Development and characterisation of neutralising monoclonal antibody to the SARS-coronavirus (original) (raw)
Abstract
There is a global need to elucidate protective antigens expressed by the SARS-coronavirus (SARS-CoV). Monoclonal antibody reagents that recognise specific antigens on SARS-CoV are needed urgently. In this report, the development and immunochemical characterisation of a panel of murine monoclonal antibodies (mAbs) against the SARS-CoV is presented, based upon their specificity, binding requirements, and biological activity. Initial screening by ELISA, using highly purified virus as the coating antigen, resulted in the selection of 103 mAbs to the SARS virus. Subsequent screening steps reduced this panel to seventeen IgG mAbs. A single mAb, F26G15, is specific for the nucleoprotein as seen in Western immunoblot while five other mAbs react with the Spike protein. Two of these Spike-specific mAbs demonstrate the ability to neutralise SARS-CoV in vitro while another four Western immunoblot-negative mAbs also neutralise the virus. The utility of these mAbs for diagnostic development is demonstrated. Antibody from convalescent SARS patients, but not normal human serum, is also shown to specifically compete off binding of mAbs to whole SARS-CoV. These studies highlight the importance of using standardised assays and reagents. These mAbs will be useful for the development of diagnostic tests, studies of SARS-CoV pathogenesis and vaccine development.
Abbreviations: mAb, monoclonal antibody; SARS-CoV, human severe acute respiratory syndrome coronavirus; SDS-PAGE, sodium dodecyl sulfate-polyacrylamide gel electrophoresis
Keywords: SARS-coronavirus, Monoclonal antibody, Neutralising, Epitope, Immunochemistry
1. Introduction
The SARS-coronavirus (SARS-CoV) is recognised as the causal agent of severe acute respiratory syndrome (SARS) in humans. This virus caused nearly 800 deaths and infected more than 8000 people in various affected countries throughout the world (Stadler et al., 2003). The SARS-coronavirus Spike protein has only 20–27% pairwise identity at the amino acid level to the Spike proteins of other previously characterised coronaviruses. Recently, the genomes of SARS-CoV isolates, implicated in the 2003 Toronto outbreak, were sequenced in their entirety (Marra et al., 2003, Rota et al., 2003). The production of mAbs to the SARS-CoV virus is critical for diagnostic development, vaccine research and studies of viral pathogenesis. Assays that detect the presence of virally encoded proteins or nucleic acids may be preferable for diagnosis of SARS infections as the development of serum antibodies in infected individuals is quite protracted (Li et al., 2003a).
Coronaviruses are enveloped, single-stranded RNA viruses that replicate in the host cell cytoplasm (Fields et al., 2001). The coronaviruses form a single genus of the family Coronaviridae and the virions are large (80–160 nm in diameter) pleomorphic, but generally spherical, particles. Virions of most coronaviruses contain three major proteins: the phosphorylated nucleocapsid protein (N); a small membrane-embedded glycoprotein (M); and a large club-shaped peplomer glycoprotein (S) which appears in EM micrographs as protruding spikes 20 nm in length. The M protein is synthesised on ribosomes bound to the endoplasmic reticulum and accumulates in the Golgi apparatus. The subcellular localisation of M protein to the Golgi is believed to influence the site of virus budding in the infected cell. The S-protein mediates many of the biological properties of the virus, including attachment to cell receptors, penetration, and cell-fusion, and it is the major target for virus-neutralising antibodies (Collins et al., 1982, Talbot et al., 1984, Wege et al., 1984, Jimenez et al., 1986, Laude et al., 1986, Godet et al., 1994). A portion of the S glycoprotein that is not incorporated into budding virions is transported to the plasma membrane of the cell where it remains bound to the cell surface (Gerna et al., 1982).
Coronaviruses infect a wide range of mammalian hosts to produce a variety of disease outcomes including respiratory disease, enteritis and encephalitis. Antigenic similarities between various coronaviruses have been demonstrated to reside in the S-protein and have been used to study the evolution of this virus family (Brian et al., 1983). For most coronaviruses causing enteric and respiratory diseases the pathophysiological events leading to clinical symptoms are due to the acute cytocidal infection of the target cells. These infections can be limited by the local immune response resulting in the production of secretory antibodies specific for the S-protein (Enjuanes et al., 1995). In contrast, many coronaviruses are maintained and spread in the population as inapparent and subclinical infections. The sequence of events leading to chronic versus acute disease is unknown but likely depends on the expression of viral genes, the functional impairment of host cells, and the interaction with the host immune response.
