were fixed in 10% formalin for a minimum of 24 h, and then cryoprotected with 25% sucrose solution in 0.1 m phosphate buffer. Subsequently, slices were cut on a cryostat into 40 μm coronal sections, mounted on glass slides, and stained with cresyl violet. Recording sites were identified by the presence of the Pontamine sky blue dye spot, and the location of the stimulating electrodes was identified by the presence of a small lesion at the end of the electrode track.
Data analysis.
Only those animals with electrode placements localized to the LC, EC, Te3, and BLA were included in the analysis. Firing rate was calculated as spikes per second. An effect was labeled as a significant excitatory or inhibitory event if during the experimental manipulation (NE iontophoresis, propranolol administration, clonidine iontophoresis, footshock, or LC stimulation) the average firing rate increased or decreased by 25% or more from baseline. This was based on conservative estimates of the change necessary to yield significance in a t test given the degree of firing rate variability. Premanipulation and postmanipulation firing rates were compared using paired t tests. For neurons activated monosynaptically by EC or Te3 stimulation, the average onset latency was measured across 50 stimulation trials. The effects of NE iontophoresis on responses evoked by afferent stimulation were evaluated in the following manner: current applied to EC or Te3 was adjusted to achieve between 50 and 60% probability of evoking a spike response. Fifty single pulses at this current intensity were applied to the EC or Te3 and the number of stimuli that evoked spike discharge within the BLA was measured. This was compared with the number of afferent stimuli that evoked spike firing during NE iontophoresis. These spike probabilities were compared using paired t tests. Changes in evoked spikes were expressed as percentage increase or decrease in the probability of evoked responses during NE iontophoresis. A minimum of 3 min of spontaneous firing was allowed before delivery of a second stimulation period of 50 sweeps concomitant with NE iontophoresis.
Responses were categorized based on identification of neuronal subtype. Neurons were confirmed as projection cells if they exhibited antidromic activation from EC stimulation (n = 8). These neurons were examined separately for their response to NE iontophoresis, and pre-NE and post-NE firing rates were compared with paired t tests. Subclasses of neurons were also examined based on neuronal location. Histological verification revealed subgroups of neurons located in the lateral and basolateral subnuclei of the BLA complex. These two groups of neurons were analyzed separately for their responses to NE iontophoresis. Proportions of neurons in the lateral nucleus displaying excitatory versus inhibitory responses were compared with proportions of neurons in the basolateral nucleus displaying excitatory versus inhibitory responses using χ2 analyses.
Results
Spontaneous activity
All neurons examined in this study were verified to lie within the BLA (Fig. 1A). Accordingly, stimulation sites were verified as confined to the LC, EC, or Te3 (Fig. 1B–D). Placements outside these defined areas were not used for analysis. A total of 93 neurons were recorded from 34 rats. Of these, 35 were tested for their responses to NE alone (eight of those were antidromically activated and confirmed as projection neurons), 15 were examined for responses to Te3 (eight) or EC (seven) stimulation before or after NE iontophoresis, 5 were examined for responses to NE before and after propranolol administration, five were examined for responses to NE and clonidine, 20 were examined for responses to NE and LC stimulation, and 13 were examined for responses to NE and footshock.
Figure 1.

Diagram demonstrating sites of recording and stimulating electrodes. A, Neurons recorded from were located within the basolateral complex of the amygdala (lateral, basolateral, and basomedial nuclei). B, Stimulating electrode sites localized to the LC. C, Stimulating electrode sites localized to the EC. D, Stimulating electrode sites localized to the Te3.
Neurons from the lateral (LAT) and basolateral (BL) nuclei of the basolateral complex were examined for the above responses. There was no difference in baseline firing rate (FR) among neurons located in different nuclei of the basolateral amygdala (LAT FR, 1.04 ± 0.14 Hz; BL FR, 1.2 ± 0.24 Hz; p = 0.88, t test). The average firing rate across all neurons was 1.1 ± 0.19 Hz. The average firing rate of neurons confirmed by antidromic activation to be projection neurons (n = 8) was 0.22 ± 0.04 Hz, which was significantly less than that of spontaneously active neurons (p = 0.01, t test).
Norepinephrine inhibits spontaneous activity of BLA neurons
Iontophoresis of NE (200 μm, 40 nA) significantly decreased spontaneous activity (by 32 ± 4.1%) in the majority of neurons of the BLA (n = 24 of 35; p = 0.02, t test) (Fig. 2A,B). A smaller proportion of neurons showed a significant increase in spontaneous activity (by 136 ± 11.4%; n = 8 of 35; p = 0.02, t test) (Fig. 2C,D) with few cells showing no response to NE iontophoresis (n = 3 of 35).
Figure 2.

Norepinephrine inhibits spontaneous activity of BLA neurons. A, Firing rate histogram showing dose-dependent inhibition of spontaneous activity during microiontophoretic application of NE. B, The majority of neurons displayed an inhibition of spontaneous activity during NE application (p = 0.02). C, In some cases, NE microiontophoresis resulted in a dose-dependent excitation of neuronal activity. D, A small number of neurons showed an increase in spontaneous activity with NE application (p = 0.02). E, Neurons confirmed as projection cells via antidromic activation only displayed inhibition to NE application (p = 0.002). F, Neurons in all nuclei of the BLA displayed excitatory and inhibitory responses.
