Activity-Evoked Capacitative Ca2+ Entry: Implications in Synaptic Plasticity (original) (raw)
Abstract
The Ca2+ influx controlled by intracellular Ca2+ stores, called store-operated Ca2+ entry (SOC), occurs in various eukaryotic cells, but whether CNS neurons are endowed with SOC capability and how they may operate have been contentious issues. Using Ca2+ imaging, we present evidence for the presence of SOC in cultured hippocampal pyramidal neurons. Depletion of internal Ca2+ stores by thapsigargin caused intracellular Ca2+ elevation, which was prevented by SOC channel inhibitors 2-aminoethoxydiphenyl borate (2-APB), SKF96365, and La3+. Interestingly, these inhibitors also accelerated the decay of NMDA-induced Ca2+ transients without affecting their peak amplitude. In addition, SOC channel inhibitors attenuated tetanus-induced dendritic Ca2+ accumulation and long-term potentiation at Schaffer collateral-CA1 synapses in hippocampal slice preparations. These data suggest a novel link between ionotropic receptor-activated SOC and neuroplasticity.
Keywords: store-operated calcium entry, NMDA, glutamate receptor, long-term potentiation, hippocampus, transient receptor potential channel
Introduction
The calcium ion (Ca2+) is a ubiquitous intracellular messenger that regulates various cell functions. Like other types of cells, CNS neurons use both extracellular and intracellular sources of Ca2+, i.e., Ca2+ influx via receptor-operated Ca2+ channels such as NMDA receptors or voltage-operated Ca2+ channels such as L-type Ca2+ channels, and Ca2+ release from endoplasmic reticulum (ER) via inositol 1,4,5-triphosphate (IP3) receptors or ryanodine receptors. Although the ER is structurally continuous across the soma, axon, and dendrites in a neuron (Spacek and Harris, 1997), the Ca2+ signals display distinct spatiotemporal subcompartments (Blaustein and Golovina, 2001). By using these local signals, neurons regulate their excitability, plasticity, gene expression, and cell death (Berridge, 1998; Zucker, 1999; Mattson et al., 2000). Of equal importance then is the characterization of the replenishing mechanisms after ER Ca2+ release in neurons.
Store-operated Ca2+ entry (SOC), also termed capacitative Ca2+ influx, is regarded as a mechanism mediating ER Ca2+ replenishing (Putney, 1986). SOC produces a rise in intracellular Ca2+ concentrations ([Ca2+]i) via recruitment of extracellular Ca2+ in response to ER Ca2+ store depletion. Ca2+ influx is assumed to take place through SOC channels rather than receptor-operated or voltage-operated Ca2+ channels. SOC appears to be a universal phenomenon across cell types; however, little is known about the molecular profiles of SOC channels or how store depletion gives rise to SOC. In the CNS, SOC has been found in astrocytes (Lo et al., 2002) and neuronal cell lines (Grudt et al., 1996), but the presence of neuronal SOC remains disputed (Koizumi et al., 1999; Bouron, 2000; Emptage et al., 2001).
In the present study, we used primary cultures of two distinct neuron populations in the hippocampal formation, i.e., hippocampal pyramidal cells and dentate granule cells, to characterize neuronal SOC properties. We report that NMDA receptor activation leads to SOC in pyramidal neurons, but not in granule cells and that pharmacological blockade of SOC results in attenuation of NMDA receptor-dependent synaptic plasticity at Schaffer-CA1 transmission in hippocampal slices.
Materials and Methods
Materials. 2-Aminoethoxydiphenyl borate (2-APB) was obtained from Tokyo-Kasei (Tokyo, Japan). AP-5, lanthanum chloride, nicardipine and NMDA were from Sigma (St. Louis, MO). SKF96365 was from Tocris Cookson (Bristol, UK). Thapsigargin was purchased from Alomone Labs (Jerusalem, Israel).
Primary cultures of pyramidal and granule cells. Postnatal 3-day-old Wistar/ST rats (SLC, Shizuoka, Japan) were deeply anesthetized by ether, according to the Japanese Pharmacological Society guide for the care and use of laboratory animals. The formatio hippocampalis was dissected out and placed in ice-cold Gey's balanced salt solution. After removal of the subicular complex, the remaining part was divided into the Ammon's horn and dentate gyrus. These tissues were trypsinized and gently triturated, and isolated cells were plated at a density of 5.0 × 104 cells/cm2 onto polyethylenimine-coated coverslips. We could consistently obtain ∼2.0 × 105 pyramidal cells or granule cells from one brain. They were cultivated in 50% Neurobasal/B-27 (Invitrogen, Gaithersburg, MD) and 50% astrocyte-conditioned medium (Ikegaya and Matsuki, 2002). The culture medium was changed to the conditioned medium-free Neurobasal/B-27 supplemented with 2 μm cytosine-d-arabinofuranoside (Sigma) 24 hr after the plating. Half of the medium was replaced with fresh one every 3 d.
