Corticolimbic Dopamine Neurotransmission Is Temporally Dissociated from the Cognitive and Locomotor Effects of Phencyclidine (original) (raw)
Abstract
The behavioral syndrome produced by phencyclidine (PCP) and its analog ketamine represents a pharmacological model for some aspects of schizophrenia. Despite the multifaceted properties of these drugs, the main mechanism for their psychotomimetic and cognitive-impairing effects has been thought heretofore to involve the corticolimbic dopamine system. The present study examined the temporal relationship between alterations in corticolimbic dopamine and glutamate neurotransmission and two dopamine-dependent behavioral effects of PCP in the rodent that have relevance to the clinical phenomenology, namely, impairment of working memory, which is used to model the frontal lobe deficits associated with schizophrenia, and hyperlocomotion, which is used as a predictor of the propensity of a drug to elicit or exacerbate psychosis. PCP increased dopamine and glutamate efflux in the prefrontal cortex and nucleus accumbens, as measured by microdialysis. The increase in dopamine in both regions remained elevated well above baseline 2.5 hr after the injection, at which time the experiment was terminated. However, locomotor activity returned to baseline in <2 hr after injection. Furthermore, impaired performance in a discrete trial delayed alternation task, a rodent working memory task, was only evident up to 60 min after PCP injection; animals tested 80 min after injection, when cortical dopamine release was elevated at 300% of baseline, did not exhibit impaired performance. These findings indicate that activation of dopamine neurotransmission is not sufficient to sustain PCP-induced locomotion and impairment of working memory. Thus, effects of PCP, including a glutamatergic hyperstimulation, may be necessary to account for the psychotomimetic and cognitive-impairing effects of this drug.
Keywords: prefrontal cortex, nucleus accumbens, microdialysis, PCP, schizophrenia, working memory, glutamate, NMDA, ketamine, drug abuse
Clinical studies and case reports over the last 3 decades have consistently reported that a single exposure to phencyclidine (PCP) or ketamine produces behavioral disruptions in healthy individuals that mimic some aspects of the deficit and positive symptoms associated with schizophrenia (Luby et al., 1959; Davies and Beech, 1960; Bakker and Amini, 1961; Krystal et al., 1994; Malhotra et al., 1996). Therefore, these drugs are used routinely as clinical and animal models of this disorder, with the underlying assumption that neuronal mechanisms responsible for some PCP-induced disruptions of normal brain function may be relevant to the pathophysiology of schizophrenia.
Consistent with clinical studies, subanesthetic doses of ketamine and PCP, as well as other NMDA receptor antagonists, produce cognitive disruptions as measured by a battery of memory and learning-related cognitive tasks in the rodent (Handelmann et al., 1987; Wesierska et al., 1990; Hauber and Andersen, 1993; Verma and Moghaddam, 1996;Jentsch et al., 1997a) and primate (Byrd et al., 1987; Hudzik and Wenger, 1993; Javitt et al., 1996; Jentsch et al., 1997b). In addition, they produce stereotypy and impair social behavior in both species (Schlemmer et al., 1978; Sturgeon et al., 1979; Steinpreis et al., 1994; Sams-Dodd, 1996) and at moderate doses increase locomotor activity in the rodent (Sturgeon et al., 1979; Greenburg and Segal, 1985).
Despite the well established involvement of nondopaminergic mechanisms in the reinforcing and disruptive effects of PCP and ketamine (Keith et al., 1991; Bakshi et al., 1994; Kitaichi et al., 1994; Ogawa et al., 1994; Carlezon and Wise, 1996; Hutson and Hogg, 1996; Gleason and Shannon, 1997), a major mechanism responsible for the psychotomimetic effects of these drugs is thought to involve dopaminergic neurotransmission (Kornhuber et al., 1990; Javitt and Zukin 1991;Carlsson et al., 1993; Iversen, 1995; Steinpreis, 1996; Breier et al., 1998; Smith et al., 1998). This is based on two lines of evidence. First, PCP increases dopamine release and turnover in the prefrontal cortex and limbic striatal regions (Doherty et al., 1980; Deutch et al., 1987; Carboni et al., 1989; Steinpreis and Salamone, 1993; Hondo et al., 1994; Hertel et al., 1996; Verma and Moghaddam, 1996), thus producing a limbic hyperdopaminergic state. This mechanism supports the well established “dopamine hypothesis of schizophrenia” and is consistent with recent imaging studies describing dopamine hyperactivity in schizophrenics (Laruelle et al., 1996; Breier et al., 1997). Of note, although chronic treatment with this class of drugs reduces dopamine metabolite levels in cortical tissue (Jentsch et al., 1997a,b), it produces a sustained increase in resting extracellular levels of dopamine in the prefrontal cortex (Lindefors et al., 1997), suggesting that the cortical hyperdopaminergic effect persists after chronic treatment. Second, dopamine antagonists and/or lesions of dopamine pathways reduce the effects of PCP on two behavioral measures that are thought to have relevance to clinical symptomatology: cognitive tasks involving working memory, which are used routinely to model some aspects of frontal lobe deficits associated with schizophrenia, and locomotor activity, which is used in preclinical studies as a predictor of the propensity of a drug to induce or exacerbate psychosis in man (Castellani and Adams, 1981; Greenburg and Segal, 1985; French, 1986; Carlsson et al., 1993; Jackson et al., 1994;Ogren and Goldstein, 1994; Steinpreis et al., 1994; Krystal et al., 1995; Goldman-Rakic, 1996; Verma and Moghaddam, 1996). Based primarily on the these findings, the major emphasis of most preclinical and clinical studies that use PCP or ketamine as putative models of schizophrenia, in particular those studies that assess the etiology or treatment options for the cognitive deficits associated with schizophrenia, is the dopamine system (Johnson and Jones, 1990;Krystal et al., 1995; Verma and Moghaddam, 1996; Jentsch et al., 1997a,b; Breier et al., 1998; Smith et al., 1998).
However, impairment of working memory-related tasks by ketamine or PCP is only partially ameliorated with dopamine D2 receptor antagonists and is not affected by other dopamine antagonists (Krystal et al., 1995;Verma and Moghaddam, 1996), and locomotor effects of PCP are blocked only at high cataleptic doses of dopamine antagonists (Castellani and Adams, 1981; Ogren and Goldstein, 1994). In fact, evidence for a temporal relationship between activation of corticolimbic dopamine release and the cognitive and locomotor effects of PCP is lacking. The present study was undertaken to establish whether such a relationship exits.
Recent studies have demonstrated that ketamine and other NMDA receptor antagonists also increase extracellular levels of glutamate (Liu and Moghaddam, 1995; Moghaddam et al., 1997). Although there is controversy on the significance of extracellular glutamate measures (Timmerman and Westerink, 1997), other lines of evidence suggest that increases in the synaptic availability of glutamate and postsynaptic activation of glutamate receptors may produce cognitive, locomotive, neurotoxic, and dopaminergic effects of NMDA receptor antagonists (Hauber and Andersen, 1993; Willins et al., 1993; Bubser et al., 1995; Olney and Farber, 1995; Sharp et al., 1995; Moghaddam et al., 1997). Thus, we also performed concomitant measures of extracellular glutamate and compared this response in the prefrontal cortex and nucleus accumbens to dopaminergic and behavioral effects of PCP.
MATERIALS AND METHODS
Animal preparation. Animal use procedures were in accordance with the NIH Guide for the Care and Use of Laboratory Animals and were approved by the Yale University Animal Care and Use Committee. Male Sprague Dawley rats (275–325 gm) were used throughout the study. Microdialysis probes were implanted while the rats were under halothane anesthesia. Animals were placed in a stereotaxic frame with blunt ear bars, and a small incision (5–7 mm) was made in the skin over the skull. The wound margin was infiltrated with lidocaine. Holes were drilled for two skull screws and two concentric microdialysis probes positioned bilaterally in the prefrontal cortex [anterioposterior (AP), 3.2; lateral (L), 1.8 set at a 10° angle; ventral (V), −6.5] and in the nucleus accumbens shell (AP, 1.8; L, 1.0; V, −8.4) with respect to bregma (Paxinos and Watson, 1982). Probes and skull screws were secured in place with dental cement. After surgery, animals were placed in a clear polycarbonate cage (44 × 22 × 21 cm) with food, water, and bedding. Animals were allowed to recover for 24 hr before microdialysis samples were taken. Probes were perfused with perfusion solution (see below) at 0.5 μl/min overnight and 2.0 μl/min during the experiment.
