Functional Hemichannels in Astrocytes: A Novel Mechanism of Glutamate Release (original) (raw)
Abstract
Little is known about the expression and possible functions of unopposed gap junction hemichannels in the brain. Emerging evidence suggests that gap junction hemichannels can act as stand-alone functional channels in astrocytes. With immunocytochemistry, dye uptake, and HPLC measurements, we show that astrocytes in vitro express functional hemichannels that can mediate robust efflux of glutamate and aspartate. Functional hemichannels were confirmed by passage of extracellular lucifer yellow (LY) into astrocytes in nominal divalent cation-free solution (DCFS) and the ability to block this passage with gap junction blocking agents. Glutamate/aspartate release (or LY loading) in DCFS was blocked by multivalent cations (Ca2+, Ba2+, Sr2+, Mg2+, and La3+) and by gap junction blocking agents (carbenoxolone, octanol, heptanol, flufenamic acid, and 18α-glycyrrhetinic acid) with affinities close to those reported for blockade of gap junction intercellular communication. Glutamate efflux via hemichannels was also accompanied by greatly reduced glutamate uptake. Glutamate release in DCFS, however, was not significantly mediated by reversal of the glutamate transporter: release did not saturate and was not blocked by glutamate transporter blockers. Control experiments in DCFS precluded glutamate release by volume-sensitive anion channels, P2X7 purinergic receptor pores, or general purinergic receptor activation. Blocking intracellular Ca2+ mobilization by BAPTA-AM or thapsigargin did not inhibit glutamate release in DCFS. Divalent cation removal also induced glutamate release from intact CNS white matter (acutely isolated optic nerve) that was blocked by carbenoxolone, suggesting the existence of functional hemichannels in situ. Our results indicated that astrocyte hemichannels could influence CNS levels of extracellular glutamate with implications for normal and pathological brain function.
Keywords: astrocyte, gap junction, hemichannel, glutamate, divalent cation, glutamate release, glutamate transport
Introduction
Glutamate is the principal excitatory neurotransmitter in the brain. Astrocytes play a pivotal role in maintaining low extracellular glutamate levels; this ensures a suitable environment for synaptic transmission and prevents neuronal death from glutamate overexcitation, i.e., excitotoxicity (Olney, 1969;Choi, 1988). Glutamate transport across plasma membranes is powered by transmembrane ionic gradients of Na+, K+, and H+and the membrane potential (Szatkowski et al., 1990; Zerangue and Kavanaugh, 1996). Under normal circumstances astrocytes take up glutamate against a steep concentration gradient and can convert it to glutamine for release to the extracellular space for neuronal uptake. Astrocytes, however, contain millimolar levels of glutamate (Ottersen, 1989; Levi and Patrizio, 1992; Ye et al., 2001) and also have the capacity to release this amino acid under certain conditions. Presently, three release mechanisms have been identified: (1) reverse operation of glutamate transporter (Nicholls and Attwell, 1990); (2) release through anion channels activated by swelling (Kimelberg et al., 1990, 1995); and (3) release mediated by stimulated (e.g., by ATP) increases in [Ca2+]i (Parpura et al., 1994; Bezzi et al., 1998; Parpura and Haydon, 2000). Astrocyte glutamate release may influence synaptic transmission and signal processing (Bezzi and Volterra, 2001) and may mediate neural injury.
Gap junctions are a special class of ion channels that mediate intercellular transfer of molecules and ions. In the CNS, these channels are abundantly expressed in astrocytes and “couple” these cells together to form a functional syncytium (Ransom, 1995;Rose and Ransom, 1997; Ceelen et al., 2001). Gap junctions are composed of two aligned connexin hexamers, called connexons or gap junction hemichannels, one in each of the opposed membranes. Until recently, little attention had been paid to the possibility that hemichannels might exist as stand-alone functional channels [but see Hofer and Dermietzel (1998); Contreras et al. (2002); Stout et al. (2002)]. In the retina, however, stand-alone hemichannels have been anatomically identified and appear to mediate current flow (DeVries and Schwartz, 1992; Kamermans et al., 2001).
In light of the functional significance of glutamate, it would be important to know whether glutamate passes through hemichannels and how this is regulated. We studied these questions using cultured and_in situ_ astrocytes. Similar to reports in the literature (Hofer and Dermietzel, 1998), we demonstrated that astrocytes in culture expressed connexin 43 (Cx43) in a distribution compatible with hemichannels. Functional hemichannels were confirmed by passage of extracellular lucifer yellow (LY) into astrocytes after removal of extracellular Ca2+ and Mg2+ and the ability to block this passage with gap junction blocking agents. Glutamate and other amino acids were released from astrocytes through open hemichannels. Open hemichannels also compromised glutamate uptake into astrocytes, presumably by degrading ionic gradients on which glutamate uptake depends. Our results indicated that hemichannels in astrocytes might participate in “setting” the concentration of glutamate in brain extracellular space. Astrocytic hemichannels could be a major source of glutamate during metabolic inhibition when such channels appear to open (John et al., 1999; Contreras et al., 2002).
Materials and Methods
Cell culture. Hippocampal astrocytes were cultured from neonatal rat pups (Charles River, Wilmington, MA) as described previously (Ye et al., 2001). Cultures reached confluence within 2 weeks, and >90% of cells stained positive for the astrocyte marker glial fibrillary acidic protein (GFAP) (Sigma, St. Louis, MO). Cultures were typically used for experiments after 2–4 weeks in culture, and they were essentially free of neurons, as judged by morphology and staining with β-III tubulin (Promega, Madison, WI).
Immunocytochemistry. Cells cultured on glass coverslips were stained for connexin 43 and GFAP, as described previously (Ye et al., 1999) with slight modification. Antibodies were used at the following concentrations: 1 μg/ml affinity-purified rabbit anti-connexin 43 (Zymed, South San Francisco, CA), and 1:2000 monoclonal Cy3-conjugated GFAP. After staining, coverslips were mounted on glass slides with Vectorshield anti-fade media (Vector, Burlingame, CA) and examined using a Bio-Rad Raddion 1000 confocal microscope (Bio-Rad, Hercules, CA); the images were processed further with Adobe Photoshop.
Amino acid release. The artificial CSF (ACSF) solution used in the experiments contained (in mm): 116 NaCl, 3.0 KCl, 1.25 NaH2PO4, 23 NaHCO3, 10 glucose, 2.0 MgSO4, and 2.0 CaCl2. The latter two components were deleted in nominal divalent cation-free solution (DCFS). These HCO3−buffered solutions were equilibrated with 5% CO2/95% O2. Residual [Ca2+] in DCFS was at low micromolar levels as tested by measuring proton release during application of the Ca2+ chelator EGTA using the Marks and Maxfield method (Marks and Maxfield, 1991). To avoid significant pH changes associated with Ca2+ binding to EGTA when switching from ACFS to DCFS + EGTA, a 30 sec period of perfusion with DCFS was interposed to eliminate most of the Ca2+. Most release experiments were performed in 24-well plates (500 μl per well), in which confluent hippocampal astrocytes were first rinsed twice with ACSF to wash out culture media. In experiments testing gap junction blockers, test compounds were first applied in regular ACSF for 10 min and then rinsed twice with the final solution (e.g., DCFS + gap junction blocker) and incubated for 2–20 min before collection of extracellular solutions for measurement of amino acid release. Collected samples were centrifuged at 16,000 × g for 3 min, and the supernatants were stored at −80°C for HPLC analysis.