Neurons from the lateral and basolateral nuclei were examined for their responses to NE. No significant differences were found in the proportions of neurons that were excited and inhibited in each of these nuclei (LAT, 74% inhibited; BL, 64% inhibited; p = 0.6, χ2). Therefore, the response to NE was not dependent on the location of the neuron (Fig. 2E).
A subset of neurons was antidromically activated by electrical stimulation of EC, confirming them as projection cells. These neurons displayed only significant inhibitory responses to NE iontophoresis (by 58 ± 9%; p = 0.002, t test) (Fig. 2F).
Norepinephrine inhibits activity evoked by stimulation of entorhinal cortex and sensory association cortex.
Electrical stimulation of the EC evoked orthodromic, excitatory responses in BLA neurons (n = 7) (Fig. 3A) with an average latency to spike onset of 12.3 ± 1.9 ms. During iontophoresis of NE (10nA), the spike probability in response to EC stimulation decreased significantly (by 38 ± 5.2%; n = 7 of 7; p = 0.005) (Fig. 3B). Electrical stimulation of Te3 caused orthodromic, excitatory responses in BLA neurons (n = 8) (Fig. 3C) with an average spike onset latency of 9.7 ± 1.4 ms. During iontophoresis of NE (10 nA), the spike probability in response to Te3 stimulation decreased significantly (by 38 ± 6.0%; n = 8 of 8 neurons; p = 0.002, t test) (Fig. 3D).
Figure 3.

Norepinephrine inhibits evoked activity of BLA neurons. A, Electrophysiological recording trace of a BLA neuron exhibiting orthodromic activation after EC stimulation. B, Iontophoretic application of NE suppresses responses to EC stimulation (p = 0.005). C, BLA neurons are also activated by Te3 stimulation. D, Iontophoretic application of NE suppresses responses to Te3 stimulation (p = 0.002). Light lines in B and D illustrate individual neuronal responses; the dark line represents the average response.
A significant and consistent decrease in evoked spiking (both EC- and Te3-evoked) with application of NE emerged at lower doses of NE (10 nA) than did the decrease in spontaneous firing (40 nA). A portion of neurons (n = 6) did not recover baseline-evoked spiking probabilities to levels where it was sufficient to continue testing at higher doses of NE; however, in those that did exhibit recovery to baseline (n = 9), NE iontophoresis (40nA) caused additional inhibition of evoked spike probability (by 70 ± 11%; p = 0.00006, t test).
Norepinephrine-induced inhibition is potentiated by systemic propranolol administration and mimicked by iontophoresis of clonidine
Iontophoresis of clonidine (50 μm, 10 nA), an α-2 agonist, caused a decrease in spontaneous activity of BLA neurons (by 65 ± 7.0%; n = 5; p = 0.02, t test) (Fig. 4A). Iontophoresis of NE (40 nA, 200 μm) inhibited spontaneous activity in these same neurons (by 61 ± 6.8%; n = 5; p = 0.03, t test) (Fig. 4A,B). Therefore, NE appears to act at α-2 receptors to cause inhibition in BLA neurons.
Figure 4.
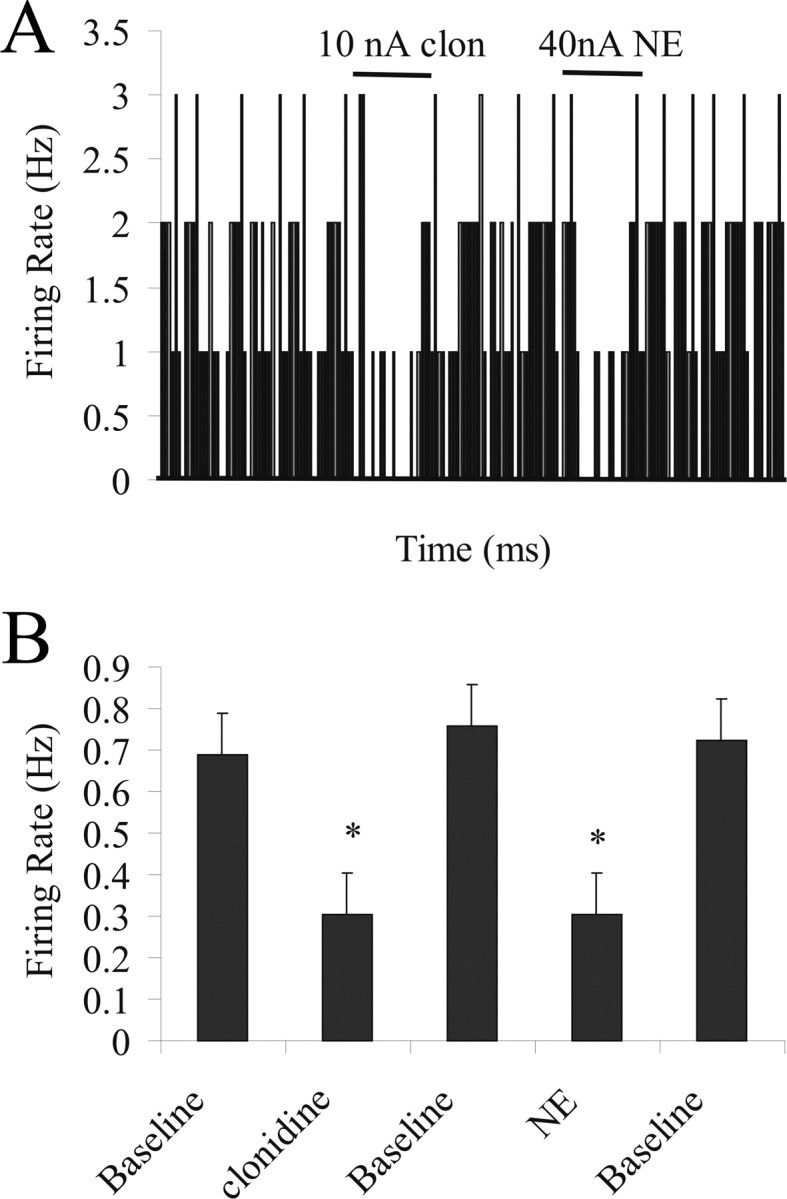
Inhibitory effects of NE are mediated via α-2 receptors. A, Firing rate histogram showing that iontophoretic application of 10 nA clonidine causes an inhibition of spontaneous activity of a BLA neuron. This same neuron exhibited inhibition of spontaneous activity in response to 40 nA NE application. B, All neurons that display inhibitory responses to clonidine iontophoresis (p = 0.02) also display inhibitory responses to NE iontophoresis (p = 0.03).