Microdialysis procedure. Concentric microdialysis probes with an exposed tip length of 3.0 mm for prefrontal cortex and 2.0 mm for nucleus accumbens were used. The perfusion solution contained (in mm): 145 NaCl, 2.7 KCl, 1.0 MgCl2, and 1.2 CaCl2. A flow rate of 2 μl/min was used during the sample collection. Samples were collected at 20 min intervals. After collection, 5 μl of the sample was removed and added to disposable autosampler vials containing homoserine (an internal standard) and was later analyzed for glutamate (see below). The rest of the sample was immediately injected onto an HPLC with electrochemical detection for analysis of dopamine.
Chromatographic analysis of amino acids and dopamine. The amount of dopamine in the dialysis samples was measured by HPLC with electrochemical detection. This system used a 10 cm narrow-bore column [2.0 mm inner diameter (i.d.); 3 μm C-18 particles] (Keystone, Bellefonte, PA) and a Bioanalytical Systems LC-4C potentiostat (BAS, West Lafayette, IN). The _E_app was +0.55 V versus Ag/AgCl reference electrode. The mobile phase consisted of 9 gm/l NaH2PO4, 640 mg/l octylsulfonic acid, 7.2% acetonitrile (v/v), 250 mg/l EDTA, and 350 μl/l triethylamine, pH 5.1. A detection limit of 5 fmol was routinely achieved.
The amount of glutamate and homoserine (internal standard) in the perfusate was determined by precolumn derivatization with 0-pthalaldehyde and mercaptoethanol, followed by HPLC with fluorescence detection. This system used an autosampler (Spectra System AS 3000; Spectra-Physics, Fremont, CA), a gradient pump (SP 8800; Spectra-Physics), and a fluorescence detector (SP 8410; Spectra-Physics). The excitation and emission wavelengths were set at 300 and 400 nm, respectively. A 10 cm, C-18, 4.2 mm i.d. analytical column (Keystone, Bellefonte, PA) was used. The mobile phase was 0.1 m phosphate buffer, pH 6.4, containing 0.01m EDTA and acetonitrile with a gradient profile of 12–22% over 12 min.
Locomotor activity and stereotypy rating. A data acquisition system (Med Associates, St. Albans, VT) was used to record the activity during the microdialysis experiments. Four pairs of photocells were spaced evenly along the length of the cage. Nonconsecutive beam breaks were totaled every 20 min, which was coordinated with the collection time of the microdialysis samples. Stereotypy was rated by a trained observer according to method of Kelley and Delfs (1994).
Discrete trial delayed alternation paradigm. Spatial delayed alternation performance was tested using a discrete trial T-maze delayed alternation paradigm (in part, adapted from Freeman and Stanton, 1992). Animals were handled and trained in a gray Plexiglas T-maze, as described previously (Verma and Moghaddam, 1996). However, instead of 10 trials of continuous alternation, which necessitates imposing long delay periods in between trials as rats become overtrained, animals were presented first with a forced run, i.e., they were given access to only one arm of the maze and rewarded (half of a morsel of Froot Loops cereal; General Mills, Kankakee, IL) after entering that arm. After a 10 sec delay, they were presented with both arms and were rewarded for entering the arm that they had not entered on the previous (forced) run. Animals were given 10 randomly chosen forced runs, followed by 10 choice runs. Animals were trained until a criterion of 80% correct choices on 2 consecutive days was achieved.
Histology. After the termination of each experiment, animals were anesthetized with chloralhydrate and perfused intracardially with saline, followed by 10% buffered formalin. Brains were removed and stored in formalin. Serial sections of the fixed brains were cut at 100 μm intervals and stained with cresyl violet. Probe placement was verified for all of the data sets used in this study.
Materials. All reagents for the HPLC mobile phase and the perfusion fluid were analytical grade and were obtained from Eastman Kodak (Rochester, NY), J. T. Baker Chemical Company (Phillipsburg, NJ), or Sigma (St. Louis, MO). PCP was purchased from Sigma.
Data analysis. For microdialysis and locomotor data, two-way ANOVA with time as the repeated measures was used to compare the drug-treated and control groups. Within each group, analyses were conducted by one-way repeated measures ANOVA. Significant main effects were analyzed further by Tukey post hoc comparison of means. For microdialysis studies, both absolute values (femtomoles per microliter not corrected for recovery) and percentage of the mean ± SEM of the three basal values obtained immediately before the treatment were analyzed. For the activity data, the nonconsecutive beam breaks in 20 min (to correspond with microdialysis sample collection) was used for analysis. The delayed alternation data were compared by multifactorial ANOVA followed by Tukey post hoc analysis. Significance was set at p ≤ 0.05.
RESULTS
Effects of PCP on dopamine and glutamate efflux in the prefrontal cortex
All microdialysis data are shown in terms of both absolute levels of dopamine and glutamate measured in the dialysate and percent increase from baseline. The latter is to facilitate the comparison of response between different regions and compounds measured.
Intraperitoneal injection of 5 mg/kg PCP produced a robust sixfold increase in extracellular levels of dopamine in the prefrontal cortex (Fig. 1). This increase in dopamine peaked at 40 min after injection and declined afterward. However, dopamine levels remained elevated significantly above baseline 140 min after the injection, at which time the experiment was terminated.
Fig. 1.

Effect of PCP (5 mg/kg, i.p.) on the extracellular levels of dopamine in the prefrontal cortex. The _top_illustrates the data in terms of percentage of baseline (mean ± SEM of the three basal values obtained immediately before the injection), and the bottom indicates the absolute dopamine concentration in the dialysis samples not corrected for probe recovery. Two-way ANOVA with time as the repeated measures indicated a significant group × time effect (p < 0.001) between the PCP (n = 7) and saline (n = 6) groups. Asterisks indicate significant differences from the saline-treated group corresponding to the same postinjection time.
PCP also produced a significant increase in extracellular glutamate levels in the prefrontal cortex (Fig. 2). This increase was apparent immediately and continued to rise for the first hour after injection. Glutamate levels remained elevated at maximal levels when the experiment was terminated 140 min after the PCP injections. The glutamate response was more variable than that observed with dopamine, which is consistent with our previous observations that dialysate glutamate levels during baseline or after pharmacological challenges (or stress) are more variable than dopamine levels.
Fig. 2.

Effect of PCP (5 mg/kg, i.p.) on the extracellular levels of glutamate in the prefrontal cortex. The _top_illustrates the data in terms of percentage of the mean ± SEM of the three basal values obtained immediately before the injection, and the bottom is the absolute dopamine concentration in the dialysis samples not corrected for probe recovery. Two-way ANOVA with time as the repeated measures indicated a significant group × time effect (p < 0.04) between the PCP (n = 7) and saline (n = 6) groups. Asterisks indicate significant differences from the saline-treated group corresponding to the same postinjection time.
Effect of PCP on discrete trial delayed alternation performance
To examine the time-dependent effect of PCP on a working memory task that is dependent on the functional integrity of the dopamine projections to the prefrontal cortex, rats were trained in a delayed alternation paradigm (Simon, 1981; Bubser and Schmidt, 1990). PCP and other NMDA antagonists produce disruption of delayed-response tasks presumably by activating dopamine neurotransmission in the prefrontal cortex (Verma and Moghaddam, 1996; Zahrt et al., 1997). Animals were trained to perform at 80% correct choice (20% error) at a delay of 10 sec on at least 2 consecutive days. They were then injected with vehicle or PCP (Fig. 3). Saline-injected animals were tested 20 or 40 min after injection. PCP-injected animals were tested at 20, 40, 60, or 80 min after injection (i.e., each rat was tested only once after PCP at one of these times after injection). Because of the profound stereotypy and hyperlocomotion apparent in some rats 20 min after receiving PCP (see below), only two rats were properly tested at 20 min after injection, both of which performed at 50% correct. Animals tested 40 and 60 min after PCP injection showed impairment in performance of this task. In contrast, an impairment was not observed in rats tested 80 min after injection.