There is a critical need to elucidate the immunologic basis for protection against SARS-CoV infection. Recently the SARS S-protein was shown to be a functional fusogen and is about 180–200 kDa in size (Xiao et al., 2003). A host cell receptor, angiotensin-converting enzyme 2 (ACE-2) was recently identified as a functional receptor for the SARS-CoV, and mediated infection of 293T cells in vitro (Li et al., 2003b). Therefore, antibody responses to the S-protein may neutralise the infectivity of the SARS-CoV. The immunogenetics of antibody responses to protective epitopes is of particular importance and will lead to a clearer understanding of the nature of protective antibody responses to SARS. Lastly, the production of protective monoclonal antibodies may lead to the development of new recombinant therapeutic antibodies in order to provide rapid protection in SARS patients. In the present work, a description of the development of murine mAbs against the SARS-CoV involved in the Toronto SARS outbreak is presented. The mAbs were analysed for pertinent immunochemical properties and for their ability to neutralise SARS-CoV in vitro_._
2. Materials and methods
2.1. Virus antigen preparation
For preparation of partially purified whole-virus antigen, SARS-CoV was expanded after plaque purification in Vero-6 cell monolayers and partially purified through a sucrose cushion. Highly purified SARS-CoV (Tor-3 strain, isolated from a patient infected in the Toronto SARS outbreak; Krokhin et al., 2003) was prepared in the same way, except the viral particles were further purified using gradient centrifugation. Briefly, 500 ml of supernatant from SARS-CoV infected Vero-6 cells was concentrated first on top of a cushion of iodixanol in a Beckman SW32 rotor (Mississauga, ON). The virus was subsequently mixed to form a suspension of 20% iodixanol and subjected to centrifugation in a Beckman NVT 90 rotor (Mississauga, ON) for 3.5 h at 400,000×g. Fractions were collected from the bottom of the self-generated gradient, tested by Western immunoblot with SARS-CoV-infected, convalescent human patient serum, and the SARS-CoV positive fractions were pooled and dialysed against phosphate buffered saline (PBS). The dialysed virus preparation was further concentrated by ultracentifugation for 1.5 h at 150,000×g.
2.2. Immunisation of mice
Immunisation of mice was performed according to NCFAD standard operating procedures under ISO17025. Five- to six-week-old female BALB/c mice (Charles River, Wilmington, MA) were injected subcutaneously (SC) with 50 μg of beta-propiolactone-inactivated, partially purified SARS-CoV (Tor-3 strain) with an equal part of complete Freund’s adjuvant (H37-Ra, CFA) from Difco (BD, Oakville, ON) on day 1. On day 30 the mice received 50 μg of partially purified SARS-CoV S.C. in incomplete Freund’s adjuvant (IFA) in a total volume of 100 μl. On days 48 and 63, the mice received 5 μg of the same antigen in a total volume of 100 μl SC with IFA. The mice received a final booster injection with 5 μg of highly purified SARS-CoV in 200 μl PBS to the intra-peritoneal cavity 3 days prior to hybridoma fusion. Mice were euthanised by anaesthesia overdose and exsanguinated by cardiac puncture. The spleens were subsequently excised under aseptic conditions.
2.3. Preparation of infected cell lysates
Infected Vero cells were scraped off of 162 cm2 Corning tissue culture flasks (Corning, NY) and clarified by centrifugation. A borate saline mixture (0.05 M boric acid, 0.12 M, NaCl, 0.024 M NaOH) was used to wash the cell pellet twice and the pellet was resuspended in 2 ml borate saline +1% Triton X-100 for each T162 flask. The pellet was kept at 4 °C using a water bath and sonicated for ten minutes at 50% power. The debris was pelleted via centrifugation at 10,000×g for 10 min and the supernatant was collected and stored at −20 °C in aliquots for later use.