[Ca 2+] i imaging. Changes in [Ca2+]i at somatic or dendritic regions were detected by a standard microfluorometrical technique with fura-2, as previously described (Baba et al., 2002). At day 7-9 in vitro, cells were incubated in 5 μm fura-2 AM (Wako Chemicals, Osaka, Japan) and 0.02% cremophor EL (Sigma) at 37°C for 30 min, followed by a rinse with balanced salt solution consisting of (in mm): 130 NaCl, 5 KCl, 1.8 CaCl2, 20 HEPES, and 10 glucose. Unless used for field stimulation, the solution was supplemented with 1 μm tetrodotoxin to prevent firing. The cells were constantly perfused with the same solution at 37°C and were illuminated by a xenon light source to monitor the ratio of the fluorescence intensity of fura-2 (F) excited at 340 and 360 nm at 1-20 Hz. Δ_F_340/360 relative to baseline was analyzed as indicative of [Ca2+]i changes with an AQUACOSMOS system (Hamamatsu Photonics, Hamamatsu, Japan). Decay kinetics of [Ca2+]i transients were fitted using the exponential fitting algorithms in Igor. Drugs were applied at 0.3 ml/min through a local perfusion pipette positioned at 200 μm from the cells.
Outside-out recording. NMDA channel currents were recorded by outside-out patches isolated from cultured pyramidal cells using an Axopatch 200B amplifier (Axon Instruments, Foster City, CA). Micropipettes (10-20 MΩ) were filled with an internal solution containing (in mm): 140 KMeSO4, 10 NaCl, 10 HEPES, and 10 EGTA, pH 7.2. The external bath solution consisted of (in mm): 150 NaCl, 5 KCl, 2 CaCl2,10 glucose, and 10 HEPES, pH 7.3 at 24°C. Recording was performed at -60 mV in the presence of 10 μm NMDA. After each experiment, we applied AP-5 to reject data that did not purely reflect NMDA receptor-medicated currents. The single-channel open probability was determined from the ratio of the time spent in the open state to the duration of recording: _P_o = (_t_1 + _t_2 +... + _t_n)/_N_ttot, where t is the amount of time that n channels are open, and the N is the maximum number of levels observed in the patch.
Electrophysiological recording. Transverse hippocampal slices (400 μm thickness) were prepared from the brains of 17- to 27-day-old Wistar/ST rats (SLC) in ice-cold artificial CSF, consisting of (in mm): 124 NaCl, 25 NaHCO3, 3 KCl, 1.24 KH2PO4, 1.4 MgSO4, 2.2 CaCl2, and 10 glucose, as described previously (Ueno et al., 2002). The slices were attached onto a MED-P515A probe (Alpha MED Sciences, Chuo-ku, Tokyo, Japan) and perfused with in a 95% O2 and 5% CO2-saturated artificial CSF for at least 1 hr at 32°C. One of 64 planar microelectrodes was used to stimulate the Schaffer collaterals every 30 sec (100 μsec bipolar rectangular pulses), and field EPSPs (fEPSPs) evoked in CA1 stratum radiatum were recorded using a MED64 multichannel recording system (Tsukamoto et al., 2003). Stimulus intensity was set to produce fEPSP with a half-maximal slope (15-50 μA), and synaptic strength was evaluated by measuring changes in the fEPSP slopes.
All data are expressed as means ± SEM.
Results
Pyramidal and granule neurons were prepared from Ammon's horns and dentate gyri, respectively, from postnatal 3-day-old rat pups. After 7-9 d in vitro, [Ca2+]i was monitored with fura-2 imaging. The cells were incubated in Ca2+-free conditions for 5 min and then treated for 5 min with 1 μm thapsigargin, an ER Ca2+-ATPase inhibitor, to deplete the ER stores. Consistent with a previous report showing that baseline ER stores are low, using little of their storage capacity, in hippocampal neurons (Irving and Collingridge, 1998), thapsigargin induced a minimal rise in [Ca2+]i (Fig. 1_A_). When 1.8 mm Ca2+ is subsequently replaced in bath saline, both neuron populations displayed prolonged [Ca2+]i increases in all 446 (pyramidal) and 285 (granule) cells tested (Fig. 1_A,C_). The amplitude of the [Ca2+]i plateau did not differ between neuron types (Fig. 1_C_). Neurons untreated with thapsigargin showed no apparent [Ca2+]i changes after Ca2+ replacement (Fig. 1_A_).
Figure 1.
