The role of evolution in the emergence of infectious diseases (original) (raw)
Abstract
It is unclear when, where and how novel pathogens such as human immunodeficiency virus (HIV), monkeypox and severe acute respiratory syndrome (SARS) will cross the barriers that separate their natural reservoirs from human populations and ignite the epidemic spread of novel infectious diseases. New pathogens are believed to emerge from animal reservoirs when ecological changes increase the pathogen's opportunities to enter the human population1 and to generate subsequent human-to-human transmission2. Effective human-to-human transmission requires that the pathogen's basic reproductive number, _R_0, should exceed one, where _R_0 is the average number of secondary infections arising from one infected individual in a completely susceptible population3. However, an increase in _R_0, even when insufficient to generate an epidemic, nonetheless increases the number of subsequently infected individuals. Here we show that, as a consequence of this, the probability of pathogen evolution to _R_0 > 1 and subsequent disease emergence can increase markedly.
Supplementary information
The online version of this article (doi:10.1038/nature02104) contains supplementary material, which is available to authorized users.
Main
The emergence of a disease combines two elements: the introduction of the pathogen into the human population and its subsequent spread and maintenance within the population. Ecological factors such as human behaviour can influence both of these elements, and consequently ecology has been recognized to have an important role in the emergence of disease1,2,4. In contrast, evolutionary factors including the adaptation of the pathogen to growth within humans and the subsequent transmission of the pathogen between humans are mostly considered in terms of changes in the virulence of the pathogen, and are often thought to have a lesser role in the initial emergence of pathogens4. One exception5 suggests that immunocompromised individuals might provide “stepping stones” for the evolution of pathogens.
The successful emergence of a pathogen requires _R_0 to exceed one in the new host. Only then can an introduction trigger emergence2. (Here we use _R_0 to refer to spread in human populations, not in the natural reservoir.) If _R_0 for a potential pathogen exceeds one, this scenario represents an epidemic waiting to happen. By contrast, when _R_0 is initially less than one, infections will inevitably die out and there will be no epidemic unless genetic or ecological changes drive _R_0 above one.
There are a number of ways in which _R_0 can increase. Ecological changes such as changes in host density or behaviour can increase _R_0, as can genetic changes in the pathogen population or in the population of its new host. Genetic changes in the pathogen can arise either through ‘coincidental’ processes such as neutral drift or coevolution of the pathogen and its reservoir host, or through adaptive evolution of the pathogen during chains of transmission in humans. Genetic changes of the new host might be more likely for domesticated or endangered species than for humans.
Here we show that factors, such as ecological changes, that increase the _R_0 value of the potential pathogen to a level not sufficient to cause an epidemic (that is, _R_0 remains less than one) can greatly increase the length of the stochastic chains of disease transmission. These long transmission chains provide an opportunity for the pathogen to adapt to human hosts, and thus for the disease to emerge.
Our model is illustrated in Fig. 1. Introductions occur stochastically from the natural reservoir of the pathogen, and each primary case is followed by stochastic transmission that generates a variable number of subsequent infections in the human population. We assume that the number of secondary cases follows a Poisson distribution with a mean equal to _R_0. Each introduction thus forms branched chains of transmission, which stutter to extinction if _R_0 < 1, and the pathogen cannot evolve (µ = 0). The probability that the pathogen evolves and a secondary infection is caused by the mutant is equal to µ for each of the secondary infections (we note that µ incorporates not only the pathogen's mutation rate, but also its dynamics within the host and its transmissibility). We use multi-type branching processes6,7,8,9 to describe the initial spread of the infection, incorporating the evolution of the pathogen.
Figure 1. Schematic for the emergence of an infectious disease.