Systemic administration of propranolol (i.v., 0.5 mg/kg) caused a decrease in spontaneous activity of BLA neurons (by 46 ± 5.2%; n = 5; p = 0.03, t test). Furthermore, iontophoresis of NE (5–40 nA, 200 μm) caused greater inhibition of BLA neuronal activity at lower doses than before propranolol (at 10 nA NE; controls, 45 ± 6.2% inhibition; after propranolol, 99 ± 11.2% inhibition; n = 5; p = 0.007, t test) (Fig. 5).
Figure 5.

Inhibitory effects of NE are potentiated under β receptor blockade. Propranolol administration potentiates the dose-dependent inhibitory effects of NE iontophoresis.
Footshock and LC stimulation cause similar responses as those produced by NE iontophoresis
Electrical stimulation of the LC caused a significant inhibition in the spontaneous activity of a subset of neurons of the BLA (by 59 ± 8.1%; n = 11 of 20; p = 0.03, t test) (Fig. 6A,C). Of these neurons, six were tested for their response to NE iontophoresis, and all six displayed NE-induced inhibition (by 51 ± 12%; n = 6 of 6; p = 0.004, t test) (Fig. 6B,C). Of the remaining neurons, five were tested for blockade of response with systemic yohimbine administration. Whereas three of five neurons displayed a partial blockade of inhibitory effects and a slight, but nonsignificant excitation (10 ± 2%; p = 0.41, t test) (Fig. 6C), the remaining neurons showed a decrease in spontaneous activity in response to yohimbine administration (presumably because of blockade of presynaptic NE autoreceptors leading to increases in NE release (Broderick, 1997), causing a floor effect from which the presence of further inhibition by LC stimulation could not be detected. LC stimulation also caused excitation in a subset of BLA neurons (by 62 ± 8.2%; n = 9 of 20; p = 0.01, t test) (Fig. 6D,F). Of these neurons, four were tested for response to NE iontophoresis, and three were also excited by NE (by 104 ± 18%; n = 3 of 4; p = 0.001, t test) (Fig. 6E,F). One neuron tested did not respond significantly to NE iontophoresis. In the remaining five neurons excited by LC stimulation, the ability of systemically administered propranolol to block the excitation was examined. After propranolol, the previous LC-induced excitation (by 61.5 ± 7.2%; n = 5 of 5; p = 0.04, t test) was blocked. In fact, after propranolol, LC stimulation now caused a slight, but not significant inhibition of BLA neuronal activity (18.6 ± 4.3%; n = 5 of 5; p = 0.24, t test).
Figure 6.

LC stimulation and NE iontophoresis cause similar effects on BLA neurons. A, Firing rate histogram of a neuron showing inhibition in response to LC stimulation. B, This same neuron shows inhibition in response to NE iontophoresis. C, All neurons that display inhibitory responses to LC stimulation (p = 0.03) also display inhibitory responses to NE iontophoresis (p = 0.004). D, Example of a neuron showing excitation in response to LC stimulation. E, This same neuron shows excitation in response to NE iontophoresis. F, The majority of neurons that display excitatory responses to LC stimulation (p = 0.01) also display excitatory responses to NE iontophoresis (p = 0.01).