Fig. 3.
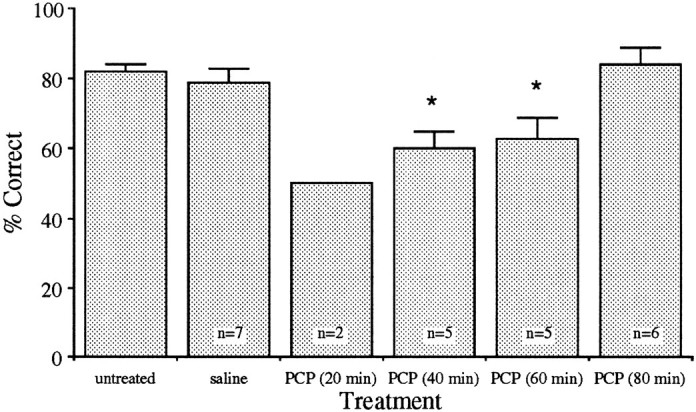
Time-dependent effect of intraperitoneal injection of PCP on discrete trial delayed alternation performance. Only two rats were properly tested at 20 min after injection, both of which performed at 50% correct. (Three other rats tested at this time ran up and down the main alley of the maze and did not enter the side arms or remained in the start box exhibiting profound head rolling.) Animals tested 40 and 60 min after PCP injection showed significant impairment compared with the saline-treated group (p < 0.05). Rats tested 80 min after PCP injection did not display an impairment.
Figure 4 displays the PCP-induced relative increase in dopamine and glutamate levels in the prefrontal cortex, along with the percent of error exhibited during the delayed-alternation task corresponding to the same time after PCP injection. As shown, 20–60 min after injection an increase in percent of error is accompanied by an increase in glutamate and dopamine efflux. However, 80 min after injection when the delayed alternation performance is no longer impaired, dopamine and glutamate levels in the prefrontal cortex, as well as dopamine levels in the nucleus accumbens (see below), remain elevated well above baseline.
Fig. 4.

Temporal comparison of the increase in extracellular levels of dopamine and glutamate (displayed as percent of preinjection value) and performance in delayed alternation paradigm corresponding to the time that the neurochemical analysis was performed after PCP injection.
Effect of PCP on dopamine and glutamate efflux in the nucleus accumbens
Intraperitoneal injection of 5 mg/kg PCP produced a significant increase of nearly 300% in extracellular levels of dopamine in the nucleus accumbens (Fig. 5). Similar to the effect seen in the prefrontal cortex, the increase in dopamine peaked at 40 min after injection and remained significantly elevated above baseline for the duration of the experiment. The relative magnitude of this increase compared with baseline was significantly smaller (p < 0.01) than that observed in the prefrontal cortex.
Fig. 5.

Effect of PCP (5 mg/kg, i.p.) on the extracellular levels of dopamine in the nucleus accumbens. The _top_illustrates the data in terms of percentage of the mean ± SEM of the three basal values obtained immediately before the injection, and the bottom is the absolute dopamine concentration in the dialysis samples not corrected for probe recovery. Two-way ANOVA with time as the repeated measures indicated a significant group × time effect (p < 0.01) between the PCP (n = 8) and saline (n = 8) groups. Asterisks indicate significant differences from the saline-treated group corresponding to the same postinjection time.
PCP also produced a significant increase in extracellular glutamate levels in the nucleus accumbens compared with baseline (Fig.6). This increase peaked immediately and declined toward baseline 40 min after injection. Whereas glutamate levels remained elevated above baseline (i.e., preinjection value), because of the variability in both saline- and PCP-treated data groups, the response to PCP was significantly different from saline-injected rats only for the first two samples collected after PCP injection.
Fig. 6.

Effect of PCP (5 mg/kg, i.p.;n = 6) or saline (n = 6) on the extracellular levels of glutamate in the nucleus accumbens. The_top_ illustrates the data in terms of percentage of the mean ± SEM of the three basal values obtained immediately before the injection, and the bottom is the absolute dopamine concentration in the dialysis samples not corrected for probe recovery. PCP produced a significant increase in glutamate levels relative to preinjection values (p < 0.05, using ANOVA with time as the repeated measures). Asterisks indicate significant differences from preinjection values.
Effect of PCP on locomotor activity and stereotypy
PCP elicited an immediate and profound hyperlocomotion and stereotypy (Fig. 7). The locomotor count peaked in the first 20 min after injection and declined to baseline levels 100 min after injection. The repetitive behaviors exhibited after receiving PCP included head rolling, circling, peddling, hand rubbing, sniffing, and mouth movements. The time course of the stereotypy followed that of the locomotor response. The highest stereotypy score occurred during the first 20 min after injection. The head-rolling response subsided in most animals 40–60 min after injection. All other repetitive behaviors subsided 100–120 min after injection.
Fig. 7.

Effect of PCP and saline injection on locomotor activity (top) and stereotypy score (bottom). Two-way ANOVA with time as the repeated measures indicated a significant group × time effect (p < 0.001) between the PCP (n = 8) and saline (n = 8) groups. Asterisks indicate significant differences from the saline-treated group corresponding to the same postinjection time.
Dopamine projections to the nucleus accumbens have been strongly associated with PCP-induced locomotion because intra-accumbens application of PCP elicits locomotion (McCullough and Salamone, 1992), and lesions of the dopaminergic projections to the nucleus accumbens block the locomotor stimulating effects of PCP (French, 1986;Steinpreis and Salamone, 1993; Ouagazzal et al., 1994). Therefore, in the present study the magnitude and duration of the effects of PCP on locomotor activity and dopamine release in the nucleus accumbens were compared. Figure 8 demonstrates correlational analysis and the temporal comparison of the locomotion and extracellular dopamine data. Although a significant correlation between these two measures was obtained (Fig. 8, top), there was a dissociation in the time course of the response in that despite a sustained elevation in extracellular levels of dopamine, nearly double the resting levels, the locomotor activity declined toward preinjection resting levels. A similar analysis performed with locomotor activity and extracellular levels of glutamate in response to PCP is demonstrated in Figure 9. Similar to that observed with dopamine, a significant correlation was apparent between these two measures; however, despite elevated extracellular glutamate levels 2 hr after PCP injection (relative to preinjection values), the increase in locomotion subsided.
Fig. 8.

Correlational analysis for concomitant 20 min measurements (top) and the temporal relationship (bottom) between the locomotor activity and extracellular levels of dopamine in the nucleus accumbens after PCP.
Fig. 9.

Correlational analysis for concomitant 20 min measurements (top) and the temporal relationship (bottom) between the locomotor activity and extracellular levels of glutamate in the nucleus accumbens after PCP.
DISCUSSION
Subanesthetic doses of PCP and its analog ketamine increase dopamine release in rodents (Hondo et al., 1994; Verma and Moghaddam, 1996) and humans (Breier et al., 1998; Smith et al., 1998), and dopamine antagonists ameliorate the behavioral disruption caused by these drugs, which may have relevance to schizophrenic symptomatology (Freed et al., 1980; Ogren and Goldstein, 1994; Krystal et al., 1995;Verma and Moghaddam, 1996). Hence, dopamine hyperfunction has been associated with the psychotomimetic properties of these drugs. The present study examined whether activation of dopamine release in the prefrontal cortex and nucleus accumbens is temporally related to those behavioral effects of PCP that have relevance to the clinical phenomenology: impairment of working memory, which is used to model the frontal lobe deficits associated with schizophrenia, and hyperlocomotion, which is used as a predictor of the propensity of a drug to elicit or exacerbate psychosis. In light of recent findings that the dopamine-releasing effects of the NMDA receptor antagonists may be in part attributable to activation of glutamatergic neurotransmission (Moghaddam et al., 1997), the time course of dopaminergic and behavioral effects of PCP was also compared with glutamate efflux.
