Bidirectional Modulation of GABA Release by Presynaptic Glutamate Receptor 5 Kainate Receptors in the Basolateral Amygdala (original) (raw)
Abstract
The activation of kainate receptors modulates GABAergic synaptic transmission, but the mechanisms are currently a matter of intense debate. In the basolateral amygdala (BLA), the glutamate receptor 5 (GluR5) subunit of kainate receptors is heavily expressed, and GluR5 antagonists block a novel form of synaptic plasticity; yet little is known about the role of GluR5-containing kainate receptors in the physiology of the amygdala. Here we show that GluR5 agonists bidirectionally modulate the strength of synaptic transmission from GABAergic interneurons to pyramidal cells in a concentration-dependent manner. Low concentrations of (RS)-_S_-amino-3-(3-hydroxy-5-tert-butylisoxazol-4-yl) (ATPA) (0.3 μm) or glutamate (5 μm) reduced the number of failures of GABAergic synaptic transmission and enhanced the frequency of miniature IPSCs (mIPSCs). High concentrations of ATPA (10 μm) or glutamate (200 μm) increased the number of synaptic failures and reduced the frequency of mIPSCs. The facilitation or suppression of GABAergic transmission by the GluR5 agonists did not require activation of voltage-gated calcium channels or presynaptic GABAB receptors. It was also found that extracellular, endogenous glutamate tonically reduces the rate of failures of GABAergic transmission. These results suggest that the terminals of GABAergic neurons in the BLA carry two subtypes of GluR5-containing kainate receptors, which have different agonist affinities and activate opposing mechanisms of action. The GluR5-mediated, bidirectional modulation of GABA release by glutamate in the BLA may play an important role in the regulation of synaptic plasticity and neuronal excitability in this structure, under normal and pathological conditions.
Keywords: GluR5, presynaptic kainate receptors, glutamate diffusion, GABA release, inhibitory synaptic transmission, amygdala
Introduction
Glutamate, the principal excitatory neurotransmitter in the vertebrate CNS, mediates fast synaptic transmission through three classes of ionotropic receptors: NMDA, AMPA, and kainate receptors. Although the roles of NMDA and AMPA receptors on synaptic transmission and plasticity have been studied extensively, the functions of kainate receptors have only recently begun to be unveiled. In addition to mediating excitatory synaptic transmission in some brain regions (Castillo et al., 1997; Vignes and Collingridge, 1997; Cossart et al., 1998; Li and Rogawski, 1998; Kidd and Isaac, 1999), kainate receptors have been shown to modulate the release of glutamate and GABA (for review, see Frerking and Nicoll, 2000; Kullmann, 2001). The modulation of GABA release by kainate receptors has been studied primarily in the hippocampus, where significant discrepancies have become apparent with regard to the nature of this regulation. A consistent finding is that kainate receptor activation enhances action potential-dependent, spontaneous GABA release (Cossart et al., 1998;Frerking et al., 1998; Mulle et al., 2000; Rodriguez-Moreno et al., 2000; Schmitz et al., 2000; Jiang et al., 2001; Semyanov and Kullmann, 2001). However, there is intense controversy with regard to the effects of kainate on evoked- or action potential-independent GABA release, the latter revealed by recording miniature IPSCs (mIPSCs) in the presence of TTX. Thus, in interneuron to CA1 pyramidal-cell synapses, depression (Rodriguez-Moreno et al., 1997) or no effect (Frerking et al., 1999; Cossart et al., 2001) of kainate on the frequency of mIPSCs has been reported. In the same type of synapses, most investigators have observed the depression of evoked GABAergic synaptic transmission after the application of kainate (Clarke et al., 1997; Rodriguez-Moreno et al., 1997, 2000; Frerking et al., 1999; Mulle et al., 2000), whereas others have seen both facilitation and depression, depending on the concentration of kainate (Jiang et al., 2001). The facilitation of GABAergic transmission by kainate has also been observed in hypothalamic neurons (Liu et al., 1999), whereas both facilitation and depression, depending on the concentration of kainate, were observed in dorsal horn neurons (Kerchner et al., 2001). The factors contributing to these discrepancies are unclear. The concentration of kainate seems to play an important role in the direction of the modulation of GABA release (Jiang et al., 2001;Kerchner et al., 2001), although this does not appear to hold true for interneuron-to-interneuron synapses, where an increase in mIPSC frequency has been observed with either low (Cossart et al., 2001) or high (Mulle et al., 2000) kainate concentrations. Thus, the type of synapses investigated may also play a role, particularly because an increase in the frequency of mIPSCs by kainate has been observed in interneuron-to-interneuron synapses (Mulle et al., 2000; Cossart et al., 2001) but not in interneuron-to-pyramidal-cell synapses (Rodriguez-Moreno et al., 1997; Frerking et al., 1999; Cossart et al., 2001). Yet others have seen no effect of kainate on mIPSCs recorded from interneurons (Frerking et al., 1998; Semyanov and Kullmann, 2001).
Considerable effort has also been made to determine the sites of action of kainate receptor agonists, but the results have not been conclusive (Kullmann, 2001). Thus, the facilitation of GABAergic transmission has been attributed to a direct effect of kainate at the presynaptic terminals of interneurons (Mulle et al., 2000; Cossart et al., 2001) and/or to depolarization of interneurons via axonal and somatodendritic kainate receptors (Semyanov and Kullmann, 2001). The depression of GABAergic transmission has also been attributed to a direct presynaptic effect of kainate (Rodriguez-Moreno and Lerma, 1998; Rodriguez-Moreno et al., 2000). However, others have argued that the kainate-induced depression of evoked GABAergic transmission is secondary to a dramatic enhancement in spontaneous GABA release induced by kainate, with accumulated GABA acting presynaptically on GABAB inhibitory receptors (Frerking et al., 1999; Kerchner et al., 2001) and/or postsynaptically by desensitizing GABAA receptors or by decreasing pyramidal-cell input resistance, thereby shunting evoked IPSCs (eIPSCs) (Frerking et al., 1999).
One difficulty in reconciling the results from different laboratories and reaching definitive conclusions is that there are different subtypes of kainate receptors that may be located in different types of cells or neuronal compartments and that may serve different functions (Chittajallu et al., 1999). In many studies, the subtype of receptors activated by kainate is not identified, because specific agonists/antagonists are currently available only for the glutamate receptor 5 (GluR5) subtype. When the GluR5 kainate receptor has been investigated, the specific GluR5 agonist (RS)-_S_-amino-3-(3-hydroxy-5-tert-butylisoxazol-4-yl) (ATPA) (Clarke et al., 1997) has been found to enhance action potential-dependent, spontaneous GABA release (Cossart et al., 1998;Rodriguez-Moreno et al., 2000), whereas it suppresses evoked GABA release (Clarke et al., 1997; Rodriguez-Moreno et al., 2000) in interneuron-to-pyramidal-cell synapses in the CA1 hippocampal area. ATPA also depresses eIPSCs recorded from neocortical pyramidal cells (Ali et al., 2001) but has virtually no effect in dorsal horn neurons (Kerchner et al., 2001). In interneuron-to-interneuron synapses, ATPA has no effect on mIPSC frequency (Cossart et al., 2001). The results reported by Mulle et al. (2000) in GluR5-deficient mice are consistent with the lack of involvement of the GluR5 subunit in the regulation of GABA release in interneuron-to-interneuron synapses, whereas Mulle et al. (2000) suggest that in interneuron-to-pyramidal-cell synapses, the GluR5 subunit together with the GluR6 subunit mediates suppression of evoked GABA release.
In the basolateral amygdala (BLA), the GluR5 subunit is highly expressed, and GluR5-containing kainate receptors seem to play a significant role, because they participate in synaptic transmission (Li and Rogawski, 1998) as well as in synaptic plasticity (Li et al., 1998,2001). However, the precise location and function of GluR5 kainate receptors are unknown. In this study, we applied the whole-cell patch-clamp technique to investigate whether GluR5 kainate receptors modulate the GABAergic inhibition of identified pyramidal neurons in the basolateral amygdala.
Materials and Methods
Electrophysiology. Coronal slices containing the amygdala were prepared from 15- to 22-d-old male Sprague Dawley rats. The rats were anesthetized with halothane and then decapitated. The brain was rapidly removed and placed in ice-cold artificial CSF (ACSF) composed of (in mm): 125 NaCl, 2.5 KCl, 2.0 CaCl2, 1.0 MgCl2, 25 NaHCO3, 1.25 NaH2PO4, and 11 glucose, bubbled with 95% O2 and 5% CO2 to maintain a pH of 7.4. A block containing the amygdala region was prepared by rostral and caudal coronal cuts, and 400-μm-thick slices were cut using a Vibratome(series 1000; Technical Products International, St. Louis, MO). Slices were kept in a holding chamber containing oxygenated ACSF at room temperature, and recordings were initiated ≥1 hr after slice preparation.