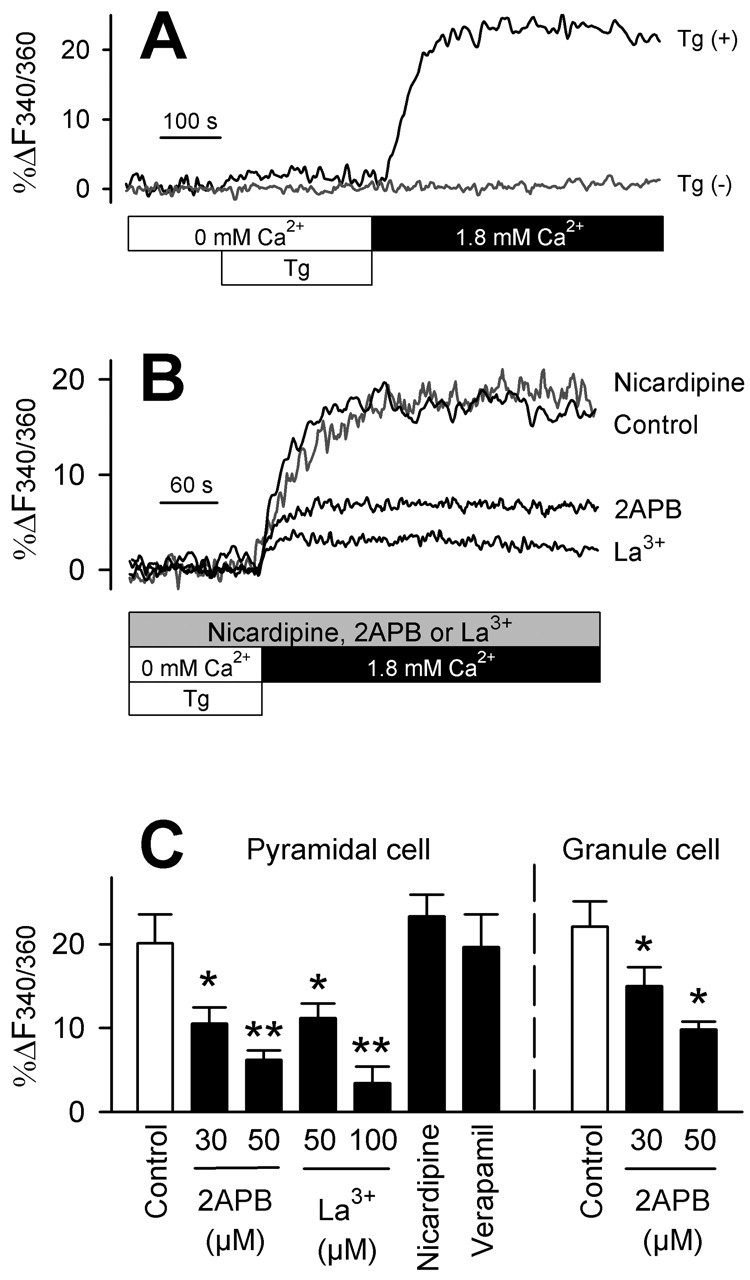
Both hippocampal and dentate neurons display SOC. A, Representative traces of somatic [Ca2+]i dynamics obtained from a pyramidal neuron. A [Ca2+]i rise was evoked by pretreatment with 1 μm thapsigargin (Tg (+)) for 5 min in Ca2+-free conditions (open bars) and subsequent bath addition of 1.8 mm Ca2+ (closed bar). B, The [Ca2+]i rise was prevented by 30 μm 2-APB, 100 μm La3+, but not by 5 μm nicardipine. These agents were continuously applied from 5 min before thapsigargin. C, Summary of the effects of 2-APB, La3+, 5 μm nicardipine, and 10 μm verapamil on SOC in cultured pyramidal and granule neurons. The ordinate indicates the average amplitude of capacitative [Ca2+]i plateaus as an increase in _F_340/360 ratios (%). *p < 0.05, **p < 0.01 versus corresponding control: Fisher's protected least significant difference after one-way ANOVA (n = 11-34 neurons from 3-8 independent experiments).
To identify the source of Ca2+ entry, we tested the effect of several types of Ca2+ channel inhibitors. 2-APB is known to selectively block SOC channels at concentrations of tens of micromolar, whereas at higher doses it inhibits IP3 receptor channels (Gregory et al., 2001; Iwasaki et al., 2001; Kukkonen et al., 2001; Bootman et al., 2002). 2-APB efficiently prevented the [Ca2+]i plateau, after Ca2+ replacement, at 30-50 μm (Fig. 1_B,C_). The results were mimicked by 50-100 μm La3+, a broad spectrum inhibitor of Ca2+ channels, including SOC channels (Fig. 1_B,C_). The L-type Ca2+ channel inhibitors nicardipine (5 μm) or verapamil (10 μm) were ineffective (Fig. 1_B,C_).
Taken together, the [Ca2+]i plateau evoked by thapsigargin-induced store depletion is dependent on external Ca2+ and is sensitive to 2-APB and La3+ but not to nicardipine or verapamil. These pharmacological results are consistent with SOC. We therefore conclude that the SOC pathway exists in both hippocampal pyramidal and dentate granule neurons.
We sought to determine whether more physiological stimuli can activate neuronal SOC. Recent studies indicate that NMDA receptor activation might cause ER Ca2+ release (Simpson et al., 1995; Emptage et al., 1999) and that Ca2+ entry through NMDA receptor channels acts to refill ER stores (Rae et al., 2000). We thus hypothesized a possible link between NMDA receptors and SOC.