Footshock administration (5 mA, 0.5 ms) caused a significant inhibition in spontaneous firing of BLA neurons (by 75 ± 11%; n = 8 of 13; p = 0.003, t test) (Fig. 7A). All neurons inhibited by footshock also displayed inhibitory responses to iontophoresis of NE (by 44 ± 9%; n = 8 of 8; p = 0.0004, t test) (Fig. 7B,C). Footshock administration also caused excitation of BLA neurons (by 212 ± 29%; n = 5 of 13; p = 0.003) (Fig. 7D). Of these neurons, the majority also displayed excitatory responses to NE iontophoresis (by 118 ± 23%; n = 4 of 5; p = 0.002, t test) (Fig. 7E,F). One neuron tested did not respond significantly to NE iontophoresis.
Figure 7.

Footshock stimulation and NE iontophoresis cause similar effects in BLA neurons. A, Firing rate histogram of a neuron showing inhibition in response to footshock. B, This same neuron shows inhibition in response to NE iontophoresis. C, All neurons that display inhibitory responses to footshock (p = 0.003) also display inhibitory responses to NE iontophoresis (p = 0.004). D, Example of a neuron showing excitation in response to footshock. E, This same neuron shows excitation in response to NE iontophoresis. F, The majority of neurons that display excitatory responses to footshock administration (p = 0.003) also display excitatory responses to NE iontophoresis (p = 0.002).
Discussion
This study demonstrates that the NE input to the BLA exerts potent α-2-mediated inhibition of spontaneous activity and attenuates evoked activity as well. NE delivered locally via iontophoresis exerts potent inhibitory effects on spontaneous activity in the BLA, and this inhibition is mimicked by an α-2 receptor agonist. NE also has smaller excitatory effects mediated by β receptors. The effects of both footshock and LC stimulation on BLA neurons can be reproduced by NE iontophoresis. Furthermore, the effects of LC stimulation can be partially blocked with noradrenergic receptor antagonists, with the β antagonist attenuating excitatory effects of LC stimulation and the α-2 antagonist attenuating the inhibitory effects. NE also potently inhibits activity evoked by stimulation of EC and Te3, which is consistent with an overall inhibitory effect of NE in the BLA. This study provides the first in vivo electrophysiological data to complement substantial behavioral studies examining the role of NE in the BLA, and suggests the LC-BLA circuitry as an important player in the development of maladaptive responses to stress and stress-related disorders.
These data are also consistent with in vitro electrophysiological studies performed in the BLA. NE enhances excitatory neurotransmission via β-adrenergic receptors (Gean et al., 1992, Huang et al., 1996, Ferry et al., 1997) while producing inhibitory effects via α-2 receptors (Ferry et al., 1997). NE also disrupts plasticity via actions at α-2 receptors (DeBock et al., 2003). We demonstrate here that the inhibitory actions of NE were mimicked by administration of the α-2 agonist clonidine and blocked by the α-2 antagonist yohimbine, suggesting that NE has its inhibitory effects at least in part via α-2 receptors. Furthermore, NE in vivo produces a tonic excitatory effect on BLA neurons via β receptors which is lost with β receptor blockade, causing a decrease in spontaneous firing rate. In fact, the excitation caused by NE, though present in a fewer number of neurons (n = 8 of 35) than the inhibition (n = 24 of 35), was much greater in magnitude (136%) than the inhibition (32%). Furthermore, blockade of β receptors leads to a stronger inhibition of neuronal activity by NE iontophoresis, presumably because of blockade of the excitatory actions of NE mediated via β-receptors, thereby unveiling a more potent inhibition via α-2 receptors.
Initial examinations of baseline firing rates and responses to NE iontophoresis suggested a possible correlation between firing rate and NE response. Firing rate has previously been used as one criterion of neuronal subtype within the BLA. Our data would suggest that whereas slower firing neurons (presumed projection cells) are excited, faster firing cells (putative interneurons) are inhibited. However, this did not correlate reliably with putative neuron subtypes for three reasons: (1) there was substantial overlap in the firing rates between fast and slow firing neurons, and we did not select for neurons that would fall into two clearly differentiable populations, (2) there were examples in each category of fast firing neurons that were excited and slow firing neurons that were inhibited, and (3) neurons that were conclusively identified as projection neurons via antidromic activation were inhibited. These data indicate that although there may be trends for certain cell types to be excited vs inhibited, neuronal subtype does not account entirely for the variety of the responses to NE.
There is evidence for the presence of α-2 and β receptors within the BLA. α-2 receptors function as autoreceptors for the NE system (Grigg et al., 1996; Arima et al., 1998), and therefore presynaptic inhibitory α-2 receptors could also account for the inhibitory effects of NE and clonidine. However, there is some evidence of postsynaptic α-2 receptors in the BLA as well (U'Prichard et al., 1980). The present studies cannot distinguish between these two mechanisms. Although not conclusive, the observation that LC stimulation produces the same inhibitory response as NE iontophoresis is consistent with a predominantly postsynaptic α-2 adrenergic inhibition of BLA neuronal activity.