For whole-cell recordings, slices were transferred to a submersion-type recording chamber, where they were continuously perfused with oxygenated ACSF at a rate of 3–4 ml/min. Neurons were visualized with an upright microscope (Nikon Eclipse E600fn; Nikon, Tokyo, Japan) using Nomarski-type differential interference optics through a 60× water immersion objective. All experiments were performed at room temperature (28°C). Tight-seal (>1 GΩ) whole-cell recordings were obtained from the cell body of neurons in the BLA region. Patch electrodes were fabricated from borosilicate glass and had a resistance of 1.5–5.0 MΩ when filled with a solution containing (in mm): 135 Cs-gluconate, 10 MgCl2, 0.1 CaCl2, 1 EGTA, 10 HEPES, 2 Na-ATP, 0.2 Na3GTP, and 0.4% Lucifer yellow, pH 7.3 (285–290 mOsm). Neurons were voltage-clamped using an Axopatch 200B amplifier (Axon Instruments, Foster City, CA). IPSCs were pharmacologically isolated and recorded at the reversal potential for glutamatergic events (_V_hold, +10 mV). Synaptic responses were evoked with sharpened tungsten bipolar stimulating electrodes (2 μm diameter; World Precision Instruments, Sarasota, FL) placed in the BLA, 50 μm from the recording electrode. Minimal stimulation was applied at 0.1 Hz, using a photoelectric stimulus isolation unit with a constant current output (PSIU6; Grass Instruments, West Warwick, RI).
Stimulation of a single presynaptic axon was confirmed by the intensity-threshold test (Raastad et al., 1992; Isaac et al., 1996;Bolshakov et al., 1997). The mean IPSC amplitude showed a steep all-or-none threshold as a function of stimulating current intensity. Increasing current intensity by 40–60% above the threshold for evoking an IPSC had no effect on the IPSC amplitude, indicating the stimulation of a single presynaptic axon. Access resistance (5–24 MΩ) was regularly monitored during recordings, and cells were rejected if it changed by >15% during the experiment. The signals were filtered at 2 kHz, digitized (Digidata 1322A; Axon Instruments), and stored on a computer using pClamp8 software (Axon Instruments). The peak amplitude, 10–90% rise time, and decay time constant of IPSCs were analyzed off-line using pClamp8 software and the Mini Analysis Program (Synaptosoft, Inc., Leonia, NJ). The visual method was used to identify failures and to calculate failure rates (Stevens and Wang, 1994). The percentage of failures was calculated from at least 30 stimulus pulses before, during, and after agonist application. mIPSCs were analyzed off-line using the Mini Analysis Program and detected by manually setting the mIPSC threshold (∼1.5 times the baseline noise amplitude) after visual inspection. In some experiments, cadmium was used to block voltage-gated calcium channels. Complete blockade of eIPSCs in the presence of 100 μm cadmium was used as a positive control for the efficacy of cadmium to block presynaptic calcium channels. In another series of experiments, postsynaptic currents were elicited by AMPA application. The fast application of AMPA was performed with a picospritzer (General Valve, Fairfield, NJ) at 3 min intervals. The pressure varied from 60 to 80 psi.
All data are presented as means ± SEM. The results were tested for statistical significance using the Student's paired _t_test.
Drugs. The following drugs were used:d-APV (Tocris Cookson, Ballwin, MO); an NMDA receptor antagonist, (+)-(2_S_)-5,5-dimethyl-2-morpholineacetic acid (SCH50911; Tocris Cookson); a GABAB receptor antagonist, bicuculline methiodide (Sigma, St. Louis, MO); a GABAA receptor antagonist, ATPA (Sigma); a GluR5 agonist (Clarke et al., 1997); and TTX (Sigma), a sodium channel blocker. The active isomer of GYKI 53655, an AMPA receptor antagonist, and the GluR5 antagonist (3_SR_,4a_RS_,6_RS_,8a_RS_)-6-[2-(iH-tetrazol-5-yl)ethyl]-1,2,3,4,4a,5,6,7,8,8a-decahydroiso-quinoline-3-carboxylic acid (LY293558; Bleakman et al., 1996) were gifts from Lilly Research Laboratories (Eli Lilly and Co., Indianapolis, IN).
Morphology. During whole-cell recordings, neurons were filled passively with 0.4% Lucifer yellow (Molecular Probes, Eugene, OR) for post hoc morphological identification. Slices were fixed for 1 hr at 4°C in PBS, pH 7.4, containing 4% paraformaldehyde; they were subsequently mounted and coverslipped in 10% PBS in glycerine. The fluorescence image of the dye-filled neurons was captured by a Leica (Nussloch, Germany) DM RXA fluorescence microscope equipped with a SPOT2 digital camera and a laser scanning confocal microscope (MRC-600; Bio-Rad, Hercules, CA).
_Protein solubilization and immunoblotting of GluR5 protein.For protein solubilization, the tissue was sonicated and subsequently solubilized in lysis buffer (10 mm Tris-HCl, pH 7.6, 5 mm EDTA, 50 mm NaCl, 30 mm sodium pyrophosphate, 50 mm sodium fluoride, 1 mmsodium orthovanadate, 1% Triton X-100, 1 mmphenylmethylsulfonyl fluoride, 5 μg/ml aprotinin, 1 μg/ml pepstatin A, and 2 μg/ml leupeptin). Cell lysates were rotated end-over-end at 4°C for 60 min, and insoluble material was pelleted at 12,000 ×_g for 30 min at 4°C. The protein concentrations of clarified tissue lysates were determined by the simplified Bradford method (Bio-Rad). The proteins were analyzed by SDS-PAGE and transferred to polyvinylidene difluoride membranes (Millipore, Bedford, MA) using a transfer unit (Multiphor Novablot; Amersham Biosciences, Clayton, NC). After transfer, blots were incubated in the blocking buffer (0.02 m Tris-HCl, pH 7.6, 0.137 m NaCl, 1% bovine serum albumin) before blotting with the first antibody. Anti-GluR5 antibody (1:1000) was purchased from Upstate Biotechnology (Lake Placid, NY). Anti-actin antibody (1:200) was purchased from Santa Cruz Biotechnology (Santa Cruz, CA). The first antibodies were incubated overnight at 4°C. The blots were washed in washing buffer (50 mmTris-HCl, pH 7.6, 200 mm NaCl, and 0.25% Tween 20) and then incubated with horseradish peroxidase-conjugated secondary antibodies (Transduction Laboratories, Lexington, KY). Enhanced chemiluminescence substrate (Amersham Biosciences, Piscataway, NJ) was used according to the manufacturer's instructions for antibody detection and exposed to x-ray film.
Results
Activation of GluR5 kainate receptors bidirectionally modulates the failure rate of GABAergic synaptic transmission
To determine whether GluR5 kainate receptors modulate action potential-dependent GABA release from BLA interneurons, we investigated the effects of ATPA (0.3–10 μm), a selective agonist for GluR5 kainate receptors (Clarke et al., 1997), on the probability of failure of GABAergic synaptic transmission evoked by electrical activation of a presynaptic axon. eIPSCs were recorded from BLA pyramidal cells at a holding potential of +10 mV, and in the presence of d-APV (50 μm), SCH50911 (20 μm), and GYKI 53655 (50 μm). As shown previously in other brain areas (Wilding and Huettner, 1995; Rammes et al., 1996), 50 μm GYKI 53655 was sufficient to completely block AMPA receptor-mediated currents in the BLA (Fig.1a), and therefore the recorded eIPSCs were not contaminated by AMPA currents. In addition, the eIPSCs were blocked by 10 μm bicuculline (Fig. 1b), which confirms that they were mediated by GABAA receptors. Application of ATPA at a 300 nm concentration decreased the number of failures of eIPSCs from 46.2 ± 8.1 to 12.4 ± 6.4% (n = 9; p < 0.01) (Fig.2_a_). At 1 μm, ATPA caused a transient reduction in the number of failures, followed by a long-lasting increase in the failure rate, from 43.4 ± 7.9 to 65.5 ± 9.1% (_n_ = 10; _p_ < 0.05) (Fig. 2_b_). At a concentration of 10 μm, ATPA caused a marked increase in the rate of failures of eIPSCs from 49.2 ± 9.8 to 92.0 ± 5.1% (_n_ = 8; _p_ < 0.01) (Fig.2_c_), which in most cells was apparent within 1 min of application of the agonist. Perfusion of the slices with ATPA-free ACSF completely reversed the effects of the agonist (Fig. 2). In addition, coapplication of ATPA (0.3–10 μm) with the selective GluR5 kainate receptor antagonist LY293558 (30 μm) had no significant effect on the number of failures of eIPSCs (59.8 ± 9.3% in 300 nm ATPA; 62.7 ± 5.9% in 1 μm ATPA; 67.1 ± 8.2% in 10 μm ATPA; _n_ = 9;_p_ > 0.1) compared with control (62.8 ± 6.2%) (Fig. 3a).
Fig. 1.