Our previous reports indicated that treatment with NMDA (10 μm, 10 sec) evokes transient [Ca2+]i elevations but that the subsequent recovery to baseline was slower in pyramidal cells than in granule cells (Baba et al., 2002) (see also Fig. 2_A_). Neither the basal _F_360 nor _F_340/360 value was different between the neuron populations, and higher concentrations of NMDA could produce larger [Ca2+]i amplitudes in both neuron types (data not shown). Thus, the different [Ca2+]i decay cannot be accounted for by a difference in indicator-loading efficiency, fluorescence saturation, or resting Ca2+ levels between both neuron classes. In the present study, we noticed that the [Ca2+]i decay in pyramidal and granule cells showed different exponential functions. Granule cells displayed a simple [Ca2+]i decrease with a monoexponential time course with a mean time constant of 24.4 ± 2.4 sec (τf) until reaching baseline. In pyramidal cells, however, the [Ca2+]i decay was best fit with a double-exponential. The [Ca2+]i initially decayed with 22.3 ± 2.6 sec of time constant (τf), similar to that measured in granule cells, however, the second exponential exhibited a 10-fold slower decay, the mean time constant τs being 212.6 ± 16.7 sec, indicating a later phase. These data imply that NMDA-induced [Ca2+]i dynamics involves different mechanisms between pyramidal and granule cells.
Figure 2.

NMDA receptor-activated SOC in hippocampal pyramidal neurons. A, Representative traces of[Ca2+]itransients evoked by local application of NMDA (10μm for 10 sec) in the absence (top) or presence (bottom) of 30μm 2-APB, which were obtained from each one pyramidal (left) or granule (right) cell. After the initial increase, the somatic[Ca2+]i decayed with a monoexponential time course with the mean time constantτf (dotted lines) in granule cells, whereas pyramidal cells exhibited double-exponential decay kinetics with the time constants τf (dotted lines) and τs (broken lines). B, Summary of the effects of 30 μm 2-APB, 3 μmSKF96365 (SKF), 100 μmLa3+, and 1 μm thapsigargin (Tg) on NMDA responses in cultured Ammon's horn and dentate gyrus neurons. The ordinate indicates the average coefficients of the fast and slow components in double-exponential[Ca2 +]i decay kinetics of NMDA responses(A f and A s, respectively). All the drugs were continuously perfused from 5 min before NMDA exposure. **p < 0.01 versus control, #p < 0.05 versus Tg: Fisher's protected least significant difference following one-way ANOVA (n = 7-64 neurons from 3-10 independent experiments). C, Effect of extracellular Ca2+ removal and 30 μm 2-APB on the _A_s component. D, No [Ca2+]i rise occurred when NMDA (10 μm for 10 sec) was applied in the absence of external Ca2+ (n = 7).
We examined the effect of 2-APB on the fast and slow exponential decay coefficients (Fig. 2_A_, _A_f, _A_s). 2-APB (30 μm) did not affect the fast component _A_f in either pyramidal or granule cells but did reduce the slow component _A_s in pyramidal cells by ∼65% (Fig. 2_B_). Some studies have suggested that 2-APB might have nonspecific effects even at such low concentrations (Wu et al., 2000; Missiaen et al., 2001), however, our results were mimicked by 3 μmSKF96365, a structurally unrelated SOC channel inhibitor (Leung and Kwan, 1999) and also by 100 μm La3+ (Fig. 2_B_), suggesting that SOC mediates the slow phase of Ca2+ decay. If this is the case, pharmacological store depletion should enhance the _A_s value. As expected, pretreatment with 1 μm thapsigargin increased _A_s by ∼45% without affecting _A_f (Fig. 2_B_). This effect also implies a relatively small contribution of ER Ca2+ release to total NMDA-induced [Ca2+]i increase. The change in _A_s, after thapsigargin, was partially attenuated by 2-APB (Fig. 2_B_). This partial blockade was probably attributable to the multipotency of thapsigargin; it not only facilitated SOC induction but likely also prevented Ca2+ reuptake by the ER, which is insensitive to 2-APB, both of which contribute to an increase in _A_s.
To determine whether 2-APB actually has no effect on Ca2+ reuptake by the ER or plasma membrane Ca2+-ATPase after NMDA-induced Ca2+ increases, we removed extracellular Ca2+ immediately after NMDA washout. Zero Ca2+ lessened the slow decay component of NMDA responses. The remaining Ca2+ component was no more reduced by 2-APB (Fig. 2_C_). Therefore, under our experimental conditions, 2-APB does not appear to effect ER Ca2+ release or Ca2+-ATPase pumps.
It is still possible that 2-APB acts directly on NMDA receptor channels. We thus performed channel current recordings by outside-out patches from cultured pyramidal cells. 2-APB did not alter the open probability or conductance of the NMDA receptor channel (Fig. 3_A_); the unit conductance was 33.3 ± 0.60 pS (control) and 35.4 ± 1.40 pS (2-APB), and the open probability was 0.243 ± 0.064 (control) and 0.247 ± 0.035 (2-APB) (p > 0.1, paired t test; mean ± SEM of five recordings). Thus, 2-APB is unlikely to alter the properties of NMDA receptor channels. These single receptor-channel recordings further suggest that the 2-APB actions observed here are mediated by SOC inhibition.
Figure 3.