Functional interpretation
NE in the BLA enhances learning and consolidation of tasks involving aversive stimuli (McGaugh, 2002). One might interpret the predominantly inhibitory effect of NE in the BLA observed here as being contradictory to the facilitative effect exerted by NE on behavior. However, these behavioral effects of NE are mediated via β receptors (McGaugh et al., 1988; Introini-Collision et al., 1991; Strange and Dolan, 2004; Ferry et al., 1999a). Our data demonstrate that the actions of NE in the BLA via β receptors are, in fact, excitatory. Furthermore, these behavioral effects have never demonstrated that facilitation of behavior involves an activation of the BLA. NE having mainly inhibitory effects in the BLA could serve two functions. First, having an overall inhibitory effect on the majority of neurons of the BLA could serve to enhance the signal-to-noise ratio of those select units that are excited, a well documented function of NE (Devilbliss and Waterhouse, 2004). This would increase transmission of those excited units onto downstream BLA targets to subserve behavioral responding. Alternatively, an inhibition of the BLA could lead to a disinhibition of the central nucleus of the amygdala (CeA), as the BLA has a predominantly inhibitory influence over the CeA (Rosenkranz et al., 2006). Disinhibition of the CeA could lead to activation of downstream brain targets involved in the behavioral response. The CeA sends excitatory, corticotropin-releasing hormone (CRH)-containing projection to the LC (Van Bockstaele et al., 2001; Bouret et al., 2003; Ramsooksingh et al., 2003) and paraventricular nucleus of the hypothalamus (PVN) (Gray et al., 1989), two regions heavily involved in the production of the stress response and stress-related behaviors. Afferent regulation of the LC by CRH is involved in such responses and behaviors (Berridge and Waterhouse, 2003).
In addition to its inhibitory effects on spontaneous BLA neuronal activity, NE also caused a decrease in activity evoked by stimulation of EC and Te3. This contrasts with the observation in other brain regions that NE alters the signal-to-noise ratio by decreasing baseline firing but increasing evoked responses (Funke and Eysel, 1993; McLean and Waterhouse, 1994; Ego-Stengel et al., 2002). At face value, this would appear to be inconsistent with stress-related learning, in which the EC modulates the learning of certain behaviors through an interaction with the BLA (Ferry et al., 1999b). However, it is possible that NE modulation of these inputs is altered during learned associations. Therefore, in the absence of learned associations, the NE system may function to suppress activity within circuits in which fear-conditioned behavior has not taken place.
These studies examine the activation of LC and consequent effects in the BLA. However, as reviewed in the Introduction, the LC also has diffuse projections to many regions involved in the stress response. Thus, studies have shown that NE has both inhibitory and excitatory actions in prefrontal cortex (Kovacs and Hernadi, 2002) and the hippocampus (Curet and de Montigny, 1988; Madison and Nicoll, 1988; Bergles et al., 1996). We have demonstrated here that NE causes mainly inhibition, but also some excitation in the BLA as well. We propose two mechanisms by which NE actions in the BLA may nonetheless produce a facilitatory action on the stress response. First, although the majority of neurons were inhibited by NE, those excited comprised an appreciable portion of responses (23%), and the magnitude of the excitatory response (136%) was much greater than that of the inhibitory response (32%). One scenario would be that the neurons that are potently activated by NE may represent a subset of neurons that project to downstream regions mediating fear-related responses such as freezing, although suppressing information sent to regions mediating competing responses, such as more cortical regions. Alternatively, as described above, inhibition of BLA neurons could lead to a disinhibition of CeA neurons, providing positive feedback to the LC and a facilitatory influence over the stress response in other postsynaptic targets of the LC.
The stress response often functions as an adaptive mechanism by which organisms react to potentially threatening or harmful stimuli with appropriate behavioral responses. Noradrenergic modulation of structures involved in these stress circuitries plays a critical role in these responses. However, extensive exposure to stress is a common factor associated with the development and/or enhancement of pathology and disease. Activation of the hypothalamic–pituitary–adrenal axis by novel stressors is enhanced after exposure to chronic stress; an effect relying on NE modulation of the bed nucleus of the stria terminalis and the PVN (Cecchi et al., 2002, Ma and Morilak, 2005) and may result in maladaptive behavioral responses to stress. Lesions of the BLA inhibit HPA responses to acute stress (Bhatnagar and Dallman, 1998), and the BLA is a potential site involved in sensitization to stressors (Bhatnagar et al., 2004). Preliminary data suggests that chronic stress exposure alters NE actions within the BLA (Buffalari and Grace, 2006). These and other data combined with the data reported here suggest that the BLA is an important component in this circuit that may be susceptible to stress-induced changes leading to sensitization and the development of stress-related pathologies. A more complete understanding of how NE alters transmission in regions like the BLA that serve as important modulators of the stress response can lead to insights into how transmission within these circuits modulates stress-related behaviors.
Footnotes
This work was supported by United States Public Health Service–National Institute on Drug Abuse Grant DA15408 (A.A.G.) and a Mellon Fellowship (D.M.B.). We thank Christy Smolak and Nicole MacMurdo for their expert histological processing of tissue and technical assistance, Brian Lowry for the development of the data acquisition and analysis software, and Dr. Hank P. Jedema and Dr. Amiel Rosenkranz for help in thoughtful discussion and input regarding these data.