Postsynaptic currents evoked in the presence of 50 μm GYKI 53655, 50 μmd-APV, and 20 μm SCH59011 are GABAA receptor-mediated IPSCs. a, GYKI 53655, at a concentration of 50 μm, completely blocks AMPA-evoked postsynaptic currents. Sample traces of postsynaptic currents evoked by puff application of 200 μm AMPA in the presence of 1 μm TTX, 50 μm bicuculline, 50 μmd-APV, and 20 μm SCH59011 (holding potential, −70 mV) are shown. These currents were reduced by 52.1 ± 5.2% (n = 8) in the presence of 1 μm GYKI 53655 and were reversibly eliminated by 50 μm GYKI 53655. The horizontal bar above the current traces denotes the duration of the puff. The graph shows pooled data from eight neurons. **p < 0.01. b, Postsynaptic currents recorded in the presence of 50 μm GYKI 53655, 50 μmd-APV, and 20 μm SCH59011 were blocked by 10 μm bicuculline. Superimposed traces are evoked currents recorded from a BLA pyramidal neuron (holding potential, +10 mV). The graph shows pooled data (means ± SEM) from nine neurons. **p < 0.01.
Fig. 2.
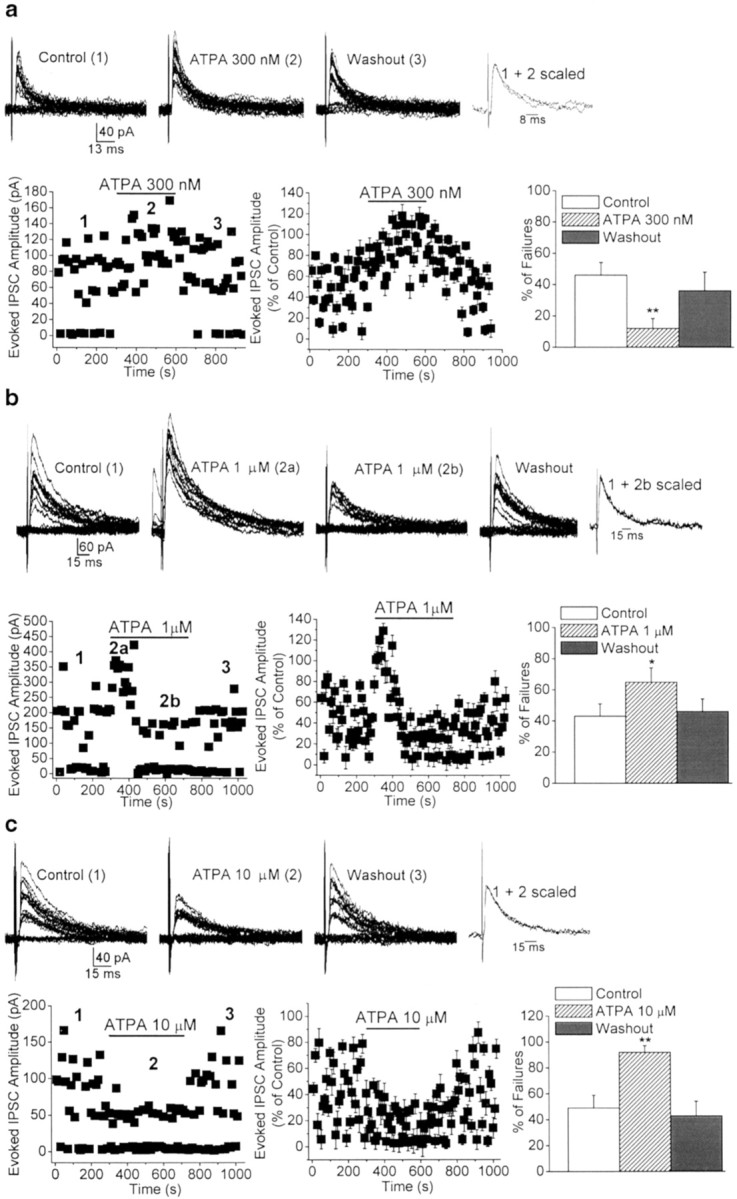
Dose-dependent, bidirectional modulation of the rate of failures of evoked GABAergic synaptic transmission by the GluR5 agonist ATPA. a–c, Superimposed traces are eIPSCs recorded from three different BLA pyramidal neurons before, during, and after bath application of different concentrations of ATPA. The scaled superimposed traces show that the effects of ATPA were not accompanied by changes in the kinetics of the eIPSCs. The plots show the time course of the effects of ATPA on the amplitude and number of failures of eIPSCs. Bar graphs are pooled data (means ± SEM) of the percentage of failures before, during, and after the application of ATPA. eIPSCs were recorded in the presence of GYKI 53655 (50 μm), d-APV (50 μm), and SCH50911 (20 μm), at a holding potential of +10 mV.a, At 300 nm, ATPA significantly decreased the percentage of failures of eIPSCs (n = 9; **p < 0.01). b, At 1 μm, ATPA caused a transient reduction (2a) followed by a long-lasting increase (2b) in the number of failures (n = 10; *p < 0.05). c, At 10 μm, ATPA produced a marked increase in the percentage of failures of eIPSCs (n = 8; **p< 0.01). Perfusion of the slices with ATPA-free ACSF completely reversed the effects of the agonist.
Fig. 3.
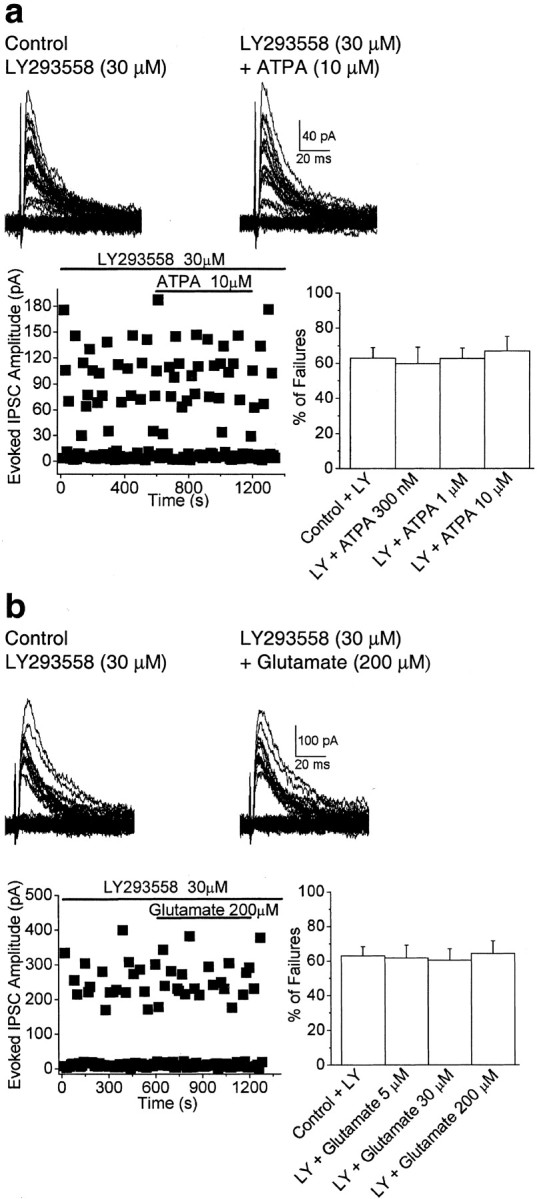
The effects of ATPA and glutamate on the rate of failures of GABAergic synaptic transmission are blocked by LY293558 (30 μm). a, b, Superimposed traces are eIPSCs recorded from pyramidal neurons in the presence of LY293558. Application of ATPA or glutamate had no effect on the failure rate of the eIPSCs. The plots show the amplitude of the eIPSCs versus time, before and during application of ATPA (a) or glutamate (b). Bar graphs are pooled data (means ± SEM) of the percentage of failures in the presence or absence of the GluR5 agonists. eIPSCs were recorded in the presence of GYKI 53655 (50 μm), d-APV (50 μm), and SCH50911 (20 μm), at a holding potential of +10 mV.
To investigate whether the effects of ATPA could be mimicked by the endogenous agonist, we tested whether glutamate was capable of inducing similar changes on the rate of failures of evoked inhibitory synaptic transmission. In the presence of GYKI 53655 (50 μm), d-APV (50 μm), SCH50911 (20 μm), and 7-(hydroxyimino)cyclopropa[b]chromen-1a-carboxylate ethyl ester (CPCCOEt; 30 μm) to block AMPA, NMDA, GABAB, and group I metabotropic GluR receptors, respectively, glutamate (5–200 μm) also produced a dose-dependent, bidirectional effect on evoked inhibitory synaptic transmission. A reduction in the number of failures of eIPSCs was observed at 5 μm glutamate, from a 37.0 ± 10.4 to a 9.1 ± 7.2% failure rate (n = 6;p < 0.01) (Fig.4_a_), and enhancement was seen at higher concentrations; thus, the failure rate was increased from 52.4 ± 8.8 to 74.3 ± 7.1% with 30 μm glutamate (_n_ = 6;_p_ < 0.01) (Fig. 4_b_) and from 42.2 ± 10.3 to 96.9 ± 3.9% with 200 μmglutamate (_n_ = 5; _p_ < 0.01) (Fig.4_c_). LY293558 completely prevented the effects of glutamate (5–200 μm). In the presence of LY293558 (30 μm), the failure rate was 61.9 ± 7.4% in 5 μm glutamate, 60.5 ± 6.7% in 30 μm glutamate, and 64.5 ± 7.3% in 200 μm glutamate (_n_ = 6;_p_ > 0.1) compared with control levels (63.1 ± 5.2%) (Fig. 3b).