PCP-mediated neurochemical effects in the prefrontal cortex and nucleus accumbens
In agreement with previous studies, PCP increased dopamine release in the prefrontal cortex and nucleus accumbens (Carboni et al., 1989;Bristow et al., 1993; Steinpreis and Salamone, 1993; Hondo et al., 1994; Hertel et al., 1996; Nishijima et al., 1996). This increase was larger in magnitude in the prefrontal cortex than in the nucleus accumbens, but the duration of effect was similar in both regions.
This is the first report of PCP increasing glutamate efflux in the prefrontal cortex. This observation is consistent with the effect of ketamine and other NMDA receptor antagonists, which increases glutamate efflux in a dose-dependent and TTX-sensitive manner (Liu and Moghaddam, 1995; Moghaddam et al., 1997). Glutamate levels in the nucleus accumbens also increased in response to PCP. This effect was more variable and shorter in duration than the effect seen in the prefrontal cortex.
Although PCP, at least at high anesthetic doses, has an affinity for the dopamine transporter (Javitt and Zukin, 1991), the increase in extracellular levels of cortical dopamine observed at the moderate dose of 5 mg/kg is unlikely to be attributable to blockade of dopamine uptake. The axonal distribution of the dopamine transporter protein in the rodent medial prefrontal cortex is highly restricted compared with striatal regions (Sesack et al., 1998). Consistent with this pattern of distribution, dopamine uptake blockers, such as cocaine, are relatively ineffective in increasing extracellular levels of dopamine in the prefrontal cortex (Moghaddam and Bunney, 1989), although they profoundly increase these levels in the nucleus accumbens and the striatum (Hurd and Ungerstedt, 1989). In contrast, the pattern of increase in dopamine release after 5 mg/kg PCP is opposite to the axonal density of the dopamine transporter; extracellular dopamine levels increase to nearly 600% above baseline in the prefrontal cortex, 300% above baseline in the nucleus accumbens, and 50% above baseline in the striatum (Nishijima et al., 1996; our unpublished observations). Thus, it is unlikely that blockade of dopamine uptake contributes significantly to the increase in extracellular dopamine levels produced by moderate doses of PCP.
In anesthetized preparations, systemic administration of PCP and other NMDA antagonists increase the firing rate of dopamine neurons (French, 1994), suggesting that these drugs enhance impulse-dependent dopamine release. However, in awake rats, application of NMDA receptor antagonists to dopamine cell body regions reduces dopamine release in the nucleus accumbens (Karreman et al., 1996; Westerink et al., 1996) and prefrontal cortex (Takahata and Moghaddam, 1998), indicating that the increase in dopamine release after systemic NMDA antagonists (in the absence of anesthesia) is mediated by mechanisms that regulate presynaptic release of dopamine. This is consistent with reports that local application of PCP in the prefrontal cortex dose-dependently increases dopamine release in this region (Hondo et al., 1994). One mechanism that may mediate this local effect and account for the preferential stimulatory effect of PCP (and other NMDA receptor antagonists, including MK801; Wedzony et al., 1994) on cortical dopamine release may involve a hyperglutamatergic state at non-NMDA receptors. The present study and previous reports (Liu and Moghaddam, 1995; Moghaddam et al., 1997) indicate that PCP and other NMDA receptor antagonists increase extracellular levels of glutamate. The mechanism for and the functional significance of this increase is not well established. However, reports that AMPA and kainate receptor antagonists reduce the increase in locomotion (Hauber and Andersen, 1993; Willins et al., 1993; Bubser et al., 1995), cognitive deficits (Moghaddam et al., 1997), neurodegeneration (Olney and Farber, 1995;Sharp et al., 1995), and cortical dopamine release (Moghaddam et al., 1997) associated with ketamine, MK801, or PCP suggest that an increase in the synaptic availability of glutamate and subsequent activation of non-NMDA glutamate receptors may be associated with these drugs. Interestingly, dopamine projections to the prefrontal cortex are more sensitive to AMPA and kainate receptor regulation, both at the cell body (Takahata and Moghaddam, 1998) and the terminal level (Jedema and Moghaddam, 1996) than projections to the ventral striatum, suggesting that glutamate hyperfunction at non-NMDA receptors may preferentially increase dopamine release in the prefrontal cortex compared with subcortical regions.
The pattern of response in extracellular glutamate levels in the prefrontal cortex after PCP had a different profile than that observed for dopamine; whereas dopamine levels peaked at 40 min and declined afterward, glutamate levels continued to rise above baseline during the first hour after injection and remained maximally elevated for the remainder of the experiment. The different temporal response pattern between the increase in extracellular glutamate and dopamine, in particular, a decline in dopamine levels while the glutamate levels were sustained at maximal levels, questions the idea that activation of glutamate release (and subsequent stimulation of AMPA and kainate receptors) may be a contributing factor to dopaminergic activation. However, both AMPA and kainate receptors desensitize rapidly in response to repeated stimulation (Kiskin et al., 1986; Otis et al., 1996; Jones et al., 1997; Wilding and Huettner, 1997). Hence, despite a sustained increase in glutamate levels after PCP, a decline in dopamine release may be expected if activation of AMPA or kainate receptors plays a part in stimulating this release.
Comparison of PCP-mediated cognitive and neurochemical effects
PCP impaired performance of a discrete trial delayed alternation task, a rodent working memory task that is dependent on the functional integrity of the prefrontal cortex (J. M. Aultman and B. Moghaddam, unpublished observations). This finding is consistent with other reports that acute administration of noncompetitive NMDA antagonists produces deficits in frontal lobe-dependent tasks in rodents and humans (Hauber and Andersen, 1993; Krystal et al., 1994; Verma and Moghaddam, 1996). This impairment was apparent when animals were tested at 20, 40, or 60 min after PCP injection. However, animals that were tested 80 min after the injection, at which time both dopamine and glutamate levels in the prefrontal cortex (and the nucleus accumbens) were elevated over 200% of basal values, did not display an impairment. These findings suggest that activation of dopamine neurotransmission is not sufficient to produce the working memory deficit associated with PCP.
The dissociation between maze performance and activated dopamine release was unexpected, because working memory impairment produced by ketamine is ameliorated by pretreatment with dopamine D2 receptor antagonists (Krystal et al., 1995; Verma and Moghaddam, 1996) and evidence for rapid desensitization of postsynaptic D2 receptors is generally lacking (Filtz et al., 1993; White, 1996; Ng et al., 1997). Based on the present findings, it may be postulated that reduction of ketamine- and PCP-mediated cognitive deficits by D2 receptor antagonists may involve indirect mechanisms, whereby D2 receptors localized presynaptically on nondopaminergic terminals may regulate the release of other neurotransmitters, such as acetylcholine or glutamate, that sustain this behavioral disruption.
It should be emphasized that the present study does not discount a role for dopamine in normal processes that subserve working memory. A convincing body of work has established that phasic changes in dopamine neurotransmission (as well as glutamatergic neurotransmission) occur in the prefrontal cortex during behavioral tasks that involve working memory (Williams and Goldman-Rakic, 1993; Wilson et al., 1994). Although phasic activation of dopamine receptors may be a necessary component subserving working memory, the present study suggests that a sustained overactivation of dopamine neurotransmission by PCP is not sufficient to deteriorate this function.
Comparison of PCP-mediated locomotor and neurochemical effects
Local infusion of PCP into the nucleus accumbens increases locomotor activity (McCullough and Salamone, 1992), and lesions of dopamine innervation of the nucleus accumbens (French, 1986; Steinpreis and Salamone, 1993) or dopamine D2 receptor antagonists (Freed et al., 1980; Castellani and Adams, 1981; Kitaichi et al., 1994; Ogren and Goldstein, 1994) reduce PCP-induced locomotion. These findings have strongly implicated mesoaccumbens dopamine neurotransmission in the locomotor activating effects of PCP. However, the present observation of a temporal dissociation between increases in dopamine release in nucleus accumbens (or prefrontal cortex) and locomotor activation does not support the idea that activation of dopamine neurotransmission by PCP is sufficient to sustain locomotor activity.