Special precaution was necessary to prevent loss of volatile long-chain alcohols, such as heptanol, from solutions, thereby reducing their effective concentration. These alcohols were mixed with DCFS immediately before use. Because of low solubility in HCO3− buffered solution, La3+ experiments were performed in HEPES-buffered DCFS containing (in mm): 126 NaCl, 3.0 KCl, 1.25 NaH2PO4, 25 HEPES (acid), and 10 glucose, with pH adjusted to 7.40 using NaOH. At a given pH, glutamate release was similar in HEPES or bicarbonate-buffered DCFS.
To test the effects of hypotonic solution, 75 mm NaCl was omitted from the ACSF. To differentiate the effects of hypo-osmolarity from the effects of lowering [Na+]o on glutamate release, an additional control was performed by substituting 75 mm NaCl with 75 mm choline-Cl.
Dye uptake and quantification. Dye uptake experiments were performed using procedures parallel to those described above for amino acid release. Astrocytes were incubated with 1 mmLY for 10 min under test conditions and then rinsed five times with regular ACSF. For quantification of dye uptake, cells were lysed in distilled water, and the amount of LY was determined using a Fluorometer (Turner Designs, Sunnyvale, CA), with excitation filter of 420 ± 10 nm and emission >475 nm. Intracellular LY concentrations were obtained by dividing the total amount of intracellular LY by cell volume. Cell volume was calculated from protein content on the basis of the principle that 1 mg of cell protein occupies a volume of 10 μl.
Transport of glutamate and related compounds. Astrocytic capacity for glutamate uptake was determined by measuring the rate at which astrocytes depleted extracellular glutamate. Generally the experiments lasted 30 min, with depletion measured at several time points. Glutamate uptake rate was also determined by measuring cytoplasmic uptake (i.e., cytoplasmic content) of an inert glutamate-uptake substrate such asdl-threo-β-hydroxyaspartate (THA). To test the effect of intracellular glutamate content on glutamate release, astrocytes were typically preloaded for 20 min with various amounts of glutamate; under these conditions, the intracellular conversion of glutamate to glutamine was outpaced by glutamate uptake.
Measurement of amino acids and related compounds. Amino acids, THA, and (1_R_,3_S_)-1-aminocyclopentane-1,3-dicarboxylic acid (RS-ACPD) were pre-column-derivatized with_o_-phthalialdehyde (Sigma), separated, and measured as described previously (Ye et al., 2001) using HPLC. The drugs 4,4′-diisothiocyanatostilbene-2,2′-disulfonic acid (DIDS) and 4-acetamido-4′-isothiocyanatostilbene-2,2′-disulfonic acid (SITS), used to block anion channels, both autofluoresce. When these drugs were used, background fluorescence was subtracted in a manner that corresponded to the appropriate drug concentration. The amount of amino acid release was calculated from the volume of the solution and normalized to the protein content of the cultures. To measure protein contents and cytoplasmic amino acid contents, cultured cells were dissolved in 0.3 m NaOH and neutralized with HCl. Aliquots were used to measure protein concentration using the Bio-Rad protein assay kit or to measure amino acid concentration by HPLC analysis. In the latter case, proteins were first removed from the sample by a Microcon (Millipore) centrifugal filter device with a pore size of 3000 Da.
Statistics. All data were expressed as mean ± SE. Statistical differences were calculated by one-way ANOVA.
Results
Functional hemichannels in astrocytes
Astrocytes primarily express Cx43 (Dermietzel et al., 1991; Giaume et al., 1991). Immunocytochemistry showed that Cx43 was broadly distributed over cultured hippocampal astrocytes (Fig.1a,b). Strong plaque-like staining was found at sites where astrocyte membranes abutted, especially in confluent cultures, and probably represented Cx43 in gap junctions. However, a large percentage of staining was found in areas lacking cell–cell contacts. Connexins inserted into these unopposed membranes could be hemichannels; they could, of course, also represent reflexive junctions or connexins in intracellular vesicles (Wolff et al., 1998).
Fig. 1.
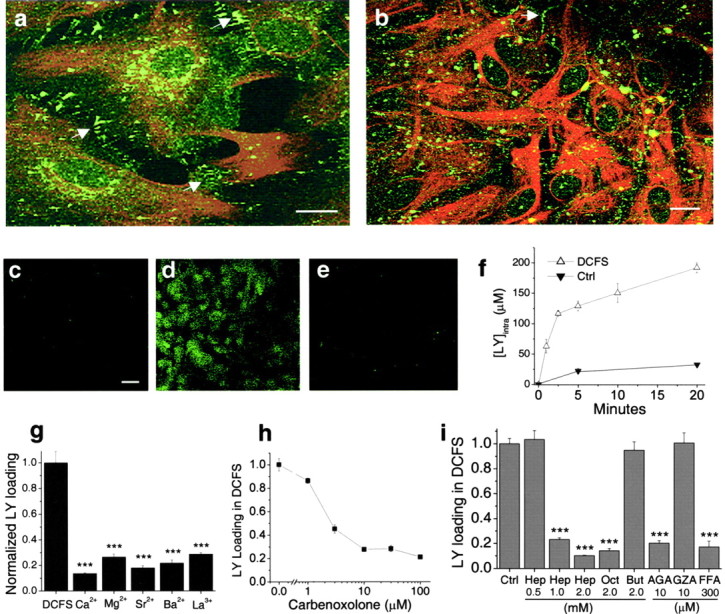
Expression and function of hemichannels in astrocytes. a, b, Cx43 (green) and GFAP (red) staining in astrocytes in low-density culture (a) and confluent culture (b). Cx43 was located both in cell–cell junctional areas (arrows) and in non-junctional areas.c_–_e, Lowering divalent cation concentrations increased lucifer yellow (LY) loading into astrocytes, which was blocked by classic gap junction blockers. c, Background LY uptake in control solution. d, LY uptake by astrocytes incubated in divalent cation-free solution (DCFS) for 10 min. e, CBX (100 μm) blocked LY loading in DCFS. f, Time course of LY (1 mm) loading in DCFS or in control solution (see Results). g, LY loading in DCFS was blocked by selectively restoring multivalent cations (2 mm each) in DCFS. h, Carbenoxolone, in a concentration-dependent manner, blocked LY loading in DCFS. i, Other gap junction blockers significantly reduced LY loading in DCFS. Oct, Octanol; Hep, heptanol; AGA, 18α-glycyrrhetinic acid; FFA, flufenamic acid. As controls, butanol (But) and glycyrrhizic acid (GZA), structurally similar to heptanol and AGA, respectively, had no effects. Scale bars: a,b, 10 μm; c_–_e, 20 μm.f_–_i, n = 6–12 for trials. ***p < 0.001 blockers versus DCFS alone (by one-way ANOVA with Dunnett's post hoc test).
Intercellular communication through normal Cx43 gap junctions can be blocked by pathologically elevated [Ca2+]i. It appears that hemichannels, as well as gap junctions (Spray et al., 1982), are gated by Ca2+ (Pfahnl and Dahl, 1999), but the Ca2+-sensitive portion of the hemichannel faces the extracellular and not the intracellular space. Ambient levels of Ca2+ are in the millimolar range, keeping hemichannels predominantly in the closed state. Lowering [Ca2+]o, therefore, opens hemichannels (Liu et al., 1995; Hofer and Dermietzel, 1998; Eskandari et al., 2002).