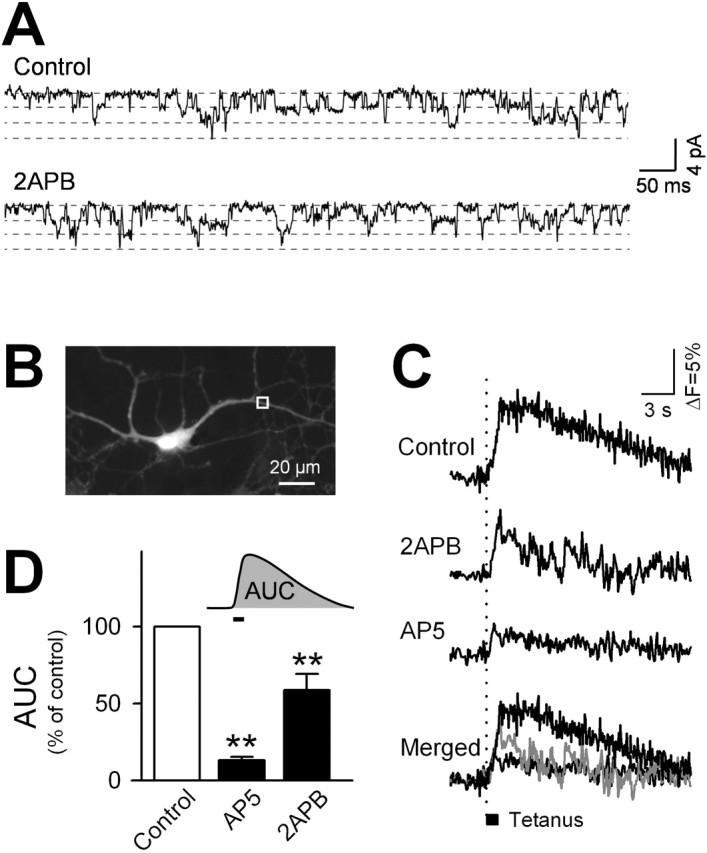
Synaptic activation induces SOC in hippocampal pyramidal cells. A, Example of the NMDA channel activity in an outside-out patch isolated from a cultured pyramidal neuron before or after application of 30μm 2-APB. B, Image of a fura-2-loaded neuron. C, Representative optical recordings of tetanus-elicited [Ca2+]i changes in a region of the neuron delimited by the box shown in_B_. Electrical field stimulation(100 Hz for 1 sec, 60 V, 200 μsecduration) was applied in the absence or presence of 30μm 2-APB or 50μm AP-5. D, Summary of the effects of AP-5 and 2-APB on tetanus-elicited [Ca2+]i elevation. The ordinate shows the average area under the curve (AUC) of Δ_F_/_F_340/360 during and after the tetanus (n = 7 neurons). **p < 0.01 versus control: Fisher's protected least significant difference after one-way ANOVA.
Our data suggest that SOC can be triggered by NMDA receptor-channel activation in hippocampal pyramidal cells and is responsible for the prolonged NMDA-mediated Ca2+ responses in these neurons. Importantly, NMDA failed to elevate [Ca2+]i when applied in the absence of extracellular Ca2+, although bath Ca2+ levels returned to normal levels immediately after NMDA washout (Fig. 2_D_). Thus, Ca2+ influx through NMDA receptor channels was required for SOC activation. NMDA receptor-mediated Ca2+ entry has been found to be essential for the induction of hippocampal long-term potentiation (LTP), a well established cellular model of synaptic plasticity that has been proposed as a substrate for memory (Bliss and Collingridge, 1993). We thus hypothesized that SOC is involved in LTP.
To address this possibility, we measured [Ca2+]i within dendritic regions (>30 μm from the soma) in response to electric field tetanic stimulation (100 Hz for 1 sec), which is known to induce LTP (Fig. 3_B_). This stimulus yielded a transient increase in dendritic [Ca2+]i that was almost completely blocked by the NMDA receptor antagonist AP-5 (50 μm) (Fig. 3_C,D_). 2-APB (30 μm) also markedly decreased these responses (Fig. 3). Therefore, tetanic stimulation induces SOC in postsynaptic dendrites in addition to the NMDA-mediated Ca_2_+ influx. Using acute rat hippocampal slices, we monitored synaptic responses of the Schaffer collateral-CA1 pathway to determine whether SOC activity contributes to LTP induction. After tetanization (100 Hz for 1 sec) of the afferents, the synaptic responses increased indicative of LTP (Fig. 4). 2-APB reduced the magnitude of LTP, whereas it did not affect pretetanus synaptic efficacy (Fig. 4). The same result was obtained with 3 μmSKF96365. These suggest that tetanus-induced SOC activation is involved in the induction of hippocampal synaptic plasticity.
Figure 4.