References
- Abercrombie ED, Jacobs BL. Single-unit response of noradrenergic neurons in the locus coeruleus of freely moving cats. I. Acutely presented stressful and nonstressful stimuli. J Neurosci. 1987;7:2837–2843. doi: 10.1523/JNEUROSCI.07-09-02837.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arima J, Kubo C, Ishibashi H, Akaike N. alpha-2-Adrenoceptor-mediated potassium currents in acutely dissociated rat locus coeruleus neurons. J Physiol. 1998;508:57–66. doi: 10.1111/j.1469-7793.1998.057br.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Asan E. The catecholaminergic innervation of the rat amygdala. Adv Anat Embryol Cell Biol. 1998;142:L1–L118. doi: 10.1007/978-3-642-72085-7. [DOI] [PubMed] [Google Scholar]
- Aston-Jones G, Chiang C, Alexinsky T. Discharge of noradrenergic locus coeruleus neurons in behaving rats and monkeys suggests a role in vigilance. Prog Brain Res. 1991;88:501–520. doi: 10.1016/s0079-6123(08)63830-3. [DOI] [PubMed] [Google Scholar]
- Bergles DE, Doze VA, Madison DV, Smith SJ. Excitatory actions of norepinephrine on multiple classes of hippocampal CA1 interneurons. J Neurosci. 1996;16:572–585. doi: 10.1523/JNEUROSCI.16-02-00572.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berretta S. Cortico-amygdala circuits: role in the conditioned stress response. Stress. 2005;8:221–232. doi: 10.1080/10253890500489395. [DOI] [PubMed] [Google Scholar]
- Berridge CW, Waterhouse BD. The locus coeruleus–noradrenergic system: modulation of behavioral state and state-dependent cognitive processes. Brain Res Brain Res Rev. 2003;42:33–84. doi: 10.1016/s0165-0173(03)00143-7. [DOI] [PubMed] [Google Scholar]
- Bhatnagar S, Dallman M. Neuroanatomical basis for facilitation of hypothalamic-pituitary-adrenal responses to a novel stressor after chronic stress. Neuroscience. 1998;84:1025–1039. doi: 10.1016/s0306-4522(97)00577-0. [DOI] [PubMed] [Google Scholar]
- Bhatnagar S, Vining C, Denski K. Regulation of chronic stress-induced changes in hypothalamic-pituitary-adrenal activity by the basolateral amygdala. Ann NY Acad Sci. 2004;1032:315–319. doi: 10.1196/annals.1314.050. [DOI] [PubMed] [Google Scholar]
- Bouret S, Duvel A, Onat S, Sara SJ. Phasic activation of locus coeruleus neurons by the central nucleus of the amygdala. J Neurosci. 2003;23:3491–3497. doi: 10.1523/JNEUROSCI.23-08-03491.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Broderick PA. Alprazolam, diazepam, yohimbine, clonidine: in vivo CA1 hippocampal norepinephrine and serotonin release profiles under chloral hydrate anesthesia. Prog Neuropsychopharmacol Biol Psychiatry. 1997;21:1117–1140. doi: 10.1016/s0278-5846(97)00103-6. [DOI] [PubMed] [Google Scholar]
- Buffalari DM, Grace AA. Noradrenergic modulation of BLA neuronal activity: alterations by chronic stress. Soc Neurosci Abstr. 2006;32:358–5. [Google Scholar]
- Cecchi M, Khoshbouei H, Morilak DA. Modulatory effects of norepinephrine in the lateral bed nucleus of the stria terminalis on behavioral and neuroendocrine responses to acute stress. Neuroscience. 2002;112:13–21. doi: 10.1016/s0306-4522(02)00062-3. [DOI] [PubMed] [Google Scholar]
- Curet O, de Montigny C. Electrophysiological characterization of adrenoceptors in the rat dorsal hippocampus. I. Receptors mediating the effect of microiontophoretically applied norepinephrine. Brain Res. 1988;475:35–46. doi: 10.1016/0006-8993(88)90196-5. [DOI] [PubMed] [Google Scholar]
- Debiec J, LeDoux JE. Noradrenergic signaling in the amygdala contributes to the reconsolidation of fear memory: treatment implications for PTSD. Ann NY Acad Sci. 2006;1071:521–524. doi: 10.1196/annals.1364.056. [DOI] [PubMed] [Google Scholar]
- DeBock F, Kurz J, Azad SC, Parsons CG, Hapfelmeier G, Zieglgansberger W, Rammes G. Alpha-2-adrenoceptor activation inhibits LTP and LTD in the basolateral amygdala: involvement of Gi/o-protein-mediated modulation of Ca2+-channels and inwardly rectifying K+ channels in LTD. Eur J Neurosci. 2003;17:1411–1424. doi: 10.1046/j.1460-9568.2003.02544.x. [DOI] [PubMed] [Google Scholar]