Gap junctions pass small dye molecules like LY, and this dye transfer serves as a marker for open gap junctions (Gutnick et al., 1981; Liu et al., 1995). As described by others (Hofer and Dermietzel, 1998), removal of extracellular Ca2+caused loading of LY into astrocytes (Fig. 1d). Astrocytes exposed to DCFS for periods up to 30 min showed no irreversible effects when returned to ACSF. To quantify the rate of dye-loading, we incubated astrocytes for variable periods with 1 mm LY in the presence or absence of divalent cations and measured intracellular LY after rinsing the cells to remove residual extracellular dye (Fig. 1f). Cellular volume was calculated from protein content. LY loading in DCFS showed a rapid initial phase lasting ∼5 min followed by slower uptake; minimal LY entered astrocytes in control solution containing normal concentrations of Ca2+ and Mg2+ (Fig. 1f).
The effects of multivalent cations and gap junction blockers on LY loading in DCFS were tested. Astrocytes were exposed to 1 mm LY for 10 min in DCFS. At 2 mm, all tested multivalent cations (i.e., Ca2+, Mg2+, Sr2+, Ba2+, La3+) significantly reduced LY uptake, with Ca2+being the most effective (Fig. 1g). La3+, a hemichannel blocker (John et al., 1999; Contreras et al., 2002), also reduced LY uptake. Carbenoxolone (CBX), a commonly used gap junctional blocker, reduced LY loading in DCFS (Fig. 1e) in a concentration-dependent manner (Fig.1h). Other gap junction blockers, including octanol, heptanol, 18α-glycyrrhetinic acid (AGA), and flufenamic acid (FFA) (Harks et al., 2001), also significantly reduced LY loading in DCFS (Fig. 1i). As controls, butanol and glycyrrhizic acid (GZA), which are structurally similar to the blockers heptanol and AGA, respectively, were applied and had no effect. Because the gap junction blockers presumably work via different mechanisms to block gap junctions (or hemichannels), their uniform effectiveness in blocking DCFS-induced dye loading minimizes the possibility that this effect occurred via some non-specific drug effect not related to hemichannel blockade.
Glutamate/aspartate efflux through open hemichannels
We determined whether glutamate was released from astrocytes through open hemichannels. Intracellular glutamate levels are typically >1 mm, whereas extracellular levels are in the submicromolar range, creating a steep gradient favoring glutamate efflux through passive pathways (Nicholls and Attwell, 1990). When astrocytes were exposed to DCFS, the extracellular glutamate level increased (Fig. 2a). Glutamate release was a direct function of [Ca2+]o (Fig.2b). The divalent cations that blocked DCFS-induced LY loading (Fig. 1g) (and see above) also reduced glutamate release in a concentration-dependent manner (Fig. 2c). The relative potency with which these divalent ions reduced glutamate release was as follows: Ca2+ > Sr2+ > Ba2+> Mg2+, which is the identical order of their effectiveness in blocking LY loading. La3+ also reduced glutamate release in a concentration-dependent manner (Fig. 2d).
Fig. 2.

Divalent cation reduction induced release of glutamate and aspartate from astrocytes. a, Time course of glutamate release in DCFS. b, Glutamate and aspartate release in DCFS were blocked in a concentration-dependent manner by Ca2+. c, Other divalent cations, including Mg2+, Sr2+, and Ba2+, mimicked the effect of Ca2+on glutamate release in DCFS but with lower affinity. d, La3+ also reduced glutamate and aspartate release in DCFS.
Other gap junction blocking agents reduced DCFS-induced glutamate release from astrocytes in a concentration-dependent manner (Fig.3). Glutamate release in DCFS was essentially restored to the levels seen in normal Ca2+- and Mg2+-containing solution by the following concentrations of gap junction blockers: octanol >1 mm, heptanol ≥1 mm, carbenoxolone >10 μm, and flufenamic acid >100 μm. In contrast, shorter chain alcohols known to be less effective gap junction blockers (Rozental et al., 2001) had no effects (butanol and cyclohexanol) or less effect (hexanol) on glutamate release (Fig. 3c). In addition, GZA, which is structurally similar to CBX and AGA but has no effect on gap junctions, had no effect on aspartate or glutamate release in DCFS (Fig. 3e). The efficacies and potencies of the gap junction blockers were similar in reducing LY loading and inhibiting glutamate release in DCFS, suggesting that both effects were mediated by a common mechanism, most likely the blockade of open hemichannels.
Fig. 3.

DCFS-induced glutamate release was blocked in a concentration-dependent manner by gap junction blockers but not by control compounds. a, Octanol. b, Heptanol. c, Shorter-chain alcohols (2 mm) had no effect or had less effect in reducing glutamate release compared with heptanol and octanol. But, Butanol; cHex, cyclohexanol; Hex, hexanol. d, Carbenoxolone. e, AGA but not GZA reduced glutamate release in DCFS. f, Flufenamic acid. n = 6–8 in all experiments; *p < 0.05, ***p < 0.001; Control (ctrl) or blockers versus DCFS (by one-way ANOVA).
Open hemichannels reduce glutamate transport
For theoretical reasons, it seemed possible that open hemichannels could retard glutamate transport in astrocytes (see Discussion). To evaluate this question we measured glutamate uptake by quantifying the decline of the exogenous extracellular glutamate concentration ([Glu]o; 50 μm). [Glu]o declined significantly slower in DCFS than in normal solution, suggesting reduced glutamate uptake when hemichannels are open. The rate of [Glu]odecline in DCFS astrocytes was substantially restored by the gap junction blocker CBX (50 μm). CBX had no effect on glutamate uptake in the control solution, in which hemichannels were presumably closed (data not shown). As anticipated, decline of [Glu]o in astrocyte cultures was associated with an increase in intracellular glutamate levels ([Glu]i) (Fig.4b). Astrocyte [Glu]i rose much faster in control solution compared with DCFS. Application of CBX augmented the rate of [Glu]i increase seen in DCFS, a result analogous to the effect of CBX on [Glu]odecline in DCFS.
Fig. 4.

Opening of hemichannels compromised glutamate uptake. a, Astrocytes in DCFS depleted exogenous glutamate (50 μm) significantly slower than astrocytes in control solution (DCFS plus 2 mm[Ca2+]o). CBX (100 μm) significantly increased glutamate uptake in DCFS.b, Challenged with 50 μm glutamate application, astrocytes in DCFS increased intracellular glutamate at a slower rate than astrocytes in control solution. This reduction was significantly rescued by CBX, suggesting that open hemichannels compromised glutamate uptake. c, Uptake of the exogenous glutamate transporter substrate THA was similarly reduced by lowering [Ca2+]o but rescued by CBX (100 μm). n = 6 for all experiments. For_c_, **p < 0.01; ***p < 0.001.
The net reduction of glutamate uptake into astrocytes in DCFS could be caused by (1) reduced uptake with unchanged release, (2) increased release with unchanged uptake, or (3) reduced uptake with increased release. Although the above results favor the latter two possibilities, they do not distinguish between them. To do this requires a more exclusive measurement for glutamate uptake; we thus tested uptake alone using the non-endogenous transporter substrate THA, which has no initial intracellular concentration. The rate of THA uptake was indeed reduced in DCFS (Fig. 4c). Therefore, the net reduction of glutamate accumulation in astrocytes caused by open hemichannels was caused by both reduced uptake and increased release.
Glutamate efflux in DCFS is essentially unrelated to reverse transport
Exposure to DCFS opened astrocytic hemichannels, causing glutamate release and compromising glutamate uptake. It remained conceivable that glutamate transport could be affected to the point of reversing operation, in which case it would actually contribute to glutamate efflux. Experiments were done to assess this possibility.