Although dopamine projections to the ventral striatum have been classically thought to play a major role in sustaining locomotor activity (Kelly and Iversen, 1976; Koob et al., 1978), the present finding of a functional dissociation between locomotor and dopaminergic effects of PCP may not be considered novel; previous findings had clearly established that NMDA receptor antagonists produce hyperlocomotion in an apparent absence of dopaminergic activity and, therefore, dopamine neurotransmission is not necessary to sustain locomotor activation by this class of drugs (Carlsson and Carlsson, 1989; Druhan et al., 1996; Waters et al., 1996) or other psychostimulants (Kuczenski and Segal, 1989; Kuczenski et al., 1997). The novel aspect of the present findings is that this is the first evidence, to our knowledge, demonstrating that activation of dopamine neurotransmission is not sufficient to sustain locomotor activity. This observation has two implications: (1) it provides additional support for the idea that the role of dopamine in regulating the ventral striatal neurons that subserve locomotion may be indirect and secondary to glutamatergic involvement (Druhan et al., 1996; Waters et al., 1996); and (2) assuming that rodent hyperlocomotion may reveal a propensity to elicit psychosis in man, the dissociation of dopaminergic and locomotor activation after PCP is in agreement with case reports and unpublished clinical trials indicating that dopamine antagonists do not improve the symptoms of psychosis produced by PCP (Perry, 1975;Burns and Lerner, 1976) or ketamine (A. K. Malhotra, personal communication).
Conclusions
The present findings demonstrate a temporal discordance between increased dopamine release in the prefrontal cortex and nucleus accumbens and two behavioral effects of PCP that previously were thought to be associated with dopaminergic activation in these regions. Specifically, <2 hr after animals received PCP, dopamine and glutamate levels remained elevated well above baseline in the nucleus accumbens and prefrontal cortex, whereas hyperlocomotion and impaired performance in a discrete delayed alternation task, a rodent working memory task, subsided. While functional expression of activated glutamate neurotransmission is expected to diminish rapidly because of desensitization of non-NMDA receptors, dopamine receptors generally do not undergo rapid desensitization. Hence, the present findings indicate that increases in synaptic availability of dopamine is not sufficient to sustain the hyperlocomotion or cognitive deficits produced by PCP and that nondopaminergic mechanisms are necessary for expression of these behavioral disruptions. These findings further suggest that targeting receptors other than dopamine receptors, in particular metabotropic glutamate receptors that may normalize the glutamatergic activation produced by PCP (and ketamine); may be a more effective strategy to reduce psychotomimetic and mnemonic effects of NMDA receptor antagonists.
Footnotes
This work was supported by National Institute of Mental Health Grants MH48404 and MH44866 and the Veterans Administration Center for Schizophrenia.
Correspondence should be addressed to Dr. Bita Moghaddam, Department of Psychiatry, Yale University School of Medicine, Veterans Administration Medical Center 116A/2, West Haven, CT 06516.
REFERENCES
- 1.Bakker CB, Amini FB. Observations on the psychotomimetic effects of sernyl. Compr Psychiatry. 1961;2:269–280. doi: 10.1016/s0010-440x(61)80033-3. [DOI] [PubMed] [Google Scholar]
- 2.Bakshi VP, Swerdlow NR, Geyer MA. Clozapine antagonizes phencyclidine-induced deficits in sensorimotor gating of the startle response. J Pharmacol Exp Ther. 1994;271:787–794. [PubMed] [Google Scholar]
- 3.Breier A, Su TP, Saunders R, Carson RE, Kolachana BS, de Bartolomeis A, Weinberger DR, Weisenfeld N, Malhorta AK, Eckelman WC, Pickar D. Schizophrenia is associated with elevated amphetamine-induced synaptic dopamine concentrations: evidence from a novel positron emission tomography method. Proc Natl Acad Sci USA. 1977;94:2569–2574. doi: 10.1073/pnas.94.6.2569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Breier A, Adler C, Weisenfeld NI, Elman I, Picken L, Malhotra AK, Pickar D. Effects of NMDA antagonism on striatal dopamine release in healthy subjects: application of a novel PET approach. Synapse. 1998;29:142–147. doi: 10.1002/(SICI)1098-2396(199806)29:2<142::AID-SYN5>3.0.CO;2-7. [DOI] [PubMed] [Google Scholar]
- 5.Bristow LJ, Hutson PH, Thorn L, Tricklebank MD. The glycine/NMDA receptor antagonist, R-(+)-HA-966, blocks activation of the mesolimbic dopaminergic system induced by phencyclidine and dizocilpine (MK-801) in rodents. Br J Pharmacol. 1993;108:1156–1163. doi: 10.1111/j.1476-5381.1993.tb13520.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bubser M, Schmidt WJ. 6-Hydroxydopamine lesion of the rat prefrontal cortex increases locomotor activity, impairs acquisition of delayed alternation tasks, but does not affect uninterrupted tasks in the radial maze. Behav Brain Res. 1990;37:157–168. doi: 10.1016/0166-4328(90)90091-r. [DOI] [PubMed] [Google Scholar]
- 7.Bubser M, Tzschentke T, Hauber W. Behavioral and neurochemical interactions of the AMPA antagonist GYKI 52466 and the non-competitive NMDA antagonist dizocilpine in rats. J Neural Transm Gen Sect. 1995;101:115–126. doi: 10.1007/BF01271550. [DOI] [PubMed] [Google Scholar]
- 8.Burns RS, Lerner LS. Perspectives: acute phencyclidine intoxication. Clin Toxicol. 1976;9:477–501. doi: 10.3109/15563657608988151. [DOI] [PubMed] [Google Scholar]
- 9.Byrd LD, Standish LJ, Howell LL. Behavioral effects of phencyclidine and ketamine alone and in combination with other drugs. Eur J Pharmacol. 1987;144:331–341. doi: 10.1016/0014-2999(87)90386-4. [DOI] [PubMed] [Google Scholar]
- 10.Carboni E, Imperato A, Perezzani L, Di Chiara G. Amphetamine, cocaine, phencyclidine and nomifensine increase extracellular dopamine concentrations preferentially in the nucleus accumbens of freely moving rats. Neuroscience. 1989;28:653–661. doi: 10.1016/0306-4522(89)90012-2. [DOI] [PubMed] [Google Scholar]
- 11.Carlezon WA, Jr, Wise RA. Rewarding actions of phencyclidine and related drugs in nucleus accumbens shell and frontal cortex. J Neurosci. 1996;16:3112–3122. doi: 10.1523/JNEUROSCI.16-09-03112.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Carlsson A, Svensson A, Carlsson ML. Future strategies in the discovery of new antipsychotic agents: focus on dopamine-glutamate interactions. In: Brunello N, Mendlewicz J, Racagni G, editors. New generation of antipsychotic drugs: novel mechanisms of action, Vol 4. Basel; Basel: 1993. pp. 118–129. [Google Scholar]
- 13.Carlsson M, Carlsson A. The NMDA antagonist MK-801 causes marked locomotor stimulation in monamine-depleted mice. J Neural Transm. 1989;75:221–226. doi: 10.1007/BF01258633. [DOI] [PubMed] [Google Scholar]