Fig. 4.
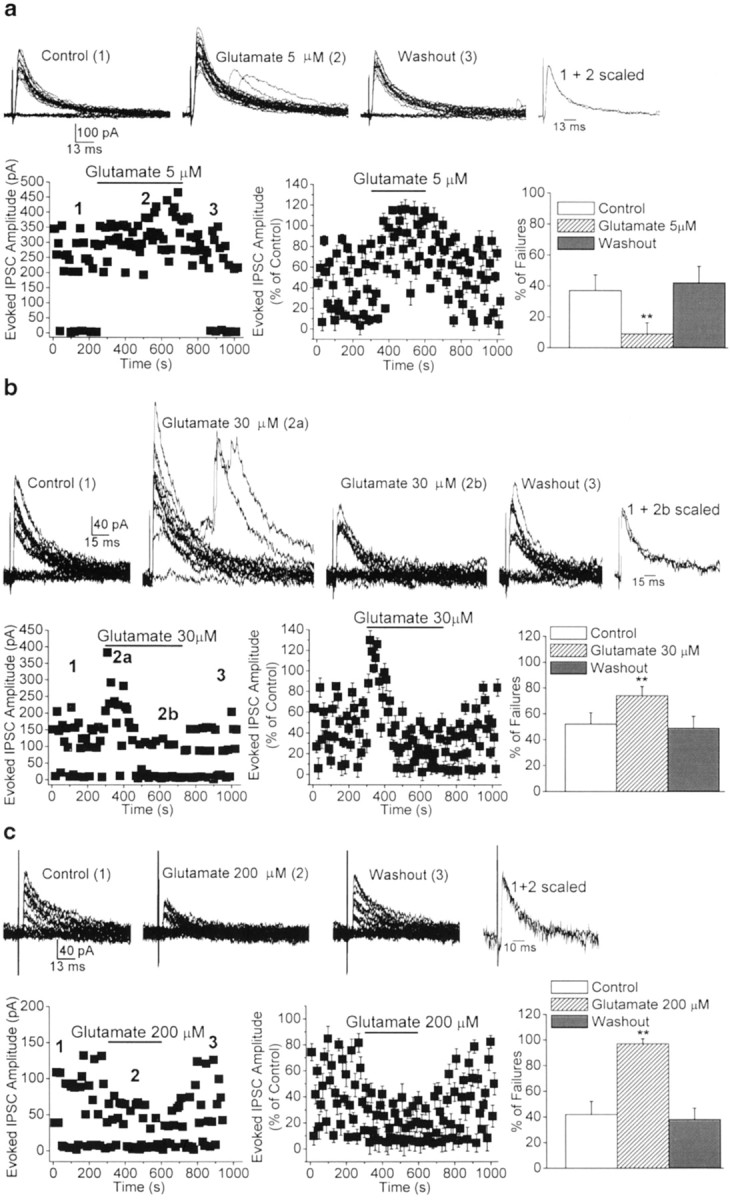
Dose-dependent, bidirectional modulation of the rate of failures of evoked GABAergic synaptic transmission by glutamate, via activation of GluR5 kainate receptors.a–c, Superimposed traces are eIPSCs recorded from three different BLA pyramidal neurons before, during, and after the application of glutamate. The scaled superimposed traces show that the effects of glutamate were not accompanied by changes in the kinetics of the eIPSCs. The plots show the time course of the effects of glutamate on the amplitude and number of failures of eIPSCs. Bar graphs are pooled data (means ± SEM) of the percentage of failures before, during, and after the application of glutamate. eIPSCs were recorded in the presence of GYKI 53655 (50 μm), d-APV (50 μm), SCH50911 (20 μm), and CPCCOEt (30 μm), at a holding potential of +10 mV. a, At 5 μm, glutamate decreased the percentage of failures of eIPSCs (n = 6; **p < 0.01).b, At 30 μm, glutamate caused a transient reduction (2a) followed by a long-lasting increase (2b) in the number of failures (n = 6; **p < 0.01). c, At 200 μm, glutamate caused a marked increase in the percentage of failures of eIPSCs (n = 5; **p < 0.01). Perfusion of the slices with glutamate-free ACSF completely reversed the effects of the agonist.
Excitation of BLA interneurons via activation of GluR5 kainate receptors
The effects of ATPA on the failure rate of GABAergic transmission could be explained by a presynaptic action of the GluR5 agonist at GABAergic terminals. However, previous work in the hippocampus has shown that kainate depolarizes GABAergic interneurons via somatodendritic (Cossart et al., 1998; Frerking et al., 1998) and axonal (Semyanov and Kullmann, 2001) kainate receptors. If similar effects are induced on BLA interneurons by GluR5 kainate receptor activation, these effects could be responsible at least for the observed reduction in the number of failures of GABAergic transmission. Indeed, when AMPA, NMDA, GABAA, and GABAB receptors were blocked by GYKI 53655 (50 μm), d-APV (50 μm), bicuculline (10 μm), and SCH50911 (20 μm), respectively, EPSCs evoked by electrical stimulation of the external capsule (3 shocks delivered at 100 Hz, every 10 sec) were recorded from BLA interneurons (Fig.5a). These evoked EPSCs were completely blocked by bath application of LY293558 (30 μm) (Fig. 5a). Thus, GluR5 kainate receptors are present on the somatodendritic regions of BLA interneurons and mediate a component of the evoked EPSCs. Furthermore, application of ATPA (0.3–10 μm) in the presence of GYKI 53655 (50 μm),d-APV (50 μm), and SCH50911 (20 μm) increased the frequency of action potential-dependent spontaneous IPSCs (sIPSCs) recorded in the soma of BLA pyramidal neurons (to 244.5 ± 61.3% by 300 nm ATPA, 572.4 ± 49.1% by 1 μm ATPA, and 908.2 ± 138.6% by 10 μm ATPA; n = 9;p < 0.01) (Fig. 5_b_). The effect persisted throughout the application of ATPA, was fully reversed after washout of the agonist, and was blocked by coapplication of LY293558 (30 μm) (Fig. 5_b_). The ATPA-induced increase in sIPSC frequency could be mimicked by glutamate. The application of glutamate (5–200 μm) in the presence of GYKI 53655 (50 μm),d-APV (50 μm), SCH50911 (20 μm), and CPCCOEt (30 μm) also increased the frequency of IPSCs recorded in the soma of BLA pyramidal neurons (to 303.8 ± 101.7% by 5 μm glutamate, 588.1 ± 112.3% by 30 μm glutamate, and 867.8 ± 144.1% by 200 μm glutamate;_n_ = 6; _p_ < 0.01). The glutamate-induced increase in sIPSC frequency was completely blocked by LY293558 (30 μm). Bath application of LY293558 (30 μm) alone did not affect the frequency of sIPSCs (5.8 ± 1.2 and 6.2 ± 1.4 Hz in the absence and in the presence of LY293558, respectively;_n_ = 8; _p_ > 0.1), suggesting that GluR5 kainate receptors on somatodendritic and/or axonal regions of BLA interneurons are not tonically activated by endogenous glutamate.
Fig. 5.

Excitation of BLA interneurons via GluR5 kainate receptors. a, Kainate receptors mediate a component of the synaptic responses of BLA interneurons. 1, EPSCs (V_hold, −60 mV) recorded from a BLA interneuron in the presence of GYKI 53655 (50 μm),d-APV (50 μm), bicuculline (10 μm), and SCH50911 (20 μm). Electrical stimulation was applied to the external capsule (3 shocks delivered at 100 Hz, every 10 sec). 2, Pooled data (means ± SEM) illustrating the blockade of evoked EPSCs, recorded as in_1, by bath application of 30 μm LY293558 (n = 8; **p < 0.01). A photomicrograph of the interneuron recorded in a1 is shown in 3. Scale bar, 50 μm. b, Activation of GluR5 kainate receptors increases the spontaneous activity of BLA interneurons. 1, Effects of different concentrations of ATPA on sIPSCs recorded from the soma of three different BLA pyramidal neurons (_V_hold, +10 mV). A photomicrograph of one of these neurons is shown in 3. Scale bar, 50 μm.2, Pooled data (means ± SEM) illustrating a dose-dependent increase in the frequency of sIPSCs induced by 300 nm (n = 6), 1 μm(n = 6), and 10 μm(n = 9) ATPA (**p < 0.01). LY293558 (30 μm), when applied alone, had no effect on the frequency of sIPSCs but prevented the ATPA-induced effect when coapplied with ATPA.