An important characteristic of transporter-mediated events is substrate saturation. The rate of glutamate uptake in cultured astrocytes neared saturation at [Glu]o >0.1 mm (Fig.5a). If the efflux of glutamate in DCFS is mediated by reverse transport it should exhibit the saturation effect. On the other hand, if efflux is mediated by large-pore hemichannels, then the rate of substrate efflux should be linear with the intracellular concentration.
Fig. 5.

Evidence favoring hemichannels as main pathway for glutamate release in DCFS. a, Saturation of glutamate transport at [Glu]o > 0.1 mm. Uptake rate was assessed during 5 min of incubation; the rate of uptake was linear in this time frame within the tested concentration range.b, c, To test the effect of intracellular glutamate concentration on glutamate release in DCFS, astrocytes were preloaded with glutamate by a 20 min incubation in the presence of exogenous glutamate (0–320 μm). Glutamate release rate was proportional to intracellular glutamate content (r = 0.991). The gap junction blocker heptanol linearly reduced glutamate release (r = 0.998).d, THA or PDC preloading did not affect glutamate release in DCFS, implying that release was unlikely to be mediated by reverse transport (see Results). In DCFS, THA and RS-ACPD were released from astrocytes at about the same rate as glutamate (see Results).e, Taurine was released in DCFS and blocked in a concentration-dependent manner by Ca2+.f, Taurine release in DCFS was also blocked in a concentration-dependent way by carbenoxolone. n = 4–6 for all experiments.
The intracellular glutamate level in astrocytes is determined by metabolic reactions, the availability of precursors, and glutamate uptake. In the presence of abundant extracellular glutamate, glutamate uptake outpaces enzymatic conversion, and intracellular glutamate levels can reach very high values. This principle allowed us to load astrocytes with glutamate to a wide range of final [Glu]i levels. After 20 min of incubation in solutions containing 0–320 μm glutamate, [Glu]i values were 30–700 nmol/mg protein (Fig. 5b), implying ∼3.0–70 mm[Glu]i (assuming 1 mg protein = 10 μl of cellular space on average). The rate of glutamate efflux from glutamate-loaded cells in DCFS increased as a linear function of [Glu]i over the entire concentration range (Fig. 5c); that is, glutamate efflux failed to show saturation kinetics that should have been seen even at control levels of [Glu]i, ∼3.0 mm. The slope of this relationship was 0.139, suggesting that 13.9% of intracellular glutamate content was released per minute.
The gap junction blocking agent heptanol (2.0 mm) substantially inhibited DCFS-induced glutamate release from glutamate-loaded astrocytes. Heptanol reduced the slope of release from 0.139 to 0.037 (Fig. 5c). Control experiments showed that 2 mm heptanol alone had no significant effects on glutamate transport under control conditions. Furthermore, other gap junction blocking agents also reduced the slope of glutamate release (data not shown).
Finally, preloading astrocytes with either of the glutamate transport inhibitors THA orl-_trans_-pyrrolidine-2,4-dicarboxylidc acid (PDC) did not alter glutamate release in DCFS (Fig. 5d). Moreover, both THA, which has a similar affinity for glutamate transporters as glutamate, and RS-ACPD, which has an affinity ∼100× lower than glutamate for the transporters (Ye et al., 2001), were released at a similar rate as glutamate.
All of these results implied that the majority of glutamate efflux in DCFS proceeded via open hemichannels. This route should also be available to other amino acids. Indeed, taurine, an abundant intracellular amino acid, was released from astrocytes in a [Ca2+]o-dependent manner (Fig. 5e), and taurine release in DCFS was blocked in a manner essentially similar to glutamate release by the hemichannel blocker CBX (Fig. 5f). Likewise, taurine release was blocked by all other tested gap junction blockers (data not shown).
Glutamate release in DCFS did not involve ATP-activated channels, volume sensitive anion channels, or mobilization of intracellular Ca2+
Although the involvement of hemichannels in the release of glutamate seemed assured by the experiments above, it remained possible that other channels or mechanisms might participate as well. It has been suggested, for example, that open hemichannels can mediate ATP release (Stout et al., 2002), which in turn may stimulate glutamate release from astrocytes (Jeremic et al., 2001). Two mechanisms are possible: (1) ATP stimulation of the astrocytic P2X7 purinergic receptor, which can form a pore large enough to mediate dye uptake and presumably glutamate release (Ballerini et al., 1996; Virginio et al., 1999) and (2) ATP stimulation of purinergic receptors causing intracellular Ca2+ release followed by glutamate release (Jeremic et al., 2001). Therefore, we tested the possible involvement of ATP in DCFS-induced glutamate release. Irreversible blockade of P2X7 receptors with 300 μmoxidized-ATP (Contreras et al., 2002) had no effect on the DCFS-mediated release of glutamate and aspartate (Fig.6a). Application of the purinergic antagonists suramin (50–200 μm) or pyridoxalphosphate-6-azophenyl-2′,4′-disulfonic acid (PPADS) (10–50 μm) (Dubyak and el-Moatassim, 1993) did not alter glutamate release in DCFS (Fig. 6b). In addition, the potent blocker of P2 receptors, Reactive blue-2, did not reduce glutamate release in DCFS (Fig. 6c). ATP-mediated glutamate release is dependent on mobilization of intracellular Ca2+. Evidence for this manner of glutamate release was sought by blocking [Ca2+]imobilization with BAPTA-AM or thapsigargin (Jeremic et al., 2001). Neither BAPTA-AM (Molecular Probes, Eugene, OR; 50 μm for 45 min preincubation) nor thapsigargin (Sigma; 1 μm for 45 min) changed the percentage of glutamate release in DCFS (Fig. 6d) (as shown in Fig. 5c, the percentage of glutamate release is a more accurate indicator of open hemichannels if intracellular glutamate homeostasis is altered; BAPTA-AM and thapsigargin did change intracellular glutamate homeostasis (data not shown). These results indicated that DCFS-mediated glutamate release was unrelated to activation of ATP receptors and did not involve the mobilization of intracellular Ca2+.
Fig. 6.

Glutamate/taurine release in DCFS was not mediated by purinergic receptor activation or osmotic swelling-activated anion channels and did not depend on intracellular Ca2+mobilization. a, Pretreatment with 300 μmoxidized-ATP (o-ATP) for 3 hr to block P2X7 receptor channels did not influence glutamate or aspartate release in DCFS.b, c, Purinergic receptor antagonists did not reduce glutamate or taurine release in DCFS: suramine (200 μm), PPADS (50 μm), RB-2 (reactive blue-2, 10–300 μm tested). d, Glutamate release in DCFS was unaffected by pretreatment with BAPTA-AM (50 μm) or thapsigargin (1 μm) for 45 min (vehicle: 0.1% DMSO). Results are shown as ratios of released/total glutamate (see Results). e, Hypotonic (−75 mm NaCl) solution increased glutamate and taurine release from astrocytes. Heptanol (Hep, 2.0 mm) or 18α-glycyrrhetinic acid (AGA, 40 μm) at blocking concentrations (Fig. 3) had no effect on release in hypotonic solution.f, The anion channel blockers DIDS (1.0 mm) and SITS (1.0 mm) did not reduce amino acid release in DCFS. n = 6 for all experiments. ***p < 0.001, isotonic versus other groups in_e_ or control [control (Ctrl) solution with 2.0 mm Ca2+ and 2.0 mmMg2+] versus other groups by ANOVA with Dunnett's test (b, c, f) or control versus DCFS by t test (d).