- Devilbliss DM, Waterhouse BD. The effects of tonic locus coeruleus output on sensory-evoked responses of ventral posterior medial thalamic and barrel field cortical neurons in the awake rat. J Neurosci. 2004;24:10773–10785. doi: 10.1523/JNEUROSCI.1573-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ego-Stengel V, Bringuier V, Schulz DE. Noradrenergic modulation of functional selectivity in the cat visual cortex: an in vivo extracellular and intracellular study. Neuroscience. 2002;111:275–289. doi: 10.1016/s0306-4522(02)00011-8. [DOI] [PubMed] [Google Scholar]
- Ferry B, Magistretti PJ, Pralong E. Noradrenaline modulates glutamate-mediated transmission in the rat basolateral amygdala in vitro. Eur J Neurosci. 1997;9:1356–1364. doi: 10.1111/j.1460-9568.1997.tb01490.x. [DOI] [PubMed] [Google Scholar]
- Ferry B, Wirth S, DiScala G. Functional interaction between entorhinal cortex and basolateral amygdala during trace conditioning of odor aversion in the rat. Behav Neurosci. 1999a;113:118–125. doi: 10.1037//0735-7044.113.1.118. [DOI] [PubMed] [Google Scholar]
- Ferry B, Roozendaal B, McGaugh JL. Basolateral amygdala noradrenergic influences on memory storage are mediated by an interaction between β and α-1 receptors. J Neurosci. 1999b;19:5119–5123. doi: 10.1523/JNEUROSCI.19-12-05119.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Funke K, Eysel UT. Modulatory effects of acetylcholine, serotonin and noradrenaline on the activity of cat perigeniculate neurons. Exp Brain Res. 1993;95:409–420. doi: 10.1007/BF00227133. [DOI] [PubMed] [Google Scholar]
- Gallagher M, Kapp BS, Musty RE, Driscoll PA. Memory formation: evidence for a specific neurochemical system in the amygdala. Science. 1977;198:423–425. doi: 10.1126/science.20664. [DOI] [PubMed] [Google Scholar]
- Galvez R, Mesches MH, McGaugh JL. Norepinephrine release in the amygdala in response to footshock stimulation. Neurobiol Learn Mem. 1996;66:253–257. doi: 10.1006/nlme.1996.0067. [DOI] [PubMed] [Google Scholar]
- Gean PW, Huang CC, Lin HJ, Tsai JJ. Sustained enhancement of NMDA receptor-mediated synaptic potential by isoproterenol in rat amygdalar slices. Brain Res. 1992;594:331–334. doi: 10.1016/0006-8993(92)91146-6. [DOI] [PubMed] [Google Scholar]
- Grant SJ, Aston-Jones G, Redmond DE., Jr Responses of primate locus coeruleus neurons to simple and complex sensory stimuli. Brain Res Bull. 1988;21:401–410. doi: 10.1016/0361-9230(88)90152-9. [DOI] [PubMed] [Google Scholar]
- Gray TS, Carney ME, Magnuson DJ. Direct projections from the central amygdaloid nucleus to the hypothalamic paraventricular nucleus: possible role in stress-induced adrenocorticotropin release. Neuroendocrinology. 1989;50:433–446. doi: 10.1159/000125260. [DOI] [PubMed] [Google Scholar]
- Grigg JJ, Kozasa T, Nakajima Y, Nakajima S. Single-channel properties of a G-protein coupled inward rectifier potassium channel in brain neurons. J Neurophysiol. 1996;75:318–328. doi: 10.1152/jn.1996.75.1.318. [DOI] [PubMed] [Google Scholar]
- Hatfield T, Spanis C, McGaugh JL. Response of amygdalar norepinephrine to footshock and GABAergic drugs using in vivo microdialysis and HPLC. Brain Res. 1999;835:340–345. doi: 10.1016/s0006-8993(99)01566-8. [DOI] [PubMed] [Google Scholar]
- Herman JP, Cullinan WE. Neurocircuitry of stress: central control of the hypothalamo-pituitary-adrenocortical axis. Trends Neurosci. 1997;20:78–84. doi: 10.1016/s0166-2236(96)10069-2. [DOI] [PubMed] [Google Scholar]
- Huang CC, Kuei-Sen H, Gean PW. Isoproterenol potentiates synaptic transmission primarily by enhancing presynaptic calcium influx via P- and/or Q-type calcium channels in the rat amygdala. J Neurosci. 1996;16:1026–1033. doi: 10.1523/JNEUROSCI.16-03-01026.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Introini-Collision IB, Miyataki B, McGaugh JL. Involvement of the amygdala in the memory-enhancing effects of clenbuterol. Psychopharmacology. 1991;104:541–544. doi: 10.1007/BF02245663. [DOI] [PubMed] [Google Scholar]
- Kovacs P, Hernadi I. Iontophoresis of lithium antagonizes noradrenergic action on prefrontal neurons of the rat. Brain Res. 2002;947:150–156. doi: 10.1016/s0006-8993(02)03150-5. [DOI] [PubMed] [Google Scholar]
- LeDoux JE. Emotion circuits in the brain. Ann Rev Neurosci. 2000;23:155–184. doi: 10.1146/annurev.neuro.23.1.155. [DOI] [PubMed] [Google Scholar]