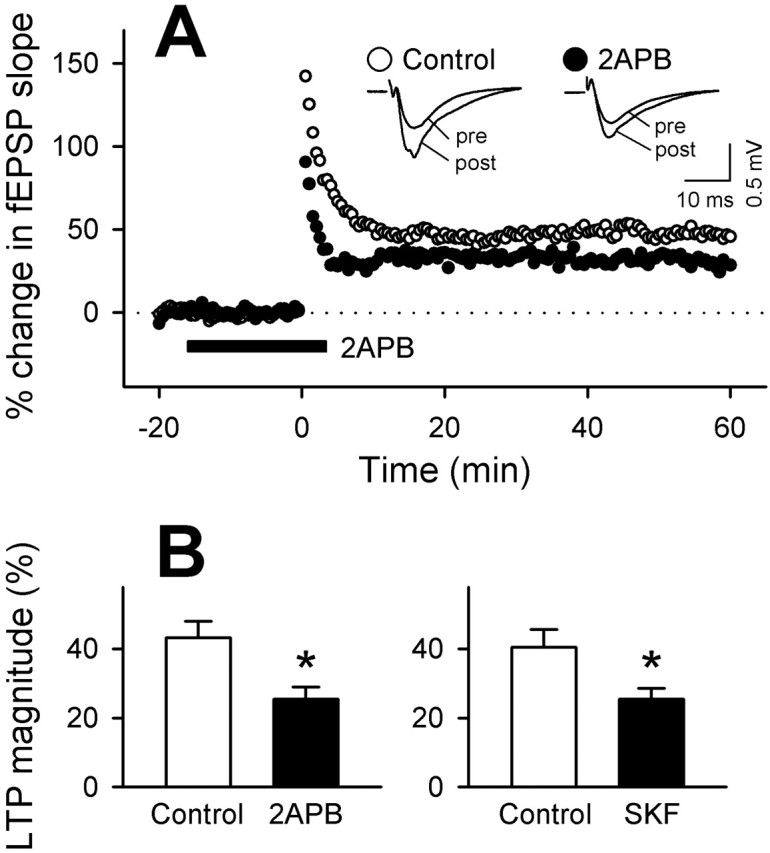
SOC channel inhibitors attenuate hippocampal CA1 LTP. A, Representative time course of changes in fEPSPs evoked at Schaffer collateral-CA1 synapses after tetanic stimulation (100 Hz for 1 sec) applied in the absence (open circles) or presence (closed circles) of 2-APB. 2-APB was applied during time -15 to 5. The insets indicate field potentials recorded at times 0 (pre) and 60(post). The fEPSP slopes are expressed as a percentage of changes from baseline. B, Summary of the effects of 30μm 2-APB and 3μmSKF96365 (SKF) on the LTP magnitude. The ordinate shows the average changes in fEPSP slopes at time 55-60 (n = 6-16 recordings). *p < 0.05 versus control: Student's t test.
Discussion
SOC is present in a wide range of cell types, and despite of the importance of ER-mediated Ca2+ signaling in neurons, the role of SOC in CNS neurons has been poorly described. We have shown that SOC is inducible in both hippocampal pyramidal and dentate granule cells, that SOC can be activated by NMDA receptor stimulation in pyramidal cells, and that SOC may play a role in synaptic plasticity of pyramidal cells.
It is intriguing to find that CNS neurons possess SOC machinery despite the presence of a spectrum of voltage-operated and receptor-operated Ca2+-permeable channels on the plasma membrane, each of which supports dynamic Ca2+ signaling in subcellular components, e.g., dendrites, spines, somata, axons, and synaptic terminals. SOC is an additional pathway for dynamic Ca2+ entry potentially playing a complementary role for intracellular Ca2+ release. This and other studies have demonstrated that thapsigargin alone can elicit only marginal [Ca2+]i increases, suggesting small releasable Ca2+ pools in neuronal ER stores (Irving and Collingridge, 1998). SOC may functionally compensate for this potential Ca2+ shortfall. Indeed, the [Ca2+]i amplitude yielded by SOC was comparable to NMDA-induced [Ca2+]i transients, and thus is likely sufficient to initiate various cellular events that the small available ER Ca2+ cannot.
We previously established a method for isolating and maintaining hippocampal pyramidal and dentate granule cells in culture and found that Ca2+ dynamics of these neuron populations differ in their decay kinetics, but we were unable to determine the source of this difference (Baba et al., 2002). The present study revealed that in pyramidal cells, an NMDA-induced [Ca2+]i transient is followed by a 2-APB/SKF96365-sensitive [Ca2+]i trail, which is blocked by La3+ and facilitated by thapsigargin. Considering that 2-APB and SKF96365 almost eliminated the difference in Ca2+ dynamics between pyramidal and granule cells, the different decay kinetics may be attributable to NMDA-induced SOC in pyramidal cells.
The lack of NMDA-induced SOC in granule cells is enigmatic. This may be attributable to differential cellular distribution of NMDA receptors and SOC channels. It is also possible that SOC activation is prevented by strong endogenous Ca2+ buffers; Ca2+-binding proteins such as calbindin are abundant in granule cells (Baba et al., 2002).
Most past studies used artificial conditions to induce SOC, i.e., protocols in which ER stores were pharmacologically forced to be empty. The use of such nonphysiological conditions has made it difficult to accurately argue how and when SOC occurs in nature. Here we have successfully induced SOC using physiological stimuli, i.e., synaptic NMDA receptor activation. Ca2+ influx through NMDA receptors triggers SOC. This Ca2+ signal may recruit signal molecules that can stimulate ER stores, such as IP3. Indeed, there have been previous indications that NMDA receptor activation may lead to Ca2+ release from ER (Simpson et al., 1995; Emptage et al., 1999). This may in turn cause store depletion, eventually activating SOC.