Astrocytes swell in hypotonic solutions and activate volume-sensitive anion channels, which have been shown to mediate the release of taurine and even glutamate (Kimelberg et al., 1990, 1995). We exposed cultured hippocampal astrocytes to hypotonic solution (by removing 75 mm NaCl from the solution). To control for the effects of [Na+]o reduction, especially on glutamate and aspartate release, 75 mm NaCl was replaced by choline-Cl in the control solution (isotonic). Hypotonic solution increased release of glutamate 3.45-fold and that of taurine 8.98-fold (Fig. 6e). Gap junction blockers (2.0 mm heptanol and 40 μmAGA) that nearly completely blocked the release of glutamate and taurine in DCFS did not reduce their release in hypotonic solution. On the other hand, commonly used anion channel blockers DIDS and SITS (0.1–1.0 mm) did not block glutamate and taurine release in DCFS (Fig. 6f). Taken together, these data suggest that the glutamate release in DCFS is not mediated by volume-sensitive anion channels.
Hemichannels in situ can be opened by divalent cation removal
Hemichannel expression could be artificially common in cultured astrocytes. The “two-dimensional” character of the culture environment might provide less opportunity for gap junction formation than in vivo, resulting in more hemichannels. To verify the existence of functional hemichannels in vivo we used the acutely isolated mouse optic nerve, a relatively simple in situ CNS preparation. The absence of synapses in CNS white matter was advantageous because it eliminated the complication of Ca2+-dependent synaptic release of glutamate.
Typically, five pairs of mouse optic nerves were placed in a brain slice perfusion chamber, and stable basal levels of glutamate release were reached after 30 min at 37°C under control conditions. Control release levels were determined over the subsequent 20 min before switching to test solutions. For comparison, all release data were normalized to the average control release levels, which fall in the range of 9.2 ± 2.0 nm (_n_= 7), 5.3 ± 1.2 nm (n = 6), and 17.6 ± 2.3 nm (n = 8) for glutamate, aspartate, and taurine, respectively. Switching to DCFS + 2.0 mm EGTA [EGTA was added in these experiments because it greatly increases the speed with which [Ca2+]o falls in the optic nerve (Brown et al., 1998)] significantly increased release of glutamate, aspartate, and taurine, which returned to basal or control levels on switching back to control solution. CBX (100 μm) blocked or reduced DCFS + EGTA-induced release of these amino acids (Fig. 7). These data suggested, at least for CNS white matter, that functional hemichannels are present in vivo.
Fig. 7.

Amino acid release from mouse optic nerve. Glutamate (a), aspartate (b), and taurine (c) were released from acutely isolated mouse optic nerves when exposed to DCFS + EGTA (see Results). Amino acid release in DCFS + EGTA was blocked by CBX (100 μm). Amino acid release was normalized to average basal release levels determined 20 min before testing solution. Normalized data were pooled from six to eight experiments.
Discussion
The novel result reported here is that astrocyte hemichannels mediated massive release of glutamate and other amino acids when opened by exposure to divalent cation-reduced solution. These channels were abundant in cultured hippocampal astrocytes and also appeared to be present in CNS white matter cells in situ. Hemichannel involvement in glutamate release was confirmed by (1) appropriate distribution of Cx43, (2) characteristic permeability of these channels to LY, (3) characteristic gating by divalent cations and gap junction blocking agents, (4) appropriate release kinetics (i.e., linear with glutamate concentration gradient and nonsaturating), (5) independence from [Ca2+]ielevation, and (6) exclusion of other channels that could have mediated glutamate release, specifically the P2X7 receptor channel and volume-sensitive anion channel.
On the basis of our results, the existence of this powerful glutamate release pathway must be considered under pathological circumstances during which hemichannels could be expected to be open (John et al., 1999; Contreras et al., 2002) and glutamate excitotoxicity is likely to be important. It also seems likely that a small fraction of these channels may be open under normal physiological conditions or be gated open in a controlled manner during normal activity (Quist et al., 2000;Bruzzone et al., 2001; Kamermans et al., 2001; Plotkin and Bellido, 2001). If true, the implications for CNS physiology, ranging from synaptic transmission to development, are obvious but remain, for now, essentially unexplored.
Hemichannel gating
Unlike gap junctions, hemichannels are directly exposed to extracellular ions and thus can be effectively gated by extracellular, as opposed to intracellular, ionic changes. Although we have not tested the capacity of astrocyte hemichannels to respond to intracellular ionic changes, it seems probable that this would be the case. If so, this would provide for a complex level of permeability control that could be very important for, and a unique feature of, the overall biology of these channels.
The effectiveness with which divalent cations blocked the release of glutamate from astrocytes in DCFS was Ca2+> Sr2+ > Ba2+ > Mg2+. This same order applied to blocking LY loading in DCFS. These results are consistent with previous observations of LY loading into cells subjected to removal of extracellular Ca2+(Liu et al., 1996; Hofer and Dermietzel, 1998). In contrast, the rat astrocytes studied by Contreras et al. (2002) did not exhibit sensitivity to [Ca2+]o. The cultured cells used in those studies were “passaged” and studied within 24 hr of replating. Astrocytes clearly change their characteristics over time in culture (Sontheimer et al., 1991), and this may be the explanation for this discrepancy. Preliminary studies indicated that glutamate release through hemichannels was also gated by H+ and K+-mediated depolarization (Z.-C. Ye and B. R. Ransom, unpublished observations), other biophysical features characteristic of Cx43 gap junctions (Spray et al., 1981).
It has been known for years that plasma membranes can become leaky to a broad spectrum of small molecules at low [Ca2+]o. (Hille, 1992). Lowering of [Ca2+]o can significantly change membrane surface potential, and this may have important effects on voltage-gated Na+channels (Campbell and Hille, 1976; Armstrong and Cota, 1991) and K+ channels (Armstrong and Lopez-Barneo, 1987; Armstrong and Miller, 1990). Our results, and those of others (Liu et al., 1996; Hofer and Dermietzel, 1998), suggest that astrocytic hemichannels can be another important group of plasma membrane proteins that are sensitive to [Ca2+]o. Open hemichannels would make membranes leaky to all major ions and degrade transmembrane ion gradients. In addition, as shown here, they would allow release of glutamate and probably other small neuroactive molecules that would exact their own effects via independent actions on neural membranes. The selectivity among the cytoplasmic molecules released via gap junctions or, by inference, via hemichannels does not correlate precisely with molecular size (Harris, 2001), but larger molecules generally diffuse through gap junctions more slowly (Imanaga et al., 1987). Our results suggested that glutamate moved through hemichannels faster than LY, on the basis of 13.9% glutamate release (Fig. 5c) compared with 6% LY loading (Fig.1f) in the first minute after exposure to DCFS. This difference could be attributable to the larger size of LY compared with glutamate. Glutamate uptake in astrocytes was greatly compromised when hemichannels were opened by the removal of divalent cations. This is most likely caused by the dissipation of transmembrane ion gradients via open hemichannels, because the effect was alleviated by hemichannel blockade by carbenoxolone (Fig. 4).
DCFS-induced glutamate release in situ
To extend our observations about the effects of opening hemichannels beyond cultured astrocytes, we applied DCFS to isolated optic nerves, a well studied CNS white matter structure (Ransom et al., 1998). The release of glutamate induced by DCFS was blocked by CBX, indicating that hemichannels could be activated in situ. We cannot state with certainty which cell type within the optic nerve released glutamate from hemichannels when exposed to DCFS, but astrocytes are abundant in this structure and express gap junctions (Butt and Ransom, 1993). This observation represented proof-of-principle that functional hemichannels can be activated_in situ_ and thus presumably in vivo.