- 14.Castellani S, Adams PM. Effects of dopaminergic drugs on phencyclidine-induced behavior in the rat. Neuropharmacology. 1981;20:371–374. doi: 10.1016/0028-3908(81)90011-3. [DOI] [PubMed] [Google Scholar]
- 15.Davies BM, Beech DR. The effect of the 1-arylcyclohexylamine (sernyl) on twelve normal volunteers. J Ment Sci. 1960;106:912–924. doi: 10.1192/bjp.106.444.912. [DOI] [PubMed] [Google Scholar]
- 16.Deutch AY, Tam S-Y, Freeman AS, Bowers MB, Jr, Roth RH. Mesolimbic and mesocortical dopamine activation induced by phencyclidine: contrasting pattern to striatal response. Eur J Pharmacol. 1987;134:257–264. doi: 10.1016/0014-2999(87)90356-6. [DOI] [PubMed] [Google Scholar]
- 17.Doherty JD, Simonovic M, So R, Meltzer HY. The effect of phencyclidine on dopamine synthesis and metabolism in rat striatum. Eur J Pharmacol. 1980;65:139–149. doi: 10.1016/0014-2999(80)90386-6. [DOI] [PubMed] [Google Scholar]
- 18.Druhan JP, Rajabi H, Stewart J. MK-801 increases locomotor activity without elevating extracellular dopamine levels in the nucleus accumbens. Synapse. 1996;24:135–146. doi: 10.1002/(SICI)1098-2396(199610)24:2<135::AID-SYN5>3.0.CO;2-G. [DOI] [PubMed] [Google Scholar]
- 19.Filtz TM, Artymyshyn RP, Guan W, Molinoff PB. Paradoxical regulation of dopamine receptors in transfected 293 cells. Mol Pharmacol. 1993;44:371–379. [PubMed] [Google Scholar]
- 20.Freed WJ, Weinberger DR, Bing LA, Wyatt RJ. Neuropharmacological studies of phencyclidine (PCP)-induced behavioral stimulation in mice. Psychopharmacology. 1980;71:291–297. doi: 10.1007/BF00433064. [DOI] [PubMed] [Google Scholar]
- 21.Freeman J, Stanton M. Medial prefrontal cortex lesions and spatial delayed alternation in the developing rat: recovery or sparing? Behav Neurosci. 1992;106:924–932. doi: 10.1037//0735-7044.106.6.924. [DOI] [PubMed] [Google Scholar]
- 22.French ED. Effects of n-allyl normetazocine (SKF 10,047), phencyclidine and other psychomotor stimulants in the rat following 6-hydroxydopamine lesion of the ventral tegmental area. Neuropharmacology. 1986;25:447–450. doi: 10.1016/0028-3908(86)90241-8. [DOI] [PubMed] [Google Scholar]
- 23.French ED. Phencyclidine and the midbrain dopamine system: electrophysiology and behavior. Neurotoxicol Teratol. 1994;4:355–362. doi: 10.1016/0892-0362(94)90023-x. [DOI] [PubMed] [Google Scholar]
- 24.Gleason SD, Shannon HE. Blockade of phencyclidine-induced hyperlocomotion by olanzapine, clozapine and serotonin receptor subtype selective antagonists in mice. Psychopharmacology. 1997;129:79–84. doi: 10.1007/s002130050165. [DOI] [PubMed] [Google Scholar]
- 25.Goldman-Rakic PS. Memory: recording experience in cells and circuits: diversity in memory research. Proc Natl Acad Sci USA. 1996;93:13435–13447. doi: 10.1073/pnas.93.24.13435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Greenburg BD, Segal DS. Acute and chronic behavioral interactions between phencyclidine (PCP) and amphetamine: evidence for a dopaminergic role in some PCP-induced behaviors. Pharmacol Biochem Behav. 1985;23:99–105. doi: 10.1016/0091-3057(85)90137-6. [DOI] [PubMed] [Google Scholar]
- 27.Handelmann GE, Contreras PC, O’Donohue TL. Selective memory impairment by phencyclidine in rats. Eur J Pharmacol. 1987;140:69–73. doi: 10.1016/0014-2999(87)90635-2. [DOI] [PubMed] [Google Scholar]
- 28.Hauber W, Andersen R. The non-NMDA glutamate receptor antagonist GYKI 52466 counteracts locomotor stimulation and anticataleptic activity induced by the NMDA antagonist dizocilpine. Naunyn-Schmiedeberg’s Arch Pharmacol. 1993;348:486–490. doi: 10.1007/BF00173207. [DOI] [PubMed] [Google Scholar]
- 29.Hertel P, Mathe JM, Nomikos GG, Iurlo M, Mathe AA, Svensson TH. Effects of D-amphetamine and phencyclidine on behavior and extracellular concentrations of neurotensin and dopamine in the ventral striatum and the medial prefrontal cortex of the rat. Behav Brain Res. 1996;72:103–114. doi: 10.1016/0166-4328(96)00138-6. [DOI] [PubMed] [Google Scholar]
- 30.Hondo H, Yonezawa Y, Nakahara T, Nakamura K, Hirano M, Uchimura H, Tashiro N. Effects of phencyclidine on dopamine release in the rat prefrontal cortex: an in vivo microdialysis study. Brain Res. 1994;633:337–342. doi: 10.1016/0006-8993(94)91558-x. [DOI] [PubMed] [Google Scholar]
- 31.Hudzik TJ, Wenger GR. Effects of drugs of abuse and cholinergic agents on delayed matching-to-sample responding in the squirrel monkey. J Pharmacol Exp Ther. 1993;265:120–127. [PubMed] [Google Scholar]
- 32.Hurd YL, Ungerstedt U. Cocaine: an in vivo microdialysis evaluation of its acute action on dopamine transmission in rat striatum. Synapse. 1989;3:48–54. doi: 10.1002/syn.890030107. [DOI] [PubMed] [Google Scholar]
- 33.Hutson PH, Hogg JE. Effects of and interactions between antagonists for different sites on the NMDA receptor complex on hippocampal and striatal acetylcholine efflux in vivo. Eur J Pharmacol. 1996;295:45–52. doi: 10.1016/0014-2999(95)00634-6. [DOI] [PubMed] [Google Scholar]
- 34.Iversen SD. Interactions between excitatory amino acids and dopamine systems in the forebrain: implications for schizophrenia and Parkinson’s disease. Behav Pharmacol. 1995;6:478–491. [PubMed] [Google Scholar]
- 35.Jackson DM, Johansson C, Lindgren LM, Bengtsson A. Dopamine receptor antagonists block amphetamine and phencyclidine-induced motor stimulation in rats. Pharmacol Biochem Behav. 1994;48:465–471. doi: 10.1016/0091-3057(94)90554-1. [DOI] [PubMed] [Google Scholar]
- 36.Javitt DC, Zukin SR. Recent advances in the phencyclidine model of schizophrenia. Am J Psychiatry. 1991;148:1301–1308. doi: 10.1176/ajp.148.10.1301. [DOI] [PubMed] [Google Scholar]
- 37.Javitt DC, Steinschneider M, Schroeder CE, Arezzo JC. Role of cortical N-methyl-d-aspartate receptors in auditory sensory memory and mismatch negativity generation: implications for schizophrenia. Proc Natl Acad Sci USA. 1996;93:11962–11967. doi: 10.1073/pnas.93.21.11962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Jedema HP, Moghaddam B. Characterization of excitatory amino acid modulation of dopamine release in the prefrontal cortex of conscious rats. J Neurochem. 1996;66:1448–1453. doi: 10.1046/j.1471-4159.1996.66041448.x. [DOI] [PubMed] [Google Scholar]
- 39.Jentsch JD, Redmond DE, Jr, Elsworth JD, Taylor JR, Youngren KD, Roth RH. Enduring cognitive deficits and cortical dopamine dysfunction in monkeys after long-term administration of phencyclidine. Science. 1997a;277:953–955. doi: 10.1126/science.277.5328.953. [DOI] [PubMed] [Google Scholar]
- 40.Jentsch JD, Tran A, Le D, Youngren KD, Roth RH. Subchronic phencyclidine administration reduces mesoprefrontal dopamine utilization and impairs prefrontal cortical-dependent cognition in the rat. Neuropsychopharmacology. 1997b;17:92–99. doi: 10.1016/S0893-133X(97)00034-1. [DOI] [PubMed] [Google Scholar]