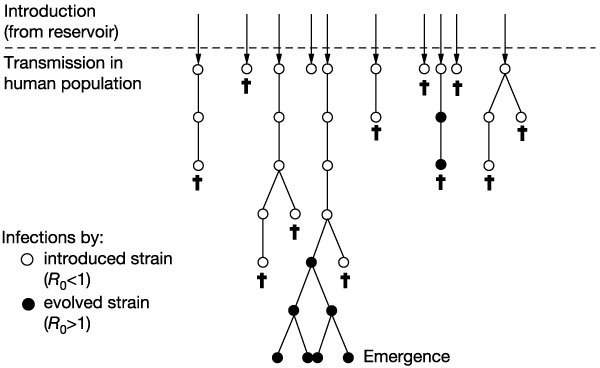
Introductions from the reservoir are followed by chains of transmission in the human population. Infections with the introduced strain (open circles) have a basic reproductive number _R_0 < 1. Pathogen evolution generates an evolved strain (filled circles) with _R_0 > 1. The infections caused by the evolved strain can go on to cause an epidemic. Daggers indicate no further transmission.
In the simplest case (see Fig. 2a) only one mutation is required for the _R_0 of the evolved pathogen to exceed one. The probability that a single introduction evolves, causing one (or more) infections with the evolved pathogen (a filled circle in Fig. 1) before it goes extinct, depends very strongly on the _R_0 of the introduced pathogen, particularly for low µ values as _R_0 approaches 1. This probability is approximately linearly dependent on the rate of evolution µ.
Figure 2. One-step evolution. A single change is required for the pathogen to evolve to _R_0 > 1.
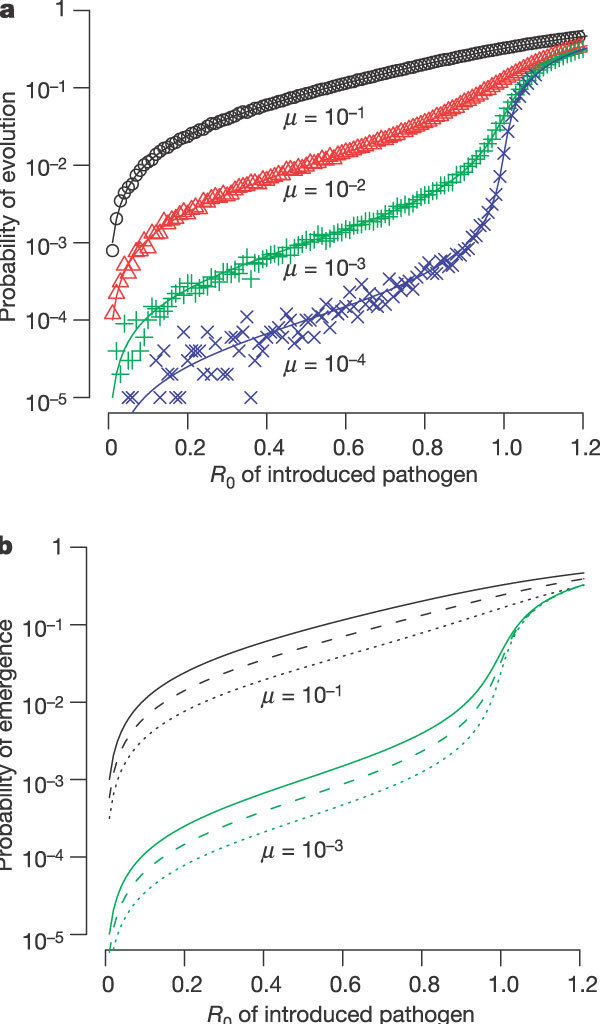
a, The probability that an introduction leads to an infection with an evolved strain of the pathogen (filled circle in Fig. 1) is highly sensitive to _R_0, and is approximately linearly dependent on the mutation rate µ. Lines correspond to numerical solutions to the branching process model (see Supplementary Information) and symbols correspond to Monte-Carlo simulations following 105 introductions. b, The probability of emergence per introduction depends on the _R_0 value of the introduced pathogen and of the evolved pathogen. The solid, dashed and dotted lines correspond to the evolved pathogen having an _R_0 of 1,000, 1.5 and 1.2 respectively.
The probability that the introduction leads to an epidemic (the ‘probability of emergence’) depends on the probability of evolution and the probability that the evolved infections do not go extinct due to stochastic effects. In Fig. 2b we plot the probability of emergence for three different _R_0 values of the evolved strain. The probability of emergence approaches the probability of evolution when _R_0 of the evolved strain is large, and is lower when the _R_0 of the evolved strain is close to 1. We find that the probability of emergence depends most strongly on the _R_0 of the introduced pathogen, increases approximately linearly with the mutation rate µ, and depends only modestly on the _R_0 of the evolved pathogen.