Activation of GluR5 kainate receptors bidirectionally modulates the frequency of mIPSCs
Because ATPA and glutamate can depolarize BLA interneurons and induce spontaneous firing (Fig. 5), the reduction in the rate of failures of eIPSCs by low concentrations of ATPA (300 nm) or glutamate (5 μm) could be attributable to the activation of GluR5 kainate receptors located on somatodendritic and/or axonal regions of the interneurons, rather than to direct effects on GABAergic terminals. Such depolarization of interneurons by ATPA could produce either more glutamate release from the stimulated axon or the recruitment of an additional axon (Semyanov and Kullmann, 2001). However, the activation of GluR5 kainate receptors by higher concentrations of ATPA (1–10 μm) or glutamate (30–200 μm), which also caused the depolarization of BLA interneurons and therefore might be expected to reduce the number of failures of eIPSCs, induced a marked increase in the rate of failures (Figs. 2b,c, 3b,c, respectively). In other CNS areas, it has been proposed that the depression of GABAergic transmission by kainate is attributable to the extracellular accumulation of spontaneously released GABA, which activates presynaptic GABAB autoreceptors and/or desensitizes postsynaptic GABAA receptors (Frerking et al., 1999; Frerking and Nicoll, 2000; Kerchner et al., 2001). However, in our studies the blockade of GABAB receptors by SCH50911 did not prevent the inhibition of evoked GABA release by high concentrations of ATPA or glutamate. In addition, there were no signs of significant desensitization of GABAA receptors during application of either ATPA or glutamate, as determined by the kinetics of the currents (rise time and decay time constant of eIPSCs) (Figs. 2,3). Accumulated extracellular GABA may also decrease pyramidal-cell input resistance, increasing the shunting of eIPSCs during electrotonic propagation from the synapse to the pyramidal cell soma, as suggested in the hippocampus (Frerking et al., 1999). In the present study, input resistance was measured from the responses to ±10 pA, 200 msec pulses applied at the resting membrane potential of pyramidal cells. Before the application of ATPA, the mean input resistance was 340 ± 18 MΩ (n = 7). ATPA, at 10 μm, caused a reduction in the input resistance of BLA pyramidal cells (to 44 ± 9% of control; n = 5; p < 0.01), but no clear correlation between the changes in the passive membrane properties and a reduction in eIPSC amplitude was observed. More importantly, 1 μm ATPA, which significantly enhanced the frequency of sIPSCs and increased the number of failures of eIPSCs, had no significant effect on the input resistance of BLA pyramidal cells (348 ± 24 MΩ in the presence of 1 μm ATPA; _n_ = 7;_p_ > 0.1). Therefore, it seems unlikely that a reduction in the input resistance of pyramidal neurons is responsible for the depression of eIPSCs by high concentrations of ATPA or glutamate.
To determine whether ATPA and glutamate acted directly on GABAergic terminals to modulate inhibitory transmission, we examined the effects of these agonists on mIPSCs, recorded in the presence of TTX (1 μm), GYKI 53655 (50 μm), d-APV (50 μm), and SCH50911 (20 μm). A change in the frequency of mIPSCs without a change in their amplitude is indicative of a change in the probability of quantal release at the presynaptic terminals. The mean frequency of mIPSCs recorded in the soma of BLA pyramidal neurons was 3.7 ± 0.8 Hz (_n_= 25). Bath application of bicuculline (10 μm) eliminated mIPSCs, confirming that these were GABAergic currents. ATPA, at 300 nm, caused a significant increase in the mean mIPSC frequency (138.9 ± 9.3% of the control values;n = 9; p < 0.01) (Fig.6_a_) that persisted throughout application of ATPA and was completely reversed after removal of the agonist. These effects of ATPA were not accompanied by any significant change in the amplitude, rise time, or decay time constant of mIPSCs (Fig. 6_a_). At 1 μm, ATPA reduced the mean frequency of mIPSCs to 74.8 ± 8.6% of control values (_n_ = 7; _p_ < 0.05) (Fig.3_b_), and at 10 μm, the mean frequency of mIPSCs was further reduced to 64.0 ± 9.4% (_n_ = 7; _p_ < 0.01) (Fig.6_c_). The reduction in the frequency of mIPSCs persisted throughout application of ATPA (1 or 10 μm) and was completely reversed after washout of the agonist. No significant changes in the amplitude, rise time, or decay time constant of mIPSCs were observed (Fig. 6_b,c_). Furthermore, bath application of LY293558 (30 μm) completely prevented the effects on the mean frequency of mIPSCs caused by ATPA (104.3 ± 7.6% of control in 300 nm ATPA, 96.2 ± 10.1% in 1 μm ATPA, and 93.5 ± 9.7% in 10 μm ATPA; _n_ = 4–7; _p_ > 0.1).
Fig. 6.

Dose-dependent, bidirectional modulation of the frequency of miniature GABAergic currents by the GluR5 agonist ATPA.a–c, Top, Samples of mIPSCs recorded from three different BLA pyramidal neurons before, during, and after the application of ATPA, at a 300 nm (a), 1 μm (b), or 10 μm(c) concentration. Recordings were obtained in the presence of TTX (1 μm), GYKI 53655 (50 μm), d-APV (50 μm), and SCH50911 (20 μm), at a holding potential of +10 mV.a–c, Bottom, The corresponding cumulative probability plots of interevent intervals and amplitudes of mIPSCs in control conditions and during the application of ATPA. Bar graphs show pooled data (means ± SEM) on the effects of ATPA on mIPSCs. At 300 nm (a), ATPA increased the frequency of mIPSCs (n = 9; **p< 0.01). At 1 μm (b), ATPA caused a small but significant reduction (n = 7; *p < 0.05), and at 10 μm(c), ATPA caused a marked reduction in the frequency of mIPSCs (n = 7; **p< 0.01). The peak amplitude, rise time, and decay time constant were not significantly affected by any of the three concentrations of ATPA. Perfusion of the slices with ATPA-free ACSF completely reversed the effects of the agonist.
Next, we tested whether the dose-dependent, bidirectional effects of ATPA on mIPSC frequency could be mimicked by the endogenous agonist glutamate. We found that in the presence of TTX (1 μm), GYKI 53655 (50 μm), d-APV (50 μm), SCH50911 (20 μm), and CPCCOEt (30 μm), glutamate (5 μm) caused a significant increase in the mean mIPSC frequency (162.9 ± 11.7% of control values; n = 3; p < 0.05). In contrast, at a concentration of 200 μm, glutamate reduced the mean frequency of mIPSCs to 61.7 ± 9.3% of control values (n = 3; p < 0.05).
The effects of ATPA or glutamate on the frequency of mIPSCs provide strong evidence for the presence of GluR5 kainate receptors on GABAergic terminals of BLA interneurons. However, it is conceivable that even in the presence of TTX, the activation of GluR5 kainate receptors located on somatodendritic and/or axonal compartments of GABAergic interneurons could induce sufficient passive depolarization of nerve terminals, activating voltage-sensitive calcium channels, calcium entry, and vesicle fusion. Thus, to test whether the enhanced rate of mIPSCs could be mediated by calcium influx through voltage-sensitive calcium channels, we recorded mIPSCs in the presence of cadmium (100 μm), GYKI 53655 (50 μm), d-APV (50 μm), and SCH50911 (20 μm). Application of ATPA (300 nm) caused a significant increase in the mean mIPSC frequency (147 ± 11.1%; n = 6; p< 0.05) (Fig. 7a) that was not significantly different from that in the absence of cadmium. Similarly, the presence of cadmium (100 μm) did not affect the reduction in the frequency of mIPSCs induced by 10 μm ATPA (55.8 ± 12.6%; _n_= 3; p < 0.05) (Fig. 7b). Thus, the effect of ATPA on the frequency of mIPSCs is mediated by GluR5 kainate receptors located on presynaptic GABAergic terminals and does not require Ca2+ influx through voltage-sensitive calcium channels.
Fig. 7.