- 41.Johnson KM, Jones SM. Neuropharmacology of phencyclidine: basic mechanisms and therapeutic potential. Annu Rev Pharmacol Toxicol. 1990;30:707–750. doi: 10.1146/annurev.pa.30.040190.003423. [DOI] [PubMed] [Google Scholar]
- 42.Jones KA, Wilding TJ, Huettner JE, Costa AM. Desensitization of kainate receptors by kainate, glutamate and diastereomers of 4-methylglutamate. Neuropharmacology. 1997;36:853–863. doi: 10.1016/s0028-3908(97)00066-x. [DOI] [PubMed] [Google Scholar]
- 43.Karreman M, Westerink BHC, Moghaddam B. Excitatory amino acid receptors in the ventral tegmental area regulate dopamine release in the ventral striatum. J Neurochem. 1996;67:601–607. doi: 10.1046/j.1471-4159.1996.67020601.x. [DOI] [PubMed] [Google Scholar]
- 44.Keith VA, Mansbach RS, Geyer MA. Failure of haloperidol to block the effects of phencyclidine and dizocilpine on prepulse inhibition of startle. Biol Psychiatry. 1991;30:557–566. doi: 10.1016/0006-3223(91)90025-h. [DOI] [PubMed] [Google Scholar]
- 45.Kelley AE, Delfs JM. Excitatory amino acid receptors mediate orafacial stereotypy elicited by dopaminergic stimulation of the ventrolateral striatum. Neuroscience. 1994;60:85–95. doi: 10.1016/0306-4522(94)90205-4. [DOI] [PubMed] [Google Scholar]
- 46.Kelly PH, Iversen SD. Selective 6-OHDA-induced destruction of mesolimbic dopamine neurons: abolition of psychostimulant-induced locomotor activity in rats. Eur J Pharmacol. 1976;40:45–56. doi: 10.1016/0014-2999(76)90352-6. [DOI] [PubMed] [Google Scholar]
- 47.Kiskin NI, Krishtal OA, Tsyndrenko AY. Excitatory amino acid receptors in hippocampal neurons: kainate fails to desensitize them. Neurosci Lett. 1986;63:225–230. doi: 10.1016/0304-3940(86)90360-5. [DOI] [PubMed] [Google Scholar]
- 48.Kitaichi K, Yamada K, Hasegawa T, Furukawa H, Nabeshima T. Effects of risperidone on phencyclidine-induced behaviors: comparison with haloperidol and ritanserin. Jpn J Pharmacol. 1994;66:181–189. doi: 10.1254/jjp.66.181. [DOI] [PubMed] [Google Scholar]
- 49.Koob GF, Riley SJ, Smith SC, Robbins TW. Effects of 6-hydroxydopamine lesions of the nucleus accumbens septi and olfactory tubercle on feeding, locomotor activity, and amphetamine anorexia in the rat. J Comp Physiol Psychol. 1978;92:917–927. doi: 10.1037/h0077542. [DOI] [PubMed] [Google Scholar]
- 50.Kornhuber J, Riederer P, Beckmann H. Neuropsychopharmacology, pp 714–720. Springer; Berlin: 1990. The dopaminergic and glutamatergic systems in schizophrenia. [Google Scholar]
- 51.Krystal JH, Karper LP, Seibyl JP, Freeman GK, Delaney R, Bremner JD, Heninger GR, Bowers MB, Jr, Charney DS. Subanesthetic effects of the noncompetitive NMDA antagonist, ketamine, in humans. Arch Gen Psychiatry. 1994;51:199–214. doi: 10.1001/archpsyc.1994.03950030035004. [DOI] [PubMed] [Google Scholar]
- 52.Krystal J, Karper L, Bennett A, Abi-Saab D, Souza C, Abi-Dargham A, Charney D. Modulating ketamine-induced thought disorder with lorazepam and haloperidol in humans [abstract]. Schizophr Res. 1995;15:156. [Google Scholar]
- 53.Kuczenski R, Segal DS. Concomitant characterization of behavioral and striatal neurotransmitter response to amphetamine using in vivo microdialysis. J Neurosci. 1989;9:2051–2065. doi: 10.1523/JNEUROSCI.09-06-02051.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Kuczenski R, Melega WP, Cho AK, Segal DS. Extracellular dopamine and amphetamine after systemic amphetamine administration: comparison to the behavioral response. J Pharm Exp Ther. 1997;282:591–596. [PubMed] [Google Scholar]
- 55.Laruelle M, Abi-Dargham A, van Dyck C, Gil R, D’Souza CD, Erdos J, McCane E, Rosenblatt W, Fingado C, Zoghbi S, Baldwin RM, Seibyl JP, Krystal JH, Charney DS, Innis RB. SPECT imaging of amphetamine-induced dopamine release in drug-free schizophrenic subjects. Proc Natl Acad Sci USA. 1996;93:9235–9240. doi: 10.1073/pnas.93.17.9235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Lindefors N, Barati S, O’Connor WT. Differential effects of single and repeated ketamine administration on dopamine, serotonin, and GABA transmission in rat medial prefrontal cortex. Brain Res. 1997;759:205–212. doi: 10.1016/s0006-8993(97)00255-2. [DOI] [PubMed] [Google Scholar]
- 57.Liu J, Moghaddam B. Regulation of glutamate efflux by excitatory amino acid receptors: evidence for tonic inhibitory and phasic excitatory regulation. J Pharm Exp Ther. 1995;274:1209–1215. [PubMed] [Google Scholar]
- 58.Luby ED, Cohen BD, Rosenbaum G, Gottlieb JS, Kelley R. Study of a new schizophrenomimetic drug—sernyl. Am Med Assoc Arch Neurol Psychiatry. 1959;81:363–369. doi: 10.1001/archneurpsyc.1959.02340150095011. [DOI] [PubMed] [Google Scholar]
- 59.Malhotra AK, Pinals DA, Weingartner H, Sirocco K, Missar CD, Pickar D, Breier A. NMDA receptor function and human cognition: the effects of ketamine in healthy volunteers. Neuropsychopharmacology. 1996;14:301–307. doi: 10.1016/0893-133X(95)00137-3. [DOI] [PubMed] [Google Scholar]
- 60.McCullough LD, Salamone JD. Anxiogenic drugs beta-CCE and FG 7142 increase extracellular dopamine levels in nucleus accumbens. Psychopharmacology. 1992;109:379–382. doi: 10.1007/BF02245888. [DOI] [PubMed] [Google Scholar]
- 61.Moghaddam B, Bunney BS. Differential effect of cocaine on extracellular dopamine levels in rat medial prefrontal cortex and nucleus accumbens: comparison to amphetamine. Synapse. 1989;4:156–161. doi: 10.1002/syn.890040209. [DOI] [PubMed] [Google Scholar]
- 62.Moghaddam B, Adams B, Verma A, Daly D. Activation of glutamatergic neurotransmission by ketamine: a novel step in the pathway from NMDA receptor blockade to dopaminergic and cognitive disruptions associated with the prefrontal cortex. J Neurosci. 1997;17:2921–2927. doi: 10.1523/JNEUROSCI.17-08-02921.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Ng GY, Varghese G, Chung HT, Trogadis J, Seeman P, O’Dowd BF, George SR. Resistance of the dopamine D2L receptor to desensitization accompanies the up-regulation of receptors on the surface of Sf9 cells. Endocrinology. 1997;138:4199–4206. doi: 10.1210/endo.138.10.5433. [DOI] [PubMed] [Google Scholar]
- 64.Nishijima K, Kashiwa A, Hashimoto A, Iwama H, Umino A, Nishikawa T. Differential effects of phencyclidine and methamphetamine on dopamine metabolism in rat frontal cortex and striatum as revealed by in vivo dialysis. Synapse. 1996;22:304–312. doi: 10.1002/(SICI)1098-2396(199604)22:4<304::AID-SYN2>3.0.CO;2-F. [DOI] [PubMed] [Google Scholar]
- 65.Ogawa SI, Okuyama S, Araki H, Otomo S. Effect of NE-100, a novel ς receptor ligand, on phencyclidine-induced cognitive dysfunction. Eur J Pharmacol. 1994;263:9–15. doi: 10.1016/0014-2999(94)90517-7. [DOI] [PubMed] [Google Scholar]
- 66.Ogren SO, Goldstein M. Phencyclidine and dizocilpine-induced hyperlocomotion are differentially mediated. Neuropsychopharmacology. 1994;11:167–177. doi: 10.1038/sj.npp.1380103. [DOI] [PubMed] [Google Scholar]