The [Glu]o in the optic nerve that resulted when hemichannels were opened by DCFS was not determined, but it probably reached a high level, on the basis of the fact that glutamate release from this structure could be measured in the massive volume of perfusate surrounding the nerve (roughly 103-fold greater than extracellular space volume). This observation has particular relevance because glutamate has been implicated in white matter injury caused by ischemia (Li et al., 1999; Baltan-Tekkok and Goldberg, 2001). The insight provided by this work is that hemichannels might contribute to the injury cascade in this tissue by mediating glutamate release during ischemia. Ischemic conditions appear to cause Cx43 dephosphorylation, leading to opening of hemichannels in astrocytes (Contreras et al., 2002).
As pointed out by Contreras et al. (2002), the actions of hemichannels and gap junctions during ischemia may be at odds with one another. If hemichannels open they would degrade ion gradients and, as shown here, cause glutamate release, events that would foster tissue damage. Gap junctions, on the other hand, might remain open during ischemia (Cotrina et al., 1998), serving to retard the degradation of ion homeostasis by equalizing ions between coupled cells (Rose and Ransom, 1997), or, in contrast, they could mediate the propagation of cell injury (Lin et al., 1998). A final complexity in this analysis is that open hemichannels would release other amino acids and small molecules that could have various consequences on tissue injury. The ubiquitous inhibitory neurotransmitter GABA, for example, could be released and has been shown to be protective of white matter under conditions of anoxic insult (Fern et al., 1995). These fascinating possibilities will require further investigation.
Our findings strongly argue for the existence of functional hemichannels that would have a role in glutamate homeostasis, both directly, by mediating glutamate release, and indirectly, by reducing glutamate uptake. If indeed a small proportion of hemichannels remained open under normal physiological conditions, they might importantly influence the local [Glu]o in the vicinity of the hemichannels and, as a consequence, influence neuronal activity. This is likely the case in the retina where horizontal cells appear to have a small percentage of their hemichannels open under physiological conditions (Kamermans et al., 2001). Importantly, a small proportion of hemichannels seems to be permeable to glutamate (i.e., open) at [Ca2+]o levels that are only slightly reduced from physiological levels (Fig.2b,c). This is consistent with the report that hemichannels can be opened by physiological fluctuations of [Ca2+]o (Quist et al., 2000; Bruzzone et al., 2001). Additional studies are necessary to establish the contribution of astrocytic hemichannel-mediated release of glutamate under normal conditions where [Ca2+]o changes would be modest.
Astrocytes may also release glutamate by exocytosis in response to elevation in [Ca2+]i (Parpura et al., 1994), and this mechanism has been shown to modulate synaptic transmission (Bezzi et al., 1998; Parpura and Haydon, 2000). Our results clearly suggest that elevation of [Ca2+]i was not responsible for glutamate release mediated by the opening of hemichannels in divalent cation-reduced solution. On the other hand, it is possible that glutamate release mediated by elevation of [Ca2+]i might involve opening of hemichannels, perhaps in addition to exocytosis. Recently, it has been shown that 5-nitro-2-(3-phenylpropylamino) benzoic acid (NPPB), a presumed selective anion channel blocker, can completely block ATP-stimulated Ca2+-dependent glutamate release (Jeremic et al., 2001). Interestingly, NPPB has been shown to be an effective blocker of Cx46 and Cx50 hemichannels (Eskandari et al., 2002), and preliminary evidence indicates that NPPB blocks both LY loading and amino acid release in DCFS (Ye and Ransom, unpublished data).
We have certainly not fully explored the factors that may contribute to hemichannel gating, including the role of depolarization (Spray et al., 1984) or selected pathological conditions, such as metabolic inhibition (John et al., 1999; Contreras et al., 2002). The availability of effective hemichannel blocking agents should permit analysis of the pathophysiological role that these channels might play in both white matter, as discussed above, and gray matter. It has been shown recently that gap junction blockers can reduce the neuronal injury caused by trauma (Frantseva et al., 2002b) or by global transient cerebral ischemia (Frantseva et al., 2002a); these effects, of course, might have been mediated by blockade of hemichannels, not gap junctions.
Footnotes
This work was supported by National Institutes of Health Grant NS 15589 and the Eastern Paralyzed Veterans Association.
Correspondence should be addressed to Dr. Bruce R. Ransom, Department of Neurology, University of Washington, School of Medicine, Room RR650, 1959 Northeast Pacific, Box 356465, Seattle, WA 98195-6465. E-mail: bransom@u.washington.edu.
References
- 1.Armstrong CM, Cota G. Calcium ion as a cofactor in Na channel gating. Proc Natl Acad Sci USA. 1991;88:6528–6531. doi: 10.1073/pnas.88.15.6528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Armstrong CM, Lopez-Barneo J. External calcium ions are required for potassium channel gating in squid neurons. Science. 1987;236:712–714. doi: 10.1126/science.2437654. [DOI] [PubMed] [Google Scholar]
- 3.Armstrong CM, Miller C. Do voltage-dependent K+ channels require Ca2+? A critical test employing a heterologous expression system. Proc Natl Acad Sci USA. 1990;87:7579–7582. doi: 10.1073/pnas.87.19.7579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ballerini P, Rathbone MP, Di Iorio P, Renzetti A, Giuliani P, D'Alimonte I, Trubiani O, Caciagli F, Ciccarelli R. Rat astroglial P2Z (P2X7) receptors regulate intracellular calcium and purine release. NeuroReport. 1996;7:2533–2537. doi: 10.1097/00001756-199611040-00026. [DOI] [PubMed] [Google Scholar]
- 5.Baltan-Tekkok S, Goldberg MP. AMPA/kainate receptor activation mediates hypoxic oligodendrocyte death and axonal injury in cerebral white matter. J Neurosci. 2001;21:4237–4248. doi: 10.1523/JNEUROSCI.21-12-04237.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bezzi P, Volterra A. A neuron-glia signaling network in the active brain. Curr Opin Neurobiol. 2001;11:387–394. doi: 10.1016/s0959-4388(00)00223-3. [DOI] [PubMed] [Google Scholar]
- 7.Bezzi P, Carmignoto G, Pasti L, Vesce S, Rossi D, Rizzini BL, Pozzan T, Volterra A. Prostaglandins stimulate calcium-dependent glutamate release in astrocytes. Nature. 1998;391:281–285. doi: 10.1038/34651. [DOI] [PubMed] [Google Scholar]