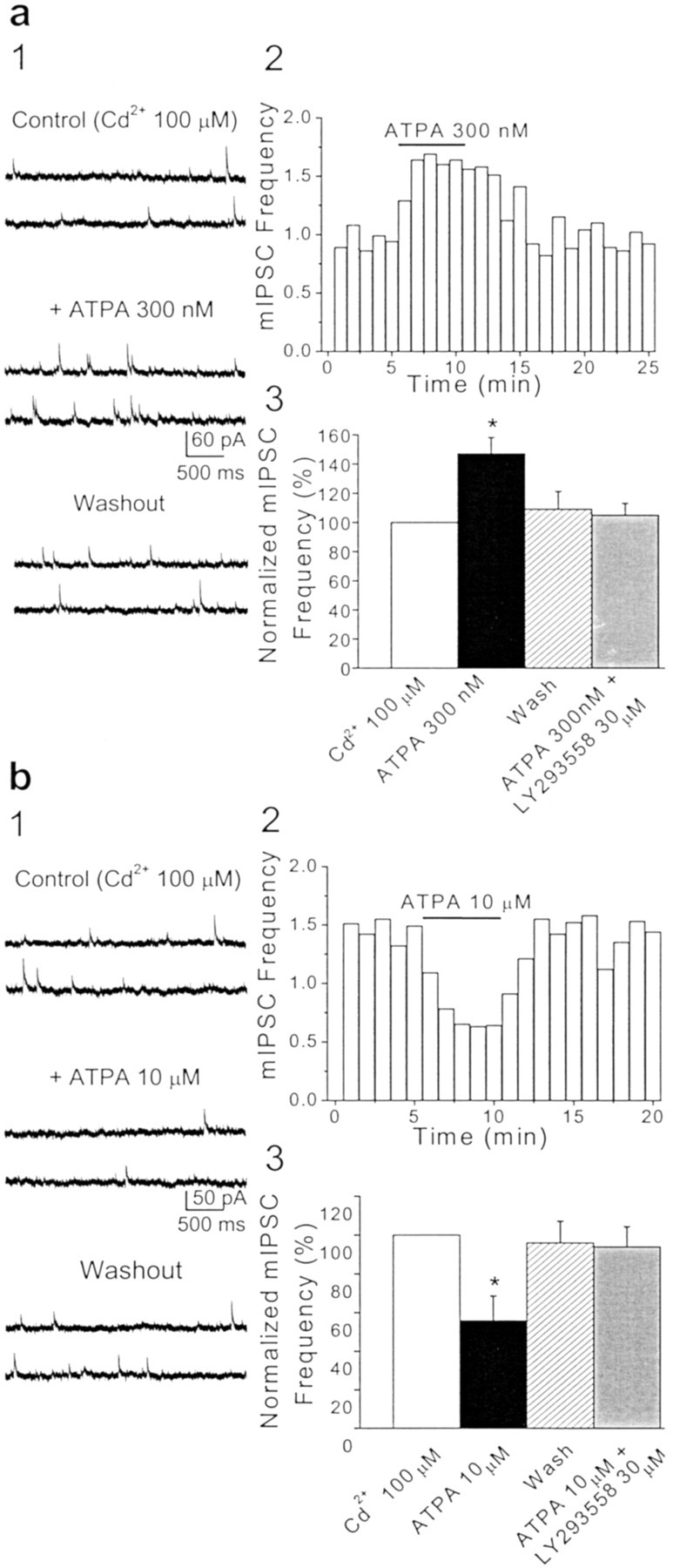
The effects of ATPA on the frequency of miniature GABAergic currents do not involve voltage-dependent calcium channels.a1, b1, Samples of mIPSCs recorded from two different BLA pyramidal neurons before, during, and after the application of 300 nm (a1) or 10 μm(b1) ATPA in the presence of Cd2+(100 μm), TTX (1 μm), GYKI 53655 (50 μm), d-APV (50 μm), and SCH50911 (20 μm), at a holding potential of +10 mV. The presence of Cd2+ in the medium did not prevent the increase (a1) or decrease (b1) in the frequency of mIPSCs by bath application of ATPA. a2, b2, Plots show the effects of 300 nm (a2) and 10 μm (b2) ATPA on the mean frequency of mIPSCs as a function of time (bin = 60 sec) (same cells as in_a1_ and b1, respectively). a3, b3, Pooled data (means ± SEM) on the effects of ATPA, applied in the presence of Cd2+, on the frequency of mIPSCs. a3, At 300 nm, ATPA increased the frequency of mIPSCs (n = 6; *p< 0.05). b3, At 10 μm, ATPA caused a marked reduction in the frequency of mIPSCs (n = 3; *p < 0.05). For each cell, the mIPSC frequency was normalized to the value of the mean mIPSC frequency before the application of ATPA. Coapplication of LY293558 (30 μm) prevented the effects of ATPA.
GluR5 kainate receptors are tonically activated by endogenous glutamate facilitating GABAergic synaptic transmission
To investigate whether presynaptic GluR5 kainate receptors of BLA interneurons are activated by endogenous glutamate, we examined the effects of bath application of LY293558 alone on the rate of failures of eIPSCs recorded from the soma of BLA pyramidal cells. LY293558 (30 μm) reversibly increased the percentage of failures of eIPSCs from 35.3 ± 11.2 to 67.0 ± 4.8% (n = 8; p < 0.01) (Fig.8). This indicates that basal levels of extracellular glutamate escaping from excitatory synapses tonically modulate the activity of nearby inhibitory synapses via presynaptic GluR5 kainate receptors. This result is in contrast to the lack of an effect of LY293558, when applied alone, on the frequency of sIPSCs. One possible explanation for this apparent discrepancy is that the frequency of sIPSCs is regulated to a greater extent by the level of activation of somatodendritic GluR5 kainate receptors, rather than by GluR5 kainate receptors located on the terminals of the interneurons. Hence, the observation that LY293558 increased the rate of failures of eIPSCs but did not affect the frequency of sIPSCs suggests either that extracellular glutamate reaches higher concentrations in the vicinity of presynaptic GluR5 kainate receptors than in the vicinity of somatodendritic GluR5 kainate receptors or that different subtypes of GluR5-containing kainate receptors are present in the somatodendritic versus the presynaptic sites, and the presynaptic GluR5 kainate receptors have a higher affinity for glutamate.
Fig. 8.

Endogenous glutamate tonically activates GluR5 kainate receptors, facilitating GABAergic synaptic transmission.a, Superimposed traces of eIPSCs recorded from a BLA pyramidal neuron before, during, and after the application of LY293558 (30 μm). Recordings were obtained in the presence of GYKI 53655 (50 μm), d-APV (50 μm), and SCH50911 (20 μm), at a holding potential of +10 mV. The scaled superimposed traces show that the effects of LY293558 (30 μm) were not accompanied by changes in the kinetics of the eIPSCs. b, The time course of the effects of LY293558 (30 μm) on the amplitude and number of failures of eIPSCs (same cell as in a). c, Pooled data (means ± SEM) illustrating a marked increase in the percentage of failures of eIPSCs induced by bath application of LY293558 (n = 8; **p < 0.01).
The GluR5 subunit is highly expressed in the basolateral amygdala
The agonist concentration-dependent, bidirectional modulation of GABA release via GluR5 receptors in the BLA is in contrast to findings in the hippocampus, where submicromolar concentrations of ATPA have no effect on evoked GABAergic transmission (Rodriguez-Moreno et al., 2000), and 1 or 10 μm ATPA induces only suppression (Clarke et al., 1997; Rodriguez-Moreno et al., 2000). In other regions, such as the dorsal horn, ATPA does not produce any significant effects on GABA release (Kerchner et al., 2001). Therefore, it appears that GluR5 kainate receptors may have different functions in different regions of the CNS; their function may depend on their location (cell type and subcellular compartment), subunit composition, and stoichiometry, as well as density. The density of the GluR5 subunit in the amygdala and particularly in the BLA, as revealed by _in situ_hybridization of GluR5 mRNA, is higher than in other CNS areas, including the hippocampus (Li et al., 2001) (Fig.9) and the dorsal horn (Tölle et al., 1993). However, the mRNA levels are suggestive but not necessarily indicative of the expression levels of the GluR5 protein itself. For this reason, we performed a Western blot analysis of GluR5 protein expression levels using a specific antibody that recognizes GluR5 isoforms (Upstate Biotechnology). We found that the amygdala has higher expression levels of the GluR5 subunit compared with other brain regions, including the hippocampus (Fig. 9). The greater expression of the GluR5 subunit in the amygdala may imply a more prominent role of GluR5 kainate receptors in this structure.
Fig. 9.
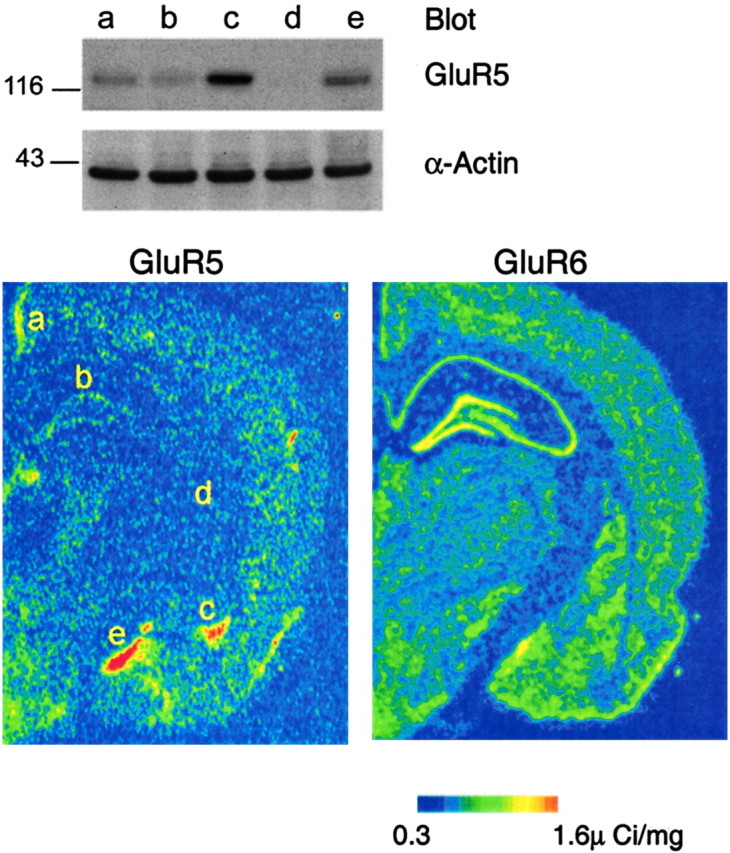
The basolateral amygdala has a high expression of the GluR5 protein. Western blot analysis of the GluR5 protein levels (top) in five brain regions:a, retrosplenial granular cortex; b, hippocampus; c, basolateral amygdala; d, medial globus pallidus; e, medial amygdala. These regions are indicated in the pseudocolor image of GluR5 mRNA in situ hybridization data from rat brain (from Li et al., 2001). An _in situ_hybridization pseudocolor image of GluR6 mRNA (from Li et al., 2001) is also shown for comparison. Although GluR6 mRNA signal is strongest in the hippocampus, GluR5 mRNA and GluR5 protein levels are highest in the amygdala. Parallel immunoblotting for α-actin was used to verify the equal loading of cell lysates. The molecular mass is indicated.