We extend the simple one-step mutation model to consider the situation in which multiple evolutionary changes are required for the pathogen to attain _R_0 > 1 in the human population. We begin with a simple scenario, which we call the jackpot model, where the _R_0 of the pathogen with the intermediate mutations is the same as that of the introduced pathogen, and where only the addition of the final mutation results in an increase in _R_0 to greater than one. As seen in Fig. 3, increasing the number of required evolutionary steps greatly reduces the probability of emergence and increases its sensitivity to changes in _R_0. The probability of emergence is approximately proportional to the mutation rate to the power of the number of evolutionary steps required (see Supplementary Information).
Figure 3. Multiple-step evolution.
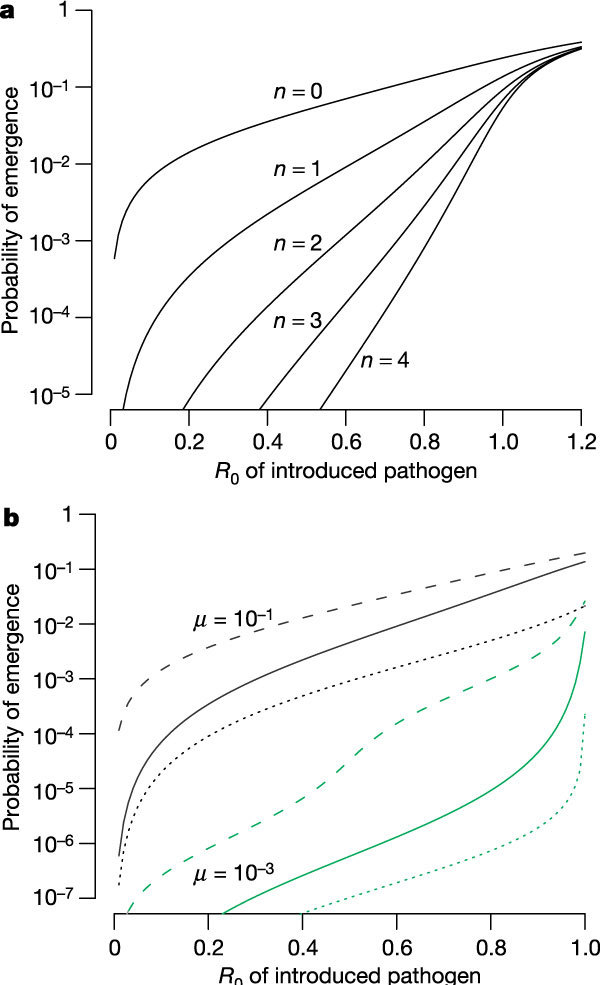
Here multiple evolutionary changes are required for evolution of the pathogen to have an _R_0 > 1. a, Jackpot model with µ = 0.1 and n intermediate changes each with _R_0 equal to that of the introduced pathogen: increasing the number of steps (n) greatly decreases the probability of evolution, and makes it more sensitive to the _R_0 of the introduced pathogen. b, Alternative multi-step models for the one-intermediate (n = 1) case. The jackpot model (solid line), additive model (dashed line) and fitness valley model (dotted line) are shown (see text for details).
The fitness landscape on which evolution occurs is important in determining the outcome10. Figure 3b illustrates this for the case of a single intermediate type. As should be expected, changing the jackpot model to an additive model, where the fitness of the intermediate is the average of the fitness of the introduced strain and fully evolved strain, increases the probability that the pathogen evolves to _R_0 > 1, whereas changing it to a fitness valley model, where the fitness of the intermediate is lower than the fitness of the introduced strain, decreases the probability of emergence.