- 8.Brown AM, Fern R, Jarvinen JP, Kaila K, Ransom BR. Changes in [Ca2+]0 during anoxia in CNS white matter. NeuroReport. 1998;9:1997–2000. doi: 10.1097/00001756-199806220-00015. [DOI] [PubMed] [Google Scholar]
- 9.Bruzzone S, Guida L, Zocchi E, Franco L, De Flora A. Connexin 43 hemi channels mediate Ca2+-regulated transmembrane NAD+ fluxes in intact cells. FASEB J. 2001;15:10–12. doi: 10.1096/fj.00-0566fje. [DOI] [PubMed] [Google Scholar]
- 10.Butt AM, Ransom BR. Morphology of astrocytes and oligodendrocytes during development in the intact rat optic nerve. J Comp Neurol. 1993;338:141–158. doi: 10.1002/cne.903380110. [DOI] [PubMed] [Google Scholar]
- 11.Campbell DT, Hille B. Kinetic and pharmacological properties of the sodium channel of frog skeletal muscle. J Gen Physiol. 1976;67:309–323. doi: 10.1085/jgp.67.3.309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ceelen PW, Lockridge A, Newman EA. Electrical coupling between glial cells in the rat retina. Glia. 2001;35:1–13. doi: 10.1002/glia.1065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Choi DW. Glutamate neurotoxicity and diseases of the nervous system. Neuron. 1988;1:623–634. doi: 10.1016/0896-6273(88)90162-6. [DOI] [PubMed] [Google Scholar]
- 14.Contreras JE, Sanchez HA, Eugenin EA, Speidel D, Theis M, Willecke K, Bukauskas FF, Bennett MV, Saez JC. Metabolic inhibition induces opening of unapposed connexin 43 gap junction hemichannels and reduces gap junctional communication in cortical astrocytes in culture. Proc Natl Acad Sci USA. 2002;99:495–500. doi: 10.1073/pnas.012589799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cotrina ML, Kang J, Lin JH, Bueno E, Hansen TW, He L, Liu Y, Nedergaard M. Astrocytic gap junctions remain open during ischemic conditions. J Neurosci. 1998;18:2520–2537. doi: 10.1523/JNEUROSCI.18-07-02520.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Dermietzel R, Hertberg EL, Kessler JA, Spray DC. Gap junctions between cultured astrocytes: immunocytochemical, molecular, and electrophysiological analysis. J Neurosci. 1991;11:1421–1432. doi: 10.1523/JNEUROSCI.11-05-01421.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.DeVries SH, Schwartz EA. Hemi-gap-junction channels in solitary horizontal cells of the catfish retina. J Physiol (Lond) 1992;445:201–230. doi: 10.1113/jphysiol.1992.sp018920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Dubyak GR, el-Moatassim C. Signal transduction via P2-purinergic receptors for extracellular ATP and other nucleotides. Am J Physiol. 1993;265:C577–606. doi: 10.1152/ajpcell.1993.265.3.C577. [DOI] [PubMed] [Google Scholar]
- 19.Eskandari S, Zampighi GA, Leung DW, Wright EM, Loo DD. Inhibition of gap junction hemichannels by chloride channel blockers. J Membr Biol. 2002;185:93–102. doi: 10.1007/s00232-001-0115-0. [DOI] [PubMed] [Google Scholar]
- 20.Fern R, Waxman SG, Ransom BR. Endogenous GABA attenuates CNS white matter dysfunction following anoxia. J Neurosci. 1995;15:699–708. doi: 10.1523/JNEUROSCI.15-01-00699.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Frantseva MV, Kokarovtseva L, Perez Velazquez JL. Ischemia-induced brain damage depends on specific gap-junctional coupling. J Cereb Blood Flow Metab. 2002a;22:453–462. doi: 10.1097/00004647-200204000-00009. [DOI] [PubMed] [Google Scholar]
- 22.Frantseva MV, Kokarovtseva L, Naus CG, Carlen PL, MacFabe D, Perez Velazquez JL. Specific gap junctions enhance the neuronal vulnerability to brain traumatic injury. J Neurosci. 2002b;22:644–653. doi: 10.1523/JNEUROSCI.22-03-00644.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Giaume C, Fromaget C, el Aoumari A, Cordier J, Glowinski J, Gros D. Gap junctions in cultured astrocytes: single-channel currents and characterization of channel-forming protein. Neuron. 1991;6:133–143. doi: 10.1016/0896-6273(91)90128-m. [DOI] [PubMed] [Google Scholar]
- 24.Gutnick MJ, Connors BW, Ransom BR. Dye-coupling between glial cells in the guinea pig neocortical slice. Brain Res. 1981;213:486–492. doi: 10.1016/0006-8993(81)90259-6. [DOI] [PubMed] [Google Scholar]
- 25.Harks EG, de Roos AD, Peters PH, de Haan LH, Brouwer A, Ypey DL, van Zoelen EJ, Theuvenet AP. Fenamates: a novel class of reversible gap junction blockers. J Pharmacol Exp Ther. 2001;298:1033–1041. [PubMed] [Google Scholar]
- 26.Harris AL. Emerging issues of connexin channels: biophysics fills the gap. Q Rev Biophys. 2001;34:325–472. doi: 10.1017/s0033583501003705. [DOI] [PubMed] [Google Scholar]
- 27.Hille B. Ionic channels of excitable membranes, Ed 2. Sinauer; Sunderland, MA: 1992. Modifiers of gating. pp. 445–471. [Google Scholar]
- 28.Hofer A, Dermietzel R. Visualization and functional blocking of gap junction hemichannels (connexons) with antibodies against external loop domains in astrocytes. Glia. 1998;24:141–154. doi: 10.1002/(sici)1098-1136(199809)24:1<141::aid-glia13>3.0.co;2-r. [DOI] [PubMed] [Google Scholar]
- 29.Imanaga I, Kameyama M, Irisawa H. Cell-to-cell diffusion of fluorescent dyes in paired ventricular cells. Am J Physiol. 1987;252:H223–232. doi: 10.1152/ajpheart.1987.252.1.H223. [DOI] [PubMed] [Google Scholar]
- 30.Jeremic A, Jeftinija K, Stevanovic J, Glavaski A, Jeftinija S. ATP stimulates calcium-dependent glutamate release from cultured astrocytes. J Neurochem. 2001;77:664–675. doi: 10.1046/j.1471-4159.2001.00272.x. [DOI] [PubMed] [Google Scholar]
- 31.John SA, Kondo R, Wang SY, Goldhaber JI, Weiss JN. Connexin-43 hemichannels opened by metabolic inhibition. J Biol Chem. 1999;274:236–240. doi: 10.1074/jbc.274.1.236. [DOI] [PubMed] [Google Scholar]
- 32.Kamermans M, Fahrenfort I, Schultz K, Janssen-Bienhold U, Sjoerdsma T, Weiler R. Hemichannel-mediated inhibition in the outer retina. Science. 2001;292:1178–1180. doi: 10.1126/science.1060101. [DOI] [PubMed] [Google Scholar]
- 33.Kimelberg HK, Goderie SK, Higman S, Pang S, Waniewski RA. Swelling-induced release of glutamate, aspartate, and taurine from astrocyte cultures. J Neurosci. 1990;10:1583–1591. doi: 10.1523/JNEUROSCI.10-05-01583.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kimelberg HK, Rutledge E, Goderie S, Charniga C. Astrocytic swelling due to hypotonic or high K+ medium causes inhibition of glutamate and aspartate uptake and increases their release. J Cereb Blood Flow Metab. 1995;15:409–416. doi: 10.1038/jcbfm.1995.51. [DOI] [PubMed] [Google Scholar]
- 35.Levi G, Patrizio M. Astrocyte heterogeneity: endogenous amino acid levels and release evoked by non-N-methyl-d-aspartate receptor agonists and by potassium-induced swelling in type-1 and type-2 astrocytes. J Neurochem. 1992;58:1943–1952. doi: 10.1111/j.1471-4159.1992.tb10073.x. [DOI] [PubMed] [Google Scholar]