Discussion
Presynaptic GluR5 kainate receptors mediate both facilitation and suppression of GABAergic transmission
The main finding of this study is that in interneuron-to-pyramidal-cell synapses in the basolateral amygdala, low concentrations of GluR5 kainate receptor agonists facilitate, whereas high concentrations suppress, GABAergic transmission. In addition, ambient concentrations of extracellular glutamate tonically facilitate inhibitory transmission. Both facilitation and suppression of inhibitory transmission are induced by a direct effect of GluR5 agonists on GABAergic terminals, as indicated by the effects of ATPA on mIPSCs, and do not require the activation of voltage-gated calcium channels or presynaptic GABAB receptors.
The most plausible interpretation of the dose-dependent, bidirectional effects of GluR5 agonists on GABAergic transmission is that the terminals of GABAergic neurons in the BLA carry two subtypes of GluR5-containing kainate receptors, which have different affinities for their agonists and activate different mechanisms of action. Based on their affinity for [3H]kainate, kainate receptor subunits can be divided into low-affinity (GluR5, GluR6, and GluR7) and high-affinity (KA1 and KA2) subunits (Chittajallu et al., 1999). Our previous work has shown that the basolateral nucleus of the amygdala also expresses the GluR6 and KA2 subunit mRNAs in addition to GluR5 mRNA (Li et al., 2001). There is also evidence that the GluR5 subunit can form functional kainate receptors with GluR6 or KA2 subunits, and both GluR5/GluR6 and GluR5/KA2 kainate receptors are sensitive to ATPA (Paternain et al., 2000). Therefore, a GluR5/KA2 and a GluR5/GluR6 subunit combination could mediate the facilitation and inhibition, respectively, of GABAergic transmission in the BLA. Consistent with the view that a GluR5/GluR6 subunit combination may mediate the suppression of GABAergic transmission in the BLA,Mulle et al. (2000) found that kainate-induced suppression of eIPSCs in the hippocampus is mediated by heteromeric kainate receptors composed of both GluR5 and GluR6 subunits.
The intracellular mechanisms activated by these receptors remain to be determined. GluR5-containing kainate receptors can be permeable to Ca2+, particularly when they contain the unedited version of the GluR5 subunit, which confers higher Ca2+ permeability (Burnashev et al., 1996;Savidge et al., 1997; Chittajallu et al., 1999). In the amygdala, ∼30% of the total GluR5 mRNA is in the unedited form (Li et al., 2001). Therefore, a substantial portion of the GluR5-containing kainate receptors in the amygdala must have a relatively high permeability to Ca2+. Thus, some of the observed effects, more likely the facilitation of GABAergic transmission, could be mediated by Ca2+ influx through GluR5 kainate receptors on GABAergic terminals. The mechanisms responsible for the suppression of GABAergic transmission by GluR5 kainate receptor activation in the amygdala may involve a metabotropic cascade, as suggested previously for the kainate receptor-mediated inhibition of transmitter release in hippocampal synaptosomes (Cunha et al., 2000), as well as in interneuron-to-pyramidal-cell synapses (Rodriguez-Moreno and Lerma, 1998) or in glutamatergic synapses (Frerking et al., 2001) in the CA1 hippocampal area.
Physiological relevance of the bidirectional modulation of GABA release by glutamate
The results presented here suggest a significant role of glutamate diffusion in the regulation of excitability in the amygdala by modulating GABA release via presynaptic, GluR5-containing kainate receptors. Low concentrations of extracellular glutamate escaping from excitatory synapses during low or moderate activity of excitatory pathways within the amygdala neuronal network can be expected to facilitate GABAergic transmission. Our data demonstrate that even ambient concentrations of extracellular glutamate released during unstimulated, basal activity of excitatory synapses can reach and activate GluR5 kainate receptors present on GABAergic terminals, increasing the efficacy of GABAergic synapses. Considering the central role of the amygdala in fear conditioning and the consolidation of emotional memories (Aggleton, 2000; McGaugh, 2002), such facilitation of GABAergic transmission may serve to prevent or dampen the excitation of the amygdala during external or internal stimuli that have only moderate emotional significance. In contrast, in response to the intense emotional stimuli that produce strong excitation of the amygdala, the amount of glutamate released may reach sufficiently high extrasynaptic concentrations to activate the low-affinity GluR5 kainate receptors on GABAergic terminals, inhibiting evoked GABAergic transmission. Because spontaneous GABA release may be enhanced (Fig.5b) at the same time that evoked GABA release is suppressed, depending on the accessibility of somatodendritic versus presynaptic GluR5 receptors to extrasynaptic glutamate, the question arises as to what would be the net effect of high concentrations of extrasynaptic glutamate on GluR5-mediated changes in GABAergic activity and, therefore, on the overall excitability of the neuronal circuits of the amygdala. High concentrations of ATPA (>5 μm) produce a dramatic increase in the excitability of the amygdala, as revealed in both intracellular (Li et al., 2001) and field potential recordings (V. Aroniadou-Anderjaska, unpublished observations). This suggests that the reduction of evoked GABAergic transmission when GluR5 receptors are exposed to high concentrations of extracellular glutamate overrides any increases in spontaneous GABA release. Such an effect could further enhance overactivity in the amygdala during intense emotional stimuli, and perhaps facilitate the “registration” of the memory trace representing the emotional event. In this respect, the GluR5-mediated disinhibitory effect of glutamate may play an important role in synaptic plasticity and memory formation in the amygdala, as well as in the development of certain stress-induced affective disorders such as post-traumatic stress syndrome. Finally, the GluR5 kainate receptor-mediated disinhibitory action of glutamate described here sheds light on the mechanisms of the epileptogenic and neurotoxic actions of kainate in the amygdala.
Footnotes
This work was supported by the Stanley Foundation (V.A.A.), by DAMD Grant 17-00-1-0110, and by Uniformed Services University of the Health Sciences Grant RO88DC (H.L.). We thank Eleanore Gamble for expert assistance.
Correspondence should be addressed to Dr. He Li, Department of Psychiatry, Uniformed Services University of the Health Sciences, 4301 Jones Bridge Road, Bethesda, MD 20814. E-mail: hli@usuhs.mil.