2.4. Generation of mAbs
Removal of mouse spleens, preparation of spleen and myeloma cells, and the fusion for hybridoma production were performed according to NCFAD standard operating procedures under ISO17025. Ampoules of the myeloma cell line P3X63Ag8.653 (ATCC, Rockville, MD) were thawed 1 week prior to fusion and grown in BD Cell Mab Quantum yield medium in the presence of 8-Azaguanine (Sigma, Oakville, ON). Cells were in log-phase growth at the time of fusion. Hybridoma fusion was performed essentially as originally described (Kohler and Milstein, 1975) with the following modifications. Briefly, spleens were harvested 3 days after a final boost with a given antigen and the splenocytes were prepared by splenic perfusion as follows. Under aseptic conditions, the spleens were perforated with a 10 cm3 syringe with a 21 gauge sterile disposable needle. The spleen cells were perfused out of the spleen with injections of serum free BD cell Mab Quantum Yield medium (BD-Pharmingen, Oakville, ON). Two identically immunised mouse spleens were used to produce these hybridoma clones. The fusion was performed using the P3X63Ag8.653 myeloma line in log-phase growth. PEG1500 (1 ml; Roche, Basel, SW) was added drop-wise over 1 min while gently tapping the tube containing the thoroughly washed myeloma-splenocyte pellet. The PEG 1500 was slowly diluted out over three minutes with serum free BD-Cell Mab Quantum Yield medium. The cells were resuspended and mixed into 90 ml of Stemcell Clonacell Medium D (HAT) (Vancouver, BC) containing 5 ml Origen hybridoma cloning factor (HCF) (IGEN, Gaithersburg, MD) and plated out according to the manufacturer’s instructions. The plates were incubated at 37 °C under a 5% CO2 overlay for 10–18 days in humidified chambers. Visible colonies were picked from the plates after approximately 2 weeks growth and placed into 96-well plates containing 150–200 μl of complete hybridoma medium supplemented with 1× hypoxanthine thymidine (Sigma, Oakville, ON), 4% HCF and 10% FBS (Wisent). Supernatants were screened 4 days later via ELISA using purified virus as antigen. Isotyping was performed using a commercial murine isotyping dipstick test (Roche, Basel, SW) according to the manufacturer’s instructions. Hybridoma culture supernatants were concentrated 5–10 fold using Amicon stirred cell nitrogen concentrators with 30 kDa cutoff Millipore (YM-30) membranes (both from Millipore, Billerica, MA).
2.5. Immunoassays
2.5.1. Enzyme linked immunosorbent assay
Hybridoma culture supernatants were assayed for binding to highly purified SARS-CoV in an ELISA assay when the cultured cells were confluent in the culture plates. The Costar 3690 96-well 1/2 well ELISA plates (Corning, NY) were coated with either bovine serum albumin (BSA) or highly purified SARS-CoV (18–37 ng/well) in PBS overnight at 4 °C and then blocked with 0.4% BSA in PBS, for 2 h at 37 °C. The supernatant (30 μl/well) was incubated neat for 1 h at 37 °C. The ELISA plates were washed 10 times with distilled water and patted dry on a paper towel. A pan-goat anti-mouse IgG-HRP antibody (Southern Biotechnology Associates, Birmingham, Alabama) was diluted to 1:2000 in 0.2% BSA in PBS, applied to the ELISA plates for 45 min at 37 °C, and then washed as described above. Positive binding was detected with commercial ABTS used according to the manufacturer’s instructions (Roche, Basel, SW). The OD was read at 405 nm at 15 and 60 min intervals after addition of the developing reagent. Mouse immune and preimmune sera were diluted 1:2000 with 2%-BSA in PBS for use as positive and negative controls, respectively, and for the establishment of the hybridoma screening assay. Competition ELISA (C-ELISA) measured the binding of murine mAbs to highly purified SARS-CoV in the presence of human serum. Infected serum was from confirmed infected patients with well-established SARS. These samples were from the initial set of patients from which the virus was isolated. Plates were coated with highly purified SARS-CoV at 18 ng per well, and normal human serum or serum from convalescent SARS patient S3, serially diluted in 2% BSA-PBS, was allowed to react on the pre-blocked plates for 30 min. mAb F26G6 (Spike specific) or F26G15 (nucleoprotein specific) were pre-diluted to a concentration that gives approximately half-maximum optical density after 1 h development in the presence of no competing serum. The diluted mAb preparations were then applied to the wells, and the incubation and development of the C-ELISA was performed as described above.