- 36.Li S, Mealing GA, Morley P, Stys PK. Novel injury mechanism in anoxia and trauma of spinal cord white matter: glutamate release via reverse Na+-dependent glutamate transport. J Neurosci 19 1999. RC16(1–9). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lin JH, Weigel H, Cotrina ML, Liu S, Bueno E, Hansen AJ, Hansen TW, Goldman S, Nedergaard M. Gap-junction-mediated propagation and amplification of cell injury. Nat Neurosci. 1998;1:494–500. doi: 10.1038/2210. [DOI] [PubMed] [Google Scholar]
- 38.Liu TF, Li HY, Atkinson MM, Johnson RG. Intracellular lucifer yellow leakage from Novikoff cells in the presence of ATP or low extracellular Ca: evidence for hemi-gap junction channels. Methods Find Exp Clin Pharmacol. 1995;17:23–28. [PubMed] [Google Scholar]
- 39.Liu TF, Li HY, Atkinson MM, Johnson RG. Comparison of lucifer yellow leakage and cell-to-cell transfer following intracellular injection in normal and antisense Novikoff cells under treatment with low extracellular Ca2+. Methods Find Exp Clin Pharmacol. 1996;18:493–497. [PubMed] [Google Scholar]
- 40.Marks PW, Maxfield FR. Preparation of solutions with free calcium concentration in the nanomolar range using 1,2-bis(o-aminophenoxy)ethane-N,N,N′, N′-tetraacetic acid. Anal Biochem. 1991;193:61–71. doi: 10.1016/0003-2697(91)90044-t. [DOI] [PubMed] [Google Scholar]
- 41.Nicholls D, Attwell D. The release and uptake of excitatory amino acids. Trends Pharmacol Sci. 1990;11:462–468. doi: 10.1016/0165-6147(90)90129-v. [DOI] [PubMed] [Google Scholar]
- 42.Olney JW. Brain lesions, obesity, and other disturbances in mice treated with monosodium glutamate. Science. 1969;164:719–721. doi: 10.1126/science.164.3880.719. [DOI] [PubMed] [Google Scholar]
- 43.Ottersen OP. Quantitative electron microscopic immunocytochemistry of neuroactive amino acids. Anat Embryol (Berl) 1989;180:1–15. doi: 10.1007/BF00321895. [DOI] [PubMed] [Google Scholar]
- 44.Parpura V, Haydon PG. Physiological astrocytic calcium levels stimulate glutamate release to modulate adjacent neurons. Proc Natl Acad Sci USA. 2000;97:8629–8634. doi: 10.1073/pnas.97.15.8629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Parpura V, Basarsky TA, Liu F, Jeftinija K, Jeftinija S, Haydon PG. Glutamate-mediated astrocyte-neuron signalling. Nature. 1994;369:744–747. doi: 10.1038/369744a0. [DOI] [PubMed] [Google Scholar]
- 46.Pfahnl A, Dahl G. Gating of cx46 gap junction hemichannels by calcium and voltage. Pflügers Arch. 1999;437:345–353. doi: 10.1007/s004240050788. [DOI] [PubMed] [Google Scholar]
- 47.Plotkin LI, Bellido T. Bisphosphonate-induced, hemichannel-mediated, anti-apoptosis through the Src/ERK pathway: a gap junction-independent action of connexin43. Cell Commun Adhes. 2001;8:377–382. doi: 10.3109/15419060109080757. [DOI] [PubMed] [Google Scholar]
- 48.Quist AP, Rhee SK, Lin H, Lal R. Physiological role of gap-junctional hemichannels. Extracellular calcium-dependent isosmotic volume regulation. J Cell Biol. 2000;148:1063–1074. doi: 10.1083/jcb.148.5.1063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ransom BR. Gap junctions. In: Kettenmann H, Ransom BR, editors. Neuroglia. Oxford UP; New York: 1995. pp. 299–318. [Google Scholar]
- 50.Ransom BR, Waxman SG, Fern R. Molecular pathophysiology of white matter anoxic/ischemic injury. In: Barnett HJM, Bennett JPM, Stein M, Yatsu FM, editors. Stroke: pathophysiology, diagnosis, and management, Ed 3. Churchill Livingstone; New York: 1998. pp. 85–100. [Google Scholar]
- 51.Rose CR, Ransom BR. Gap junctions equalize intracellular Na+ concentration in astrocytes. Glia. 1997;20:299–307. doi: 10.1002/(sici)1098-1136(199708)20:4<299::aid-glia3>3.0.co;2-1. [DOI] [PubMed] [Google Scholar]
- 52.Rozental R, Srinivas M, Spray DC. How to close a gap junction channel. Efficacies and potencies of uncoupling agents. Methods Mol Biol. 2001;154:447–476. doi: 10.1385/1-59259-043-8:447. [DOI] [PubMed] [Google Scholar]
- 53.Sontheimer H, Ransom BR, Cornell-Bell AH, Black JA, Waxman SG. Na(+)-current expression in rat hippocampal astrocytes in vitro: alterations during development. J Neurophysiol. 1991;65:3–19. doi: 10.1152/jn.1991.65.1.3. [DOI] [PubMed] [Google Scholar]
- 54.Spray DC, Harris AL, Bennett MV. Gap junctional conductance is a simple and sensitive function of intracellular pH. Science. 1981;211:712–715. doi: 10.1126/science.6779379. [DOI] [PubMed] [Google Scholar]
- 55.Spray DC, Stern JH, Harris AL, Bennett MV. Gap junctional conductance: comparison of sensitivities to H and Ca ions. Proc Natl Acad Sci USA. 1982;79:441–445. doi: 10.1073/pnas.79.2.441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Spray DC, White RL, de Carvalho AC, Harris AL, Bennett MV. Gating of gap junction channels. Biophys J. 1984;45:219–230. doi: 10.1016/S0006-3495(84)84150-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Stout CE, Costantin JL, Naus CC, Charles AC. Intercellular calcium signaling in astrocytes via ATP release through connexin hemichannels. J Biol Chem. 2002;277:10482–10488. doi: 10.1074/jbc.M109902200. [DOI] [PubMed] [Google Scholar]
- 58.Szatkowski M, Barbour B, Attwell D. Non-vesicular release of glutamate from glial cells by reversed electrogenic glutamate uptake. Nature. 1990;348:443–446. doi: 10.1038/348443a0. [DOI] [PubMed] [Google Scholar]
- 59.Virginio C, MacKenzie A, Rassendren FA, North RA, Surprenant A. Pore dilation of neuronal P2X receptor channels. Nat Neurosci. 1999;2:315–321. doi: 10.1038/7225. [DOI] [PubMed] [Google Scholar]
- 60.Wolff JR, Stuke K, Missler M, Tytko H, Schwarz P, Rohlmann A, Chao TI. Autocellular coupling by gap junctions in cultured astrocytes: a new view on cellular autoregulation during process formation. Glia. 1998;24:121–140. doi: 10.1002/(sici)1098-1136(199809)24:1<121::aid-glia12>3.0.co;2-t. [DOI] [PubMed] [Google Scholar]
- 61.Ye ZC, Rothstein JD, Sontheimer H. Compromised glutamate transport in human glioma cells: reduction–mislocalization of sodium-dependent glutamate transporters and enhanced activity of cystine–glutamate exchange. J Neurosci. 1999;19:10767–10777. doi: 10.1523/JNEUROSCI.19-24-10767.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Ye ZC, Ransom BR, Sontheimer H. (1R, 3S)-1-aminocyclopentane-1,3-dicarboxylic acid (RS-ACPD) reduces intracellular glutamate levels in astrocytes. J Neurochem. 2001;79:756–766. doi: 10.1046/j.1471-4159.2001.00581.x. [DOI] [PubMed] [Google Scholar]
- 63.Zerangue N, Kavanaugh MP. Flux coupling in a neuronal glutamate transporter. Nature. 1996;383:634–637. doi: 10.1038/383634a0. [DOI] [PubMed] [Google Scholar]