- 67.Olney JW, Farber NB. Glutamate receptor dysfunction and schizophrenia. Arch Gen Psychiatry. 1995;52:998–1007. doi: 10.1001/archpsyc.1995.03950240016004. [DOI] [PubMed] [Google Scholar]
- 68.Otis T, Zhang S, Trussell LO. Direct measurement of AMPA receptor desensitization induced by glutamatergic synaptic transmission. J Neurosci. 1996;16:7496–7504. doi: 10.1523/JNEUROSCI.16-23-07496.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Ouagazzal A, Nieoullon A, Amalric M. Locomotor activation induced by MK-801 in the rat: postsynaptic interactions with dopamine receptors in the ventral striatum. Eur J Pharmacol. 1994;251:229–236. doi: 10.1016/0014-2999(94)90404-9. [DOI] [PubMed] [Google Scholar]
- 70.Paxinos G, Watson C. The rat brain in stereotaxic coordinates. Academic; New York: 1982. [DOI] [PubMed] [Google Scholar]
- 71.Perry DC. Pharm Chem Newsletter. 1975;4:9. [Google Scholar]
- 72.Sams-Dodd R. Phencyclidine-induced stereotyped behaviour and social isolation in the rat: a possible animal model of schizophrenia. Behav Pharmacol. 1996;7:3–23. [PubMed] [Google Scholar]
- 73.Schlemmer RF, Jr, Jackson JA, Preston KL, Bederka JP, Jr, Garver DL, Davis JM. Phencyclidine-induced stereotyped behavior in monkeys: antagonism by pimozide. Eur J Pharmacol. 1978;52:379–384. doi: 10.1016/0014-2999(78)90293-5. [DOI] [PubMed] [Google Scholar]
- 74.Sesack SR, Hawrylak VA, Guido MA, Levey AI. Dopamine axon varicosities in the rat prefrontal cortex exhibit sparse immunoreactivity for the dopamine transporter. J Neurosci. 1998;18:2697–2708. doi: 10.1523/JNEUROSCI.18-07-02697.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Sharp JW, Petersen DL, Langford MT. DNQX inhibits phencyclidine (PCP) and ketamine induction of the hsp70 heat shock gene in the rat cingulate and retrosplenial cortex. Brain Res. 1995;687:114–124. doi: 10.1016/0006-8993(95)00477-8. [DOI] [PubMed] [Google Scholar]
- 76.Simon H. Dopaminergic A10 neurons and frontal system. J Physiol (Lond) 1981;77:81–95. [PubMed] [Google Scholar]
- 77.Smith GS, Schloesser R, Brodie JD, Dewey SL, Logan J, Vitkun SA, Simkowitz P, Hurley A, Cooper R, Volkow ND, Cancro R. Glutamate modulation of dopamine measured in vivo with positron emmission tomography (PET) and 11C-raclopride in normal human subjects. Neuropsychopharmacology. 1998;18:18–25. doi: 10.1016/S0893-133X(97)00092-4. [DOI] [PubMed] [Google Scholar]
- 78.Steinpreis R. The behavioral and neurochemical effects of phencyclidine in humans and animals: some implications for modeling psychosis. Behav Brain Res. 1996;74:45–55. doi: 10.1016/0166-4328(95)00162-x. [DOI] [PubMed] [Google Scholar]
- 79.Steinpreis RE, Salamone JD. The role of the nucleus accumbens dopamine in the neurochemical and behavioral effects of phencyclidine: a microdialysis and behavioral study. Brain Res. 1993;612:263–270. doi: 10.1016/0006-8993(93)91671-e. [DOI] [PubMed] [Google Scholar]
- 80.Steinpreis RE, Sokolowski JD, Papanikolaou A, Salamone JD. The effects of haloperidol and clozapine on PCP and amphetamine induced suppression of social behavior. Pharmacol Biochem Behav. 1994;47:579–585. doi: 10.1016/0091-3057(94)90162-7. [DOI] [PubMed] [Google Scholar]
- 81.Sturgeon RD, Fessler RG, Meltzer HY. Behavioral rating scales for assessing phencyclidine-induced locomotor activity, stereotyped behavior and ataxia in rats. Eur J Pharmacol. 1979;59:169–179. doi: 10.1016/0014-2999(79)90279-6. [DOI] [PubMed] [Google Scholar]
- 82.Takahata R, Moghaddam B (1998) Glutamatergic regulation of basal and stimulus-activated dopamine release in the prefrontal cortex. J Neurochem, in press. [DOI] [PubMed]
- 83.Timmerman W, Westerink BHC. Brain microdialysis of GABA and glutamate: what does it signify? Synapse. 1997;27:242–261. doi: 10.1002/(SICI)1098-2396(199711)27:3<242::AID-SYN9>3.0.CO;2-D. [DOI] [PubMed] [Google Scholar]
- 84.Verma A, Moghaddam B. NMDA receptor antagonists impair prefrontal cortex function as assessed via spatial delayed alternation performance in rats: modulation by dopamine. J Neurosci. 1996;16:373–379. doi: 10.1523/JNEUROSCI.16-01-00373.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Waters N, Lundgren C, Hansson LO, Carlsson ML. Concurrent locomotor stimulation and decrease in dopamine release in rats and mice after treatment with the competitive NMDA receptor antagonists D-CPPene and CGS 19755. J Neural Transm. 1996;103:117–129. doi: 10.1007/BF01292621. [DOI] [PubMed] [Google Scholar]
- 86.Wedzony K, Golembiowska K, Zazula M. Differential effects of CGP 37849 and MK-801, competitive and noncompetitive NMDA antagonists, with respect to the modulation of sensorimotor gating and dopamine outflow in the prefrontal cortex of rats. Naunyn-Schmiedeberg’s Arch Pharmacol. 1994;350:555–562. doi: 10.1007/BF00173026. [DOI] [PubMed] [Google Scholar]
- 87.Wesierska M, Macias-Gonzalez R, Bures J. Differential effect of ketamine on the reference and working memory versions of the Morris water maze task. Behav Neurosci. 1990;104:74–83. doi: 10.1037//0735-7044.104.1.74. [DOI] [PubMed] [Google Scholar]
- 88.Westerink BHC, Kwint HF, deVries JB. The pharmacology of mesolimbic dopamine neurons: a dual-probe microdialysis study in the ventral tegmental area and nucleus accumbens of the rat brain. J Neurosci. 1996;16:2605–2611. doi: 10.1523/JNEUROSCI.16-08-02605.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.White F. Synaptic regulation of mesocorticolimbic dopamine neurons. Annu Rev Neurosci. 1996;19:405–436. doi: 10.1146/annurev.ne.19.030196.002201. [DOI] [PubMed] [Google Scholar]
- 90.Wilding TJ, Huettner JE. Activation and desensitization of hippocampal kainate receptors. J Neurosci. 1997;17:2713–2721. doi: 10.1523/JNEUROSCI.17-08-02713.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Williams SM, Goldman-Rakic PS. Characterization of the dopaminergic innervation of the primate frontal cortex using a dopamine-specific antibody. Cereb Cortex. 1993;3:199–222. doi: 10.1093/cercor/3.3.199. [DOI] [PubMed] [Google Scholar]
- 92.Willins DL, Narayanan S, Wallace LJ, Uretsky NJ. The role of dopamine and AMPA/kainate receptors in the nucleus accumbens in the hypermotility response to MK801. Pharmacol Biochem Behav. 1993;46:881–887. doi: 10.1016/0091-3057(93)90217-h. [DOI] [PubMed] [Google Scholar]
- 93.Wilson FAW, O’Scalaidhe SP, Goldman-Rakic PS. Functional synergism between putative γ-aminobutyrate-containing neurons and pyramidal neurons in prefrontal cortex. Proc Natl Acad Sci USA. 1994;91:4009–4013. doi: 10.1073/pnas.91.9.4009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Zahrt J, Taylor JR, Mathew RG, Arnsten AFT. Supranormal stimulation of D1 dopamine receptors in the rodent prefrontal cortex impairs spatial working memory performance. J Neurosci. 1997;17:8528–8535. doi: 10.1523/JNEUROSCI.17-21-08528.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]