2.5.2. Western immunoblots
Whole virions and SARS-CoV-infected Vero cell lysates, at a final total protein concentration of 1 μg per lane, were boiled in SDS-loading buffer for 10 min. The samples were loaded in criterion pre-cast gels (BioRad, Mississauga, ON) and electrophoresed at 200 V for 30 min. The proteins were transferred to Immobilon nylon membranes (Millipore, Billerica, MA) for 2 h at room temperature at 100 V, or overnight at 27 V at 4 °C. Blots were blocked with 3% BSA in TBS, rinsed three times with TBS, and reacted with monoclonal antibody overnight at 4 °C. The antibody supernatants were reacted neat and the concentrated supernatants were diluted 1:50 in 0.2% BSA in PBS. Blots were washed three times with TBS-Tween-20 (0.05%) for 5 min before being incubated with secondary antibody (same as above) at 1:1000 in TBS, 0.2% BSA for 1 h. The blots were washed as above and developed using DAB insoluble substrate (Pierce, Rockford, IL).
2.5.3. Immunofluorescent staining of Vero cells infected with SARS-CoV
Monolayers of SARS-infected Vero cells were stained as follows. Glass slides were coated with infected Vero cell monolayers and fixed with acetone. The slides were irradiated with 20 kilogreys from a cobalt gamma irradiator, removed from biocontainment, and then stored at −80 °C. Dilutions of antibodies and test sera were made initially in 96-well plates (BD-Falcon, Oakville, ON) in sterile phosphate-buffered saline (pH 7.3). Samples were allowed to incubate for 45 min in a 37 °C incubator, and were washed with distilled water. Fluorescein labelled Goat anti-mouse secondary antibodies (Sigma, Oakville, ON) diluted in PBS were added to the slides and incubated for 45 min at 37 °C, washed as above, and air dried. Slides were coated with mounting medium and stored at 4 °C until examined.
2.5.4. Immuno-dotblot analysis
Immuno-dotblot analysis was performed using Immobilon nylon membranes (Millipore, Billerica, MA). A total of 15 μl of SARS-CoV antigen (infected Vero cell lysate) or 5 μg of highly purified virus is coated (per spot) for 1 h at 37 °C. In an attempt to further characterise the immunochemical properties of the individual mAbs, antigen was pre-treated in several different conditions as follows: untreated native antigen; denatured by heat treatment at 95 °C for 5 min; denatured by sodium dodecyl sulphate treatment (SDS) 2% prior to coating on the membrane; denatured by both heat treatment and SDS as above together; reduced with beta-mercaptoethanol (5%) prior to coating on the membrane; denatured by temperature as above and reduced as above, together; denatured by both heat and SDS in the presence of beta-mercaptoethanol. Antigen-coated blots were blocked with 3% BSA in TBS for 2 h at 37 °C, and rinsed three times with TBS, prior to incubation with mAbs. The concentrated mAb supernatants were diluted 1:50 in 0.2% BSA in TBS (tris-buffered saline, pH 7.2) and reacted overnight at 4 °C on a rotary platform shaker with gentle agitation. Blots were washed three times with TBS-Tween-20 (0.05%) for 5 min before being incubated with secondary antibody (Goat anti-mouse IgG-hrp, Southern Biotech, same as above) at 1:2000 in TBS, 0.2% BSA for 1 h. The blots were washed as above and developed using DAB insoluble substrate (Pierce, Rockford, IL).
2.6. Virus neutralisation
The ELISA positive monoclonal antibodies were screened for neutralisation of SARS-CoV using two independent formats. The first was a standard plaque reduction assay and the second measured reduction of cytopathic effect (CPE) in a microtiter format.
2.6.1. Plaque reduction virus neutralisation assay
A standard plaque reduction neutralisation test was performed as previously described (Beaty et al., 1989) using highly purified SARS-CoV. Briefly, mixtures of pre-titred (100 PFUs) SARS-CoV and serial two-fold dilutions of hybridoma supernatant were incubated at 37 °C for 1 h and added to six well plates containing Vero cell monolayers. After incubation at 37 °C for 1 h, a nutrient-agar overlay was added and the plates were placed in a CO2 incubator at 37 °C for approximately 3 days. A second overlay was then added which contained neutral red as a vital stain. Plates were then checked periodically over the next few days for plaque formation. The highest dilution tested that produced a plaque reduction of at least 90% was defined as the titration end point.
2.6.2. CPE reduction virus neutralisation assay
A microtiter format CPE reduction assay was used to test the SARS-CoV reactive mAbs for neutralisation of SARS-CoV and transmissible gastroenteritis virus Diamond strain (kindly provided by Dr. Susy Carman, LSD, University of Guelph). Briefly, concentrated hybridoma supernatants were diluted 1:10 in cell culture medium and incubated with 100 TCID50 of either highly purified SARS-CoV (Tor-3 strain), or TGEV, for 1 h at 37 °C, for a final dilution of 1:20. The virus–antibody mix was then transferred onto cell monolayers in 96-well plates (Costar, Corning, NY). Vero V-76 cells were used for the SARS-CoV, ST cells for the TGEV. The plates were incubated until CPE developed in virus back titration controls.