- Ma S, Morilak DA. Chronic intermittent cold stress sensitizes the hypothalamic-pituitary-adrenal response to a novel acute stress by enhancing noradrenergic influence in the rat paraventricular nucleus. J Neuroendocrinol. 2005;17:761–769. doi: 10.1111/j.1365-2826.2005.01372.x. [DOI] [PubMed] [Google Scholar]
- Madison DV, Nicoll RA. Norepinephrine decreases synaptic inhibition in the rat hippocampus. Brain Res. 1988;442:131–138. doi: 10.1016/0006-8993(88)91440-0. [DOI] [PubMed] [Google Scholar]
- McGaugh JL. Memory consolidation and the amygdala: a systems perspective. Trends Neurosci. 2002;25:456–461. doi: 10.1016/s0166-2236(02)02211-7. [DOI] [PubMed] [Google Scholar]
- McGaugh JL. The amygdala modulates the consolidation of memories of emotionally arousing experiences. Annu Rev Neurosci. 2004;27:1–28. doi: 10.1146/annurev.neuro.27.070203.144157. [DOI] [PubMed] [Google Scholar]
- McGaugh JL, Introini-Collison IB, Nagahara AH. Memory-enhancing effects of naloxone: involvement of beta-noradrenergic influences in the amygdaloid complex. Brain Res. 1988;446:37–49. doi: 10.1016/0006-8993(88)91294-2. [DOI] [PubMed] [Google Scholar]
- McLean J, Waterhouse BD. Noradrenergic modulation of cat area 17 neuronal responses to moving visual stimuli. Brain Res. 1994;667:83–97. doi: 10.1016/0006-8993(94)91716-7. [DOI] [PubMed] [Google Scholar]
- Muller JF, Mascagni F, McDonald AJ. Synaptic connections of distinct interneuronal subpopulations in the rat basolateral amygdalar nucleus. J Comp Neurol. 2003;456:217–236. doi: 10.1002/cne.10435. [DOI] [PubMed] [Google Scholar]
- Paxinos G, Watson C. The rat brain in stereotaxic coordinates. Ed 3. San Diego: Academic; 1997. [DOI] [PubMed] [Google Scholar]
- Ramsooksingh MD, Jedema HP, Moore H, Grace AA. Stimulation of the central nucleus of the amygdala increases single unit activity of locus coeruleus neurons. Soc Neurosci Abstr. 2003;29:282–10. [Google Scholar]
- Rasmussen K, Morilak DA, Jacobs BL. Single unit activity of locus coeruleus neurons in the freely moving cat. I. During naturalistic behaviors and in response to simple and complex stimuli. Brain Res. 1986;371:324–334. doi: 10.1016/0006-8993(86)90370-7. [DOI] [PubMed] [Google Scholar]
- Rauch SL, Whalen PJ, Shin ML, McInerney SC, Macklin ML, Lasko NP, Orr SP, Pitman RK. Exaggerated amygdala response to masked facial stimuli in posttraumatic stress disorder: a functional MRI study. Biol Psychiatry. 2000;47:769–776. doi: 10.1016/s0006-3223(00)00828-3. [DOI] [PubMed] [Google Scholar]
- Rosenkranz JA, Buffalari DM, Grace AA. Opposing influence of basolateral amygdala and footshock stimulation on neurons of the central amygdala. Biol Psychiatry. 2006;59:801–811. doi: 10.1016/j.biopsych.2005.09.013. [DOI] [PubMed] [Google Scholar]
- Shekhar A, Sajdyk T, Keim SR, Yoder KK, Sanders SK. Role of the basolateral amygdala in panic disorder. Ann NY Acad Sci. 1999;877:747–750. doi: 10.1111/j.1749-6632.1999.tb09315.x. [DOI] [PubMed] [Google Scholar]
- Shekhar A, Truitt W, Rainnie D, Sajdyk T. Role of stress, corticotrophin releasing factor (CRF) and amygdala plasticity in chronic anxiety. Stress. 2005;8:209–219. doi: 10.1080/10253890500504557. [DOI] [PubMed] [Google Scholar]
- Strange BA, Dolan RJ. Beta-adrenergic modulation of emotional memory-evoked human amygdala and hippocampal responses. Proc Natl Acad Sci USA. 2004;101:11454–11458. doi: 10.1073/pnas.0404282101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sved AF, Cano G, Passerin AM, Rabin B. The locus coeruleus, Barrington's nucleus, and neural circuits of stress. Physiol Behav. 2002;77:737–742. doi: 10.1016/s0031-9384(02)00927-7. [DOI] [PubMed] [Google Scholar]
- U'Prichard DC, Reisine TD, Mason ST, Fibiger HC, Yamamura HI. Modulation of rat brain alpha- and beta-adrenergic receptor populations by lesion of the dorsal noradrenergic bundle. Brain Res. 1980;187:143–154. doi: 10.1016/0006-8993(80)90500-4. [DOI] [PubMed] [Google Scholar]
- Van Bockstaele EJ, Bajic D, Proudfit H, Valentino RJ. Topographic architecture of stress-related pathways targeting the noradrenergic locus coeruleus. Physiol Behav. 2001;73:273–283. doi: 10.1016/s0031-9384(01)00448-6. [DOI] [PubMed] [Google Scholar]