References
- 1.Aggleton JP. The amygdala: a functional analysis. Oxford UP; Oxford: 2000. [Google Scholar]
- 2.Ali AB, Rossier J, Staiger JF, Audinat E. Kainate receptors regulate unitary IPSCs elicited in pyramidal cells by fast-spiking interneurons in the neocortex. J Neurosci. 2001;21:2992–2999. doi: 10.1523/JNEUROSCI.21-09-02992.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bleakman R, Schoepp DD, Ballyk B, Bufton H, Sharpe EF, Thomas K, Ornstein PL, Kamboj RK. Pharmacological discrimination of GluR5 and GluR6 kainate receptor subtypes by (3S,4aR,6R,8aR)-6-[2-(1(2)H-tetrazole-5-yl)ethyl]decahydroisoquinoline-3 carboxylic-acid. Mol Pharmacol. 1996;49:581–585. [PubMed] [Google Scholar]
- 4.Bolshakov VY, Golan H, Kandel ER, Siegelbaum SA. Recruitment of new sites of synaptic transmission during the cAMP-dependent late phase of LTP at CA3-CA1 synapses in the hippocampus. Neuron. 1997;19:635–651. doi: 10.1016/s0896-6273(00)80377-3. [DOI] [PubMed] [Google Scholar]
- 5.Burnashev N, Villarroel A, Sakmann B. Dimensions and ion selectivity of recombinant AMPA and kainate receptor channels and their dependence on Q/R site residues. J Physiol (Lond) 1996;496:165–173. doi: 10.1113/jphysiol.1996.sp021674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Castillo PE, Malenka RC, Nicoll RA. Kainate receptors mediate a slow postsynaptic current in hippocampal CA3 neurons. Nature. 1997;388:182–186. doi: 10.1038/40645. [DOI] [PubMed] [Google Scholar]
- 7.Chittajallu R, Braithwaite SP, Clarke VR, Henley JM. Kainate receptors: subunits, synaptic localization, and function. Trends Pharmacol Sci. 1999;20:26–35. doi: 10.1016/s0165-6147(98)01286-3. [DOI] [PubMed] [Google Scholar]
- 8.Clarke VR, Ballyk BA, Hoo KH, Mandelzys A, Pellizzari A, Bath CP, Thomas J, Sharpe EF, Davies CH, Ornstein PL, Schoepp DD, Kamboj RK, Collingridge GL, Lodge D, Bleakman D. A hippocampal GluR5 kainate receptor regulating inhibitory synaptic transmission. Nature. 1997;389:599–603. doi: 10.1038/39315. [DOI] [PubMed] [Google Scholar]
- 9.Cossart R, Esclapez M, Hirsch JC, Bernard C, Ben Ari Y. GluR5 kainate receptor activation in interneurons increases tonic inhibition of pyramidal cells. Nat Neurosci. 1998;1:470–478. doi: 10.1038/2185. [DOI] [PubMed] [Google Scholar]
- 10.Cossart R, Tyzio R, Dinocourt C, Esclapez M, Hirsch JC, Ben Ari Y, Bernard C. Presynaptic kainate receptors that enhance the release of GABA on CA1 hippocampal interneurons. Neuron. 2001;29:497–508. doi: 10.1016/s0896-6273(01)00221-5. [DOI] [PubMed] [Google Scholar]
- 11.Cunha RA, Malva JO, Ribeiro JA. Pertussis toxin prevents presynaptic inhibition by kainate receptors of rat hippocampal [3H]GABA release. FEBS Lett. 2000;469:159–162. doi: 10.1016/s0014-5793(00)01272-2. [DOI] [PubMed] [Google Scholar]
- 12.Frerking M, Nicoll RA. Synaptic kainate receptors. Curr Opin Neurobiol. 2000;10:342–351. doi: 10.1016/s0959-4388(00)00094-5. [DOI] [PubMed] [Google Scholar]
- 13.Frerking M, Malenka RC, Nicoll RA. Synaptic activation of kainate receptors on hippocampal interneurons. Nat Neurosci. 1998;1:479–486. doi: 10.1038/2194. [DOI] [PubMed] [Google Scholar]
- 14.Frerking M, Petersen CC, Nicoll RA. Mechanisms underlying kainate receptor-mediated disinhibition in the hippocampus. Proc Natl Acad Sci USA. 1999;96:12917–12922. doi: 10.1073/pnas.96.22.12917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Frerking M, Schmitz D, Zhou Q, Johansen J, Nicoll RA. Kainate receptors depress excitatory synaptic transmission at CA3→CA1 synapses in the hippocampus via a direct presynaptic action. J Neurosci. 2001;21:2958–2966. doi: 10.1523/JNEUROSCI.21-09-02958.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Isaac JTR, Hjelmstad GO, Nicoll RA, Malenka RC. Long-term potentiation at single fiber inputs to hippocampal CA1 pyramidal cells. Proc Natl Acad Sci USA. 1996;93:8710–8715. doi: 10.1073/pnas.93.16.8710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Jiang L, Xu J, Nedergaard M, Kang J. A kainate receptor increases the efficacy of GABAergic synapses. Neuron. 2001;30:503–513. doi: 10.1016/s0896-6273(01)00298-7. [DOI] [PubMed] [Google Scholar]
- 18.Kerchner GA, Wang GD, Qiu CS, Huettner JE, Zhuo M. Direct presynaptic regulation of GABA/glycine release by kainate receptors in the dorsal horn: an ionotropic mechanism. Neuron. 2001;32:477–488. doi: 10.1016/s0896-6273(01)00479-2. [DOI] [PubMed] [Google Scholar]
- 19.Kidd FL, Isaac JT. Developmental and activity-dependent regulation of kainate receptors at thalamocortical synapses. Nature. 1999;400:569–573. doi: 10.1038/23040. [DOI] [PubMed] [Google Scholar]
- 20.Kullmann DM. Presynaptic kainate receptors in the hippocampus: slowly emerging from obscurity. Neuron. 2001;32:561–564. doi: 10.1016/s0896-6273(01)00507-4. [DOI] [PubMed] [Google Scholar]
- 21.Li H, Rogawski MA. GluR5 kainate receptor mediated synaptic transmission in rat basolateral amygdala in vitro. Neuropharmacology. 1998;37:1279–1286. doi: 10.1016/s0028-3908(98)00109-9. [DOI] [PubMed] [Google Scholar]
- 22.Li H, Weiss SR, Chuang DM, Post RM, Rogawski MA. Bidirectional synaptic plasticity in the rat basolateral amygdala: characterization of an activity-dependent switch sensitive to the presynaptic metabotropic glutamate receptor antagonist 2S-α-ethylglutamic acid. J Neurosci. 1998;18:1662–1670. doi: 10.1523/JNEUROSCI.18-05-01662.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Li H, Chen A, Xing G, Wei ML, Rogawski MA. Kainate receptor-mediated heterosynaptic facilitation in the amygdala. Nat Neurosci. 2001;4:612–620. doi: 10.1038/88432. [DOI] [PubMed] [Google Scholar]
- 24.Liu QS, Patrylo PR, Gao XB, van den Pol AN. Kainate acts at presynaptic receptors to increase GABA release from hypothalamic neurons. J Neurophysiol. 1999;82:1059–1062. doi: 10.1152/jn.1999.82.2.1059. [DOI] [PubMed] [Google Scholar]
- 25.Mulle C, Sailer A, Swanson GT, Brana C, O'Gorman S, Bettler B, Heinemann SF. Subunit composition of kainate receptors in hippocampal interneurons. Neuron. 2000;28:475–484. doi: 10.1016/s0896-6273(00)00126-4. [DOI] [PubMed] [Google Scholar]
- 26.Paternain AV, Herrera MT, Nieto MA, Lerma J. GluR5 and GluR6 kainate receptor subunits coexist in hippocampal neurons and coassemble to form functional receptors. J Neurosci. 2000;20:196–205. doi: 10.1523/JNEUROSCI.20-01-00196.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Raastad M, Storm JF, Andersen P. Putative single quantum and single fiber excitatory postsynaptic currents show similar amplitude range and variability in rat hippocampal slices. Eur J Neurosci. 1992;4:113–117. doi: 10.1111/j.1460-9568.1992.tb00114.x. [DOI] [PubMed] [Google Scholar]
- 28.Rammes G, Swandulla D, Collingridge GL, Hartmann S, Parsons CG. Interactions of 2,3-benzodiazepines and cyclothiazide at AMPA receptors: patch clamp recordings in cultured neurons and area CA1 in hippocampal slices. Br J Pharmacol. 1996;117:1209–1221. doi: 10.1111/j.1476-5381.1996.tb16718.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Rodriguez-Moreno A, Lerma J. Kainate receptor modulation of GABA release involves a metabotropic function. Neuron. 1998;20:1211–1218. doi: 10.1016/s0896-6273(00)80501-2. [DOI] [PubMed] [Google Scholar]
- 30.Rodriguez-Moreno A, Herreras O, Lerma J. Kainate receptors presynaptically downregulate GABAergic inhibition in the rat hippocampus. Neuron. 1997;19:893–901. doi: 10.1016/s0896-6273(00)80970-8. [DOI] [PubMed] [Google Scholar]
- 31.Rodriguez-Moreno A, Lopez-Garcia JC, Lerma J. Two populations of kainate receptors with separate signaling mechanisms in hippocampal interneurons. Proc Natl Acad Sci USA. 2000;97:1293–1298. doi: 10.1073/pnas.97.3.1293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Savidge JR, Bleakman D, Bristow DR. Identification of kainate receptor-mediated intracellular calcium increases in cultured rat cerebellar granule cells. J Neurochem. 1997;69:1763–1766. doi: 10.1046/j.1471-4159.1997.69041763.x. [DOI] [PubMed] [Google Scholar]
- 33.Schmitz D, Frerking M, Nicoll RA. Synaptic activation of presynaptic kainate receptors on hippocampal mossy fiber synapses. Neuron. 2000;27:327–338. doi: 10.1016/s0896-6273(00)00040-4. [DOI] [PubMed] [Google Scholar]
- 34.Semyanov A, Kullmann DM. Kainate receptor-dependent axonal depolarization and action potential initiation in interneurons. Nat Neurosci. 2001;4:718–723. doi: 10.1038/89506. [DOI] [PubMed] [Google Scholar]
- 35.Stevens CF, Wang Y. Changes in reliability of synaptic function as a mechanism for plasticity. Nature. 1994;371:704–707. doi: 10.1038/371704a0. [DOI] [PubMed] [Google Scholar]
- 36.Tölle TR, Berthele A, Zieglgansberger W, Seeburg PH, Wisden W. The differential expression of 16 NMDA and non-NMDA receptor subunits in the rat spinal cord and periaqueductal gray. J Neurosci. 1993;13:5009–5028. doi: 10.1523/JNEUROSCI.13-12-05009.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Vignes M, Collingridge GL. The synaptic activation of kainate receptors. Nature. 1997;388:179–182. doi: 10.1038/40639. [DOI] [PubMed] [Google Scholar]
- 38.Wilding TJ, Huettner JE. Differential antagonism of α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid-preferring and kainate-preferring receptors by 2,3-benzodiazepines. Mol Pharmacol. 1995;47:582–587. [PubMed] [Google Scholar]