3. Results
3.1. Development and immunochemistry of mAbs to SARS-CoV
ELISA screening on highly purified SARS-CoV identified a panel of 103 mAbs reactive to the SARS-CoV antigen preparation. Negative screening on BSA reduced this number to a panel of 17 IgG/k type mAbs (Fig. 1; Table 1). In general, binding reactivity of these mAbs is greatly affected by both the conformation and purity of the antigen as illustrated by the decreased binding of these mAbs, when tested in ELISA, to heat denatured pure SARS-CoV as antigen compared to native virus (Fig. 1). This clearly shows the importance of selecting suitable antigen for screening assays.
Fig. 1.
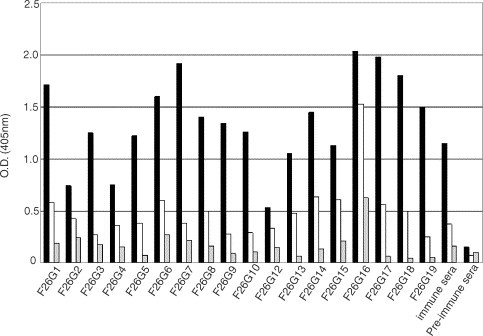
ELISA reactivity of mAbs with whole, inactivated SARS-CoV. Hybridoma supernatants were tested at a 1:4 dilution in PBS+0.2% BSA on pre-blocked plates, coated with 18 ng per well of inactivated, highly purified SARS-CoV. Positive clones were identified as having positive binding (color) in wells that were at least four-fold higher than the background level reactivity on BSA. Antigen legend: black bars—native, highly purified SARS-CoV; white bars—heat denatured, highly purified SARS-CoV; grey bars—BSA control.
Table 1.
mAbs to the SARS-CoV
| Clone | Class | Neutralising titrea | Protein targetc | Conformational requirement of epitope in immuno-dot blotd | IFAe | Epitopef | |||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| NML | NCFADb | N | H | D | HD | R | HR | A | |||||
| F26G1 | G2a/k | 0 | 0 | Spike | + | ± | + | − | + | ± | − | + | L.E |
| F26G2 | G2a/k | 0 | 0 | U | nd | nd | nd | nd | nd | nd | nd | − | C |
| F26G4 | G2a/k | 0 | 0 | U | nd | nd | nd | nd | nd | nd | nd | − | C |
| F26G5 | G2a/k | 0 | 0 | U | + | + | ± | ± | + | + | ± | L,E | |
| F26G6 | G2b/k | 0 | 0 | Spike | + | + | + | ± | + | + | + | ++ | L,E |
| F26G8 | G2a/k | 0 | 0 | Spike | + | + | + | ± | + | + | ± | + | L,E |
| F26G12 | G2a/k | 0 | 0 | U | nd | nd | nd | nd | nd | nd | nd | − | C |
| F26G13 | G2b/k | 0 | 0 | U | nd | nd | nd | nd | nd | nd | nd | ± | C,E |
| F26G14 | G2b/k | 0 | 0 | U | nd | nd | nd | nd | nd | nd | nd | + | C,E |
| F26G15 | G2b/k | 0 | 0 | nucleoprotein | − | − | − | − | − | − | − | nd | L |
| F26G16 | G1/k | 0 | 0 | U | + | − | + | − | − | − | − | − | C |
| F26G17 | G2b/k | nd | 0 | U | nd | nd | nd | nd | nd | nd | nd | nd | C |
| F26G3 | G2a/k | >1/40 | >1/20 | U | + | − | + | − | − | − | − | + | C,E,P |
| F26G7 | G2b/k | >1/80 | >1/20 | U | + | − | + | − | ± | − | − | + | C,E,P |
| F26G9 | G2a/k | >1/80 | >1/20 | u | + | − | ± | − | − | − | − | + | C,E,P |
| F26G10 | G2a/k | >1/80 | >1/20 | u | + | − | ± | − | − | − | − | ++ | C,E,P |
| F26G18 | G2b/k | nd | >1/20 | Spike | + | ± | + | − | + | + | − | nd | L,P |
| F26G19 | G2a/k | nd | >1/20 | Spike | + | − | + | − | − | − | nd | L,P |
Western immunoblot analysis identified mAbs to the SARS-CoV Spike and nucleoprotein. Five mAbs (F26G1, G6, G8, G18 and G19) react specifically with the SARS-CoV Spike protein in Western immunoblot. These mAbs recognise Spike in Western immunoblot on both highly purified virus and infected cell lysates (Fig. 2), but do not react with mock-infected cell lysates (data not shown). This result suggests that these mAbs target linear epitopes within the Spike protein. Another mAb, F26G15, bound to the nucleoprotein in Western immunoblot assays. Interestingly, the majority of the identified mAbs bound to the Spike protein and not the nucleoprotein, despite the strong sero-reactivity of the polyclonal serum of the immunised mice for the nucleoprotein (Fig. 2). The identity of the target antigen of eleven other mAbs could not be determined by Western immunoblot analysis. This observation suggests that these mAbs likely target conformational epitopes that are sensitive to the conditions employed in such analyses. Work is planned to identify the specific targets of these mAbs.