NMDA receptors play a crucial role in synaptic plasticity. Here we report that 2-APB and SKF96365 attenuated both NMDA-induced Ca2+ dynamics and LTP in hippocampal pyramidal cells. We suggest that synaptic NMDA receptor-activated SOC is involved in LTP. However, Emptage et al. (2001) reports that pharmacological depletion of ER stores evokes SOC at presynaptic terminals, thus partly determining the frequency of spontaneous transmitter release. As a result we cannot exclude the possibility that the SOC blockers prevented LTP by affecting presynaptic SOC. In particular, 2-APB-induced attenuation of post-tetanic potentiation, which is generally accepted to be presynaptic in origin (Zucker and Regehr, 2002), may be attributable to a change in the probability of neurotransmitter release.
This paper contains at least four significant implications. (1) SOC is generally considered as a store-refilling mechanism. However, we propose a more active role in CNS neurons. SOC is functionally coupled with neurotransmitter receptor-channels mediating activity-dependent Ca2+ dynamics, thus regulating synaptic efficacy. (2) Considering that Ca2+ levels in ER are kept substantially low in hippocampal neurons (Irving and Collingridge, 1998), our findings necessitate revision of prevailing concepts regarding the role of ER in CNS neurons. The stored Ca2+ is not merely a source of Ca2+ but also works to initiate SOC via its depletion. In other words, ER Ca2+ serves to prevent SOC activity under resting conditions. (3) This work also highlights the physiological significance of NMDA receptors. These receptors may be assigned a function beyond their channel kinetics and properties, because Ca2+ signals generated by NMDA receptors can be temporally and quantitatively amplified by subsequent SOC activation. This might help in temporal summation and extraction of neural information. (4) We observed SOC in both pyramidal and granule cells, however, the mechanism of SOC activation appears to be different. It is probable that the functions of neuronal SOC vary among types of neuron.
Footnotes
This work was supported in part by a grant-in-aid for Science Research from the Ministry of Education, Culture, Sports, Science, and Technology of Japan. We thank H. Jiko and A. Shimizu (Alpha MED Sciences, Chuo-ku, Tokyo, Japan) for their technical support of 8×8 multielectrode recording from hippocampal slices and Dr. Jason N. MacLean (Columbia University, New York, NY) for his critical review of this manuscript.
Correspondence should be addressed to Yuji Ikegaya, Laboratory of Chemical Pharmacology, Graduate School of Pharmaceutical Sciences, The University of Tokyo, 7-3-1 Hongo, Bunkyo-ku, Tokyo 113-0033, Japan. E-mail: ikegaya@tk.airnet.ne.jp.
Copyright © 2003 Society for Neuroscience 0270-6474/03/237737-05$15.00/0
References
- Baba A, Yamada MK, Nishiyama N, Matsuki N, Ikegaya Y ( 2002) Different Ca2+ dynamics between isolated hippocampal pyramidal cells and dentate granule cells. J Neurocytol 31: 41-48. [DOI] [PubMed] [Google Scholar]
- Berridge MJ ( 1998) Neuronal calcium signaling. Neuron 21: 13-26. [DOI] [PubMed] [Google Scholar]
- Blaustein MP, Golovina VA ( 2001) Structural complexity and functional diversity of endoplasmic reticulum Ca2+ stores. Trends Neurosci 24: 602-608. [DOI] [PubMed] [Google Scholar]
- Bliss TV, Collingridge GL ( 1993) A synaptic model of memory: long-term potentiation in the hippocampus. Nature 361: 31-39. [DOI] [PubMed] [Google Scholar]
- Bootman MD, Collins TJ, Mackenzie L, Roderick HL, Berridge MJ, Peppiatt CM ( 2002) 2-aminoethoxydiphenyl borate (2-APB) is a reliable blocker of store-operated Ca2+ entry but an inconsistent inhibitor of InsP3-induced Ca2+ release. FASEB J 16: 1145-1150. [DOI] [PubMed] [Google Scholar]
- Bouron A ( 2000) Activation of a capacitative Ca2+ entry pathway by store depletion in cultured hippocampal neurones. FEBS Lett 470: 269-272. [DOI] [PubMed] [Google Scholar]
- Emptage N, Bliss TV, Fine A ( 1999) Single synaptic events evoke NMDA receptor-mediated release of calcium from internal stores in hippocampal dendritic spines. Neuron 22: 115-124. [DOI] [PubMed] [Google Scholar]
- Emptage NJ, Reid CA, Fine A ( 2001) Calcium stores in hippocampal synaptic boutons mediate short-term plasticity, store-operated Ca2+ entry, and spontaneous transmitter release. Neuron 29: 197-208. [DOI] [PubMed] [Google Scholar]