The key characteristic distinguishing our model from the conventional view is the _R_0 of the introduced pathogen. In the conventional view the _R_0 of these infections must be greater than one, whereas in the mechanism described here it is less than one, and evolution during the stochastic chains of transmission allows _R_0 to increase above one. In the case of human infections such as HIV1,11,12, SARS13,14,15,16,17 and (potentially) monkeypox18,19,20,21, seroprevalence studies among groups at high risk of infection from the reservoir would allow evaluation of whether crossover events are usually dead ends, as we expect in our model, or whether they are associated with a large number of secondary cases. In the case of diseases emerging into non-human populations, such as the Nipah virus, which moved from bats to pigs22,23,24, it may be possible to conduct additional tests involving controlled experimental infections to estimate the _R_0 (in this case of the bat Nipah virus in pigs) and to determine whether it evolves during the course of chains of transmission. Such studies may additionally help to identify pathogens that have an ability to evolve rapidly and thus have a high potential for emergence.
The framework presented here has special relevance for pathogens that have been driven to extinction by vaccination. In the case of smallpox there are probably reservoirs of related zoonoses (such as, but not restricted to, monkeypox) from which smallpox may have originated. Although the _R_0 of monkeypox in the human population is clearly less than one, there are occasional chains of transmission in the human population18,19,20,21. As the level of herd immunity to smallpox wanes in the absence of continued vaccination, we expect an increase in _R_0 of infections with monkeypox albeit to a level still less than one (the smallpox vaccine provides about 85% cross immunity against monkeypox). Our results suggest that this increase in the effective _R_0 of monkeypox in the human population could markedly increase the probability of evolution of monkeypox, allowing it to emerge into a successful human pathogen (which, depending on the evolutionary trajectory followed, may be similar to or differ from smallpox).
The present study could be extended in a number of directions. These include explicitly incorporating the details of ecological interactions such as heterogeneity in the transmission in different areas and subpopulations2,25,26,27 and incorporating genetic diversity of the pathogen in its reservoir. Finally, we note that this framework can be applied to the more general problem of biological invasions28,29.
Methods
We describe the dynamics and evolution of emerging diseases as a multi-type branching process with the following probability-generating functions6,7,8,9:
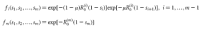
We calculated the extinction probabilities of the above process numerically. For details and definitions see Supplementary Information.
Supplementary information
Acknowledgements
We thank S. Nichol, I. Longini and D. Krakauer for discussions and comments. We acknowledge support from the NIH and DFG.
Competing interests
The authors declare that they have no competing financial interests.
References
- 1.Hahn BH, Shaw GM, De Cock KM, Sharp PM. AIDS as a zoonosis: scientific and public health implications. Science. 2000;287:607–614. doi: 10.1126/science.287.5453.607. [DOI] [PubMed] [Google Scholar]
- 2.May RM, Gupta S, McLean AR. Infectious disease dynamics: What characterizes a successful invader? Phil. Trans. R. Soc. Lond. B. 2001;356:901–910. doi: 10.1098/rstb.2001.0866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Anderson R, May R. Infectious Diseases of Humans: Dynamics and Control. 1991. [Google Scholar]
- 4.Schrag S, Wiener P. Emerging infectious disease: what are the relative roles of ecology and evolution? Trends Ecol. Evol. 1995;10:319–324. doi: 10.1016/S0169-5347(00)89118-1. [DOI] [PubMed] [Google Scholar]
- 5.Wallace B. Can “stepping stones” form stairways? Am. Nat. 1989;133:578–579. doi: 10.1086/284937. [DOI] [Google Scholar]
- 6.Athreya KB, Ney P. Branching Processes. 1972. [Google Scholar]
- 7.Demetrius L, Schuster P, Sigmund K. Polynucleotide evolution and branching processes. Bull. Math. Biol. 1985;47:239–262. doi: 10.1007/BF02460034. [DOI] [PubMed] [Google Scholar]
- 8.Stivers DN, Kimmel M, Axelrod DE. A discrete-time, multi-type generational inheritance branching process model of cell proliferation. Math. Biosci. 1996;137:25–50. doi: 10.1016/S0025-5564(96)00066-1. [DOI] [PubMed] [Google Scholar]
- 9.Wilke CO. Probability of fixation of an advantageous mutant in a viral quasispecies. Genetics. 2003;163:467–474. doi: 10.1093/genetics/163.2.467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wright S. The role of mutation, inbreeding, crossbreeding and selection in evolution. Proc. 6th Int. Cong. Genet. 1932;1:356–366. [Google Scholar]
- 11.Holmes EC. On the origin and evolution of the human immunodeficiency virus (HIV) Biol. Rev. Camb. Phil. Soc. 2001;76:239–254. doi: 10.1017/S1464793101005668. [DOI] [PubMed] [Google Scholar]
- 12.Peeters M, et al. Risk to human health from a plethora of simian immunodeficiency viruses in primate bushmeat. Emerg. Infect. Dis. 2002;8:451–457. doi: 10.3201/eid0805.010522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Guan Y, et al. Isolation and characterization of viruses related to the SARS coronavirus from animals in southern China. Science. 2003;302:276–278. doi: 10.1126/science.1087139. [DOI] [PubMed] [Google Scholar]
- 14.Cyranoski D, Abbott A. Virus detectives seek source of SARS in China's wild animals. Nature. 2003;423:467. doi: 10.1038/423467a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Enserink M. Infectious diseases. Clues to the animal origins of SARS. Science. 2003;300:1351. doi: 10.1126/science.300.5624.1351a. [DOI] [PubMed] [Google Scholar]
- 16.Riley S, et al. Transmission dynamics of the etiological agent of SARS in Hong Kong: impact of public health interventions. Science. 2003;300:1961–1966. doi: 10.1126/science.1086478. [DOI] [PubMed] [Google Scholar]
- 17.Lipsitch M, et al. Transmission dynamics and control of Severe Acute Respiratory Syndrome. Science. 2003;300:1966–1970. doi: 10.1126/science.1086616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Heymann DL, Szczeniowski M, Esteves K. Re-emergence of Monkeypox in Africa: a review of the past six years. Br. Med. Bull. 1998;54:693–702. doi: 10.1093/oxfordjournals.bmb.a011720. [DOI] [PubMed] [Google Scholar]
- 19.Fine PE, Jezek Z, Grab B, Dixon H. The transmission potential of monkeypox virus in human populations. Int. J. Epidemiol. 1988;17:643–650. doi: 10.1093/ije/17.3.643. [DOI] [PubMed] [Google Scholar]
- 20.Breman JG, Henderson DA. Poxvirus dilemmas—Monkeypox, smallpox, and biological terrorism. N. Engl. J. Med. 1998;339:556–559. doi: 10.1056/NEJM199808203390811. [DOI] [PubMed] [Google Scholar]
- 21.Hutin YJ, et al. Outbreak of human monkeypox, Democratic Republic of Congo, 1996 to 1997. Emerg. Infect. Dis. 2001;7:434–438. doi: 10.3201/eid0703.017311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Enserink M. New virus fingered in Malaysian epidemic. Science. 1999;284:407–410. doi: 10.1126/science.284.5413.407. [DOI] [PubMed] [Google Scholar]
- 23.Chua KB, et al. Nipah virus: a recently emergent deadly paramyxovirus. Science. 2000;288:1432–1435. doi: 10.1126/science.288.5470.1432. [DOI] [PubMed] [Google Scholar]
- 24.Enserink M. Emerging diseases. Malaysian researchers trace Nipah virus outbreak to bats. Science. 2000;289:518–519. doi: 10.1126/science.289.5479.518. [DOI] [PubMed] [Google Scholar]
- 25.May R, Anderson R. Parasite–host coevolution. Parasitology. 1990;100:S89–S101. doi: 10.1017/S0031182000073042. [DOI] [PubMed] [Google Scholar]
- 26.Lloyd AL, May RM. Spatial heterogeneity in epidemic models. J. Theor. Biol. 1996;179:1–11. doi: 10.1006/jtbi.1996.0042. [DOI] [PubMed] [Google Scholar]
- 27.Dobson A, Foufopoulos J. Emerging infectious pathogens of wildlife. Phil. Trans. R. Soc B. 2001;356:1001–1012. doi: 10.1098/rstb.2001.0900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Elton CS. Ecology of Invasions by Animals and Plants. 1958. [Google Scholar]
- 29.Kornberg H, Williamson M. Quantitative Biology of the Ecology of Biological Invasions. 1987. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.