- Gregory RB, Rychkov G, Barritt GJ ( 2001) Evidence that 2-aminoethyl diphenylborate is a novel inhibitor of store-operated Ca2+ channels in liver cells, and acts through a mechanism which does not involve inositol trisphosphate receptors. Biochem J 354: 285-290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grudt TJ, Usowicz MM, Henderson G ( 1996) Ca2+ entry following store depletion in SH-SY5Y neuroblastoma cells. Mol Brain Res 36: 93-100. [DOI] [PubMed] [Google Scholar]
- Ikegaya Y, Matsuki N ( 2002) Regionally selective neurotoxicity of NMDA and colchicine is independent of hippocampal neural circuitry. Neuroscience 113: 253-256. [DOI] [PubMed] [Google Scholar]
- Irving AJ, Collingridge GL ( 1998) A characterization of muscarinic receptor-mediated intracellular Ca2+ mobilization in cultured rat hippocampal neurones. J Physiol (Lond) 511: 747-759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iwasaki H, Mori Y, Hara Y, Uchida K, Zhou H, Mikoshiba K ( 2001) 2-Aminoethoxydiphenyl borate (2-APB) inhibits capacitative calcium entry independently of the function of inositol 1, 4, 5-trisphosphate receptors. Receptors Channels 7: 429-439. [PubMed] [Google Scholar]
- Koizumi S, Bootman MD, Bobanovic LK, Schell MJ, Berridge MJ, Lipp P ( 1999) Characterization of elementary Ca2+ release signals in NGF-differentiated PC12 cells and hippocampal neurons. Neuron 22: 125-137. [DOI] [PubMed] [Google Scholar]
- Kukkonen JP, Lund PE, Akerman KE ( 2001) 2-aminoethoxydiphenyl borate reveals heterogeneity in receptor-activated Ca2+ discharge and store-operated Ca2+ influx. Cell Calcium 30: 117-129. [DOI] [PubMed] [Google Scholar]
- Leung YM, Kwan CY ( 1999) Current perspectives in the pharmacological studies of store-operated Ca2+ entry blockers. Jpn J Pharmacol 81: 253-258. [DOI] [PubMed] [Google Scholar]
- Lo KJ, Luk HN, Chin TY, Chueh SH ( 2002) Store depletion-induced calcium influx in rat cerebellar astrocytes. Br J Pharmacol 135: 1383-1392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mattson MP, LaFerla FM, Chan SL, Leissring MA, Shepel PN, Geiger JD ( 2000) Calcium signaling in the ER: its role in neuronal plasticity and neurodegenerative disorders. Trends Neurosci 23: 222-229. [DOI] [PubMed] [Google Scholar]
- Missiaen L, Callewaert G, De Smedt H, Parys JB ( 2001) 2-Aminoethoxydiphenyl borate affects the inositol 1, 4, 5-trisphosphate receptor, the intracellular Ca2+ pump and the non-specific Ca2+ leak from the non-mitochondrial Ca2+ stores in permeabilized A7r5 cells. Cell Calcium 29: 111-116. [DOI] [PubMed] [Google Scholar]
- Putney JW ( 1986) A model for receptor-regulated calcium entry. Cell Calcium 7: 1-12. [DOI] [PubMed] [Google Scholar]
- Rae MG, Martin DJ, Collingridge GL, Irving AJ ( 2000) Role of Ca2+ stores in metabotropic L-glutamate receptor-mediated supralinear Ca2+ signaling in rat hippocampal neurons. J Neurosci 20: 8628-8636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simpson PB, Challiss RA, Nahorski SR ( 1995) Neuronal Ca2+ stores: activation and function. Trends Neurosci 18: 299-306. [DOI] [PubMed] [Google Scholar]
- Spacek J, Harris KM ( 1997) Three-dimensional organization of smooth endoplasmic reticulum in hippocampal CA1 dendrites and dendritic spines of the immature and mature rat. J Neurosci 17: 190-203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsukamoto M, Yasui T, Yamada MK, Nishiyama N, Matsuki N, Ikegaya Y ( 2003) Mossy fibre synaptic NMDA receptors trigger non-Hebbian long-term potentiation at entorhino-CA3 synapses in the rat. J Physiol (Lond) 546: 665-675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ueno S, Tsukamoto M, Hirano T, Kikuchi K, Yamada MK, Nishiyama N, Nagano T, Matsuki N, Ikegaya Y ( 2002) Mossy fiber Zn2+ spillover modulates heterosynaptic _N_-methyl-d-aspartate receptor activity in hippocampal CA3 circuits. J Cell Biol 158: 215-220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu J, Kamimura N, Takeo T, Suga S, Wakui M, Maruyama T, Mikoshiba K ( 2000) 2-Aminoethoxydiphenyl borate modulates kinetics of intracellular Ca2+ signals mediated by inositol 1, 4, 5-trisphosphate-sensitive Ca2+ stores in single pancreatic acinar cells of mouse. Mol Pharmacol 58: 1368-1374. [DOI] [PubMed] [Google Scholar]
- Zucker RS ( 1999) Calcium- and activity-dependent synaptic plasticity. Curr Opin Neurobiol 9: 305-313. [DOI] [PubMed] [Google Scholar]
- Zucker RS, Regehr WG ( 2002) Short-term synaptic plasticity. Annu Rev Physiol 64: 355-405. [DOI] [PubMed] [Google Scholar]