Fig. 2.
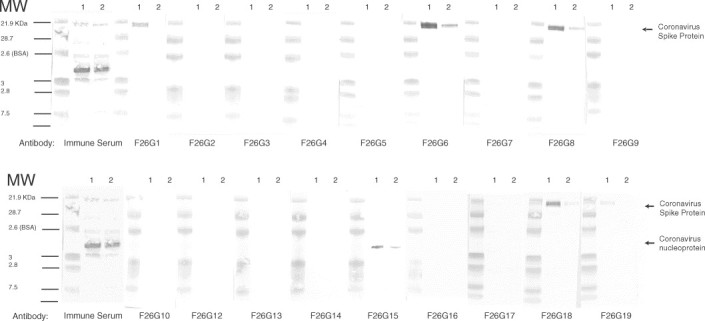
Western immunoblot reactivity of mAbs with highly purified SARS-CoV. The positive and preimmune control sera were from the corresponding immune mouse and tested at 1:2000 dilution in TBS+0.2% BSA. Lanes marked 1 were loaded with highly purified SARS-CoV, lanes marked 2 with infected Vero cell lysate. The molecular weights of the pre-stained kaleidoscope markers (BioRAD) are indicated on the left of the figure.
Immuno-dotblot analysis reveals a spectrum of conformational requirements for binding (summarised in Table 1). The effects of different denaturing treatments on the binding activity of a subset of neutralising and some non-neutralising mAbs were examined using immuno-dotblot assays on infected lysates compared to uninfected lysates. The series of conditions tested include exposure to heat, detergent, a reducing agent, and combinations thereof. Interestingly, mAb F26G15, which binds to the nucleoprotein in Western immunoblot, does not bind to the SARS-CoV antigen in immuno-dotblot under any of the conditions tested. The inherent charge of the highly phosphorylated nucleoprotein, or of the immobilon membrane used in the assays, may provide an explanation for these results. Immuno-dot blots were not performed with mAbs F26G2, F26G4, F26G12, F26G14, or F26G17, however the binding of these mAbs is considered conformational as they do not bind to SARS-CoV proteins in Western immunoblot (Fig. 2). The binding of Spike protein specific mAbs F26G1, F26G18, and F26G19 is inhibited in immuno-dotblot assays when antigens are treated with heat plus detergent, or heat plus detergent plus reducing agent. When the immuno-dotblot assay was instead performed using purified virus particles, the result was identical for nucleoprotein specific mAb F26G15, and Spike specific mAbs F26G1, F26G18, and F26G19 (data not shown). Antigen pre-treatment for Western immunoblot, which depends on the application of an electrical current, differs when compared to passive adsorption in ELISA and Immuno-dotblot assays, and clearly do not always correlate with one another. These studies illustrate the need to use multiple assays for epitope characterisation and reveal limitations of current epitope classification schemes.
The Spike protein is an immunodominant antigen when inactivated SARS-CoV is used as an antigen. Murine antibody responses to inactivated whole SARS-CoV are focused upon the SARS Spike and NP protein as shown by the specificity of the recovered mAbs. While multiple SARS-CoV proteins appear to be the target of mouse serum antibody as shown in Western blot (Fig. 2, immune sera), the majority of the mAbs recognise the Spike protein in Western immunoblot. In a SARS-CoV infection, the host immune system is exposed to a large load of nucleoprotein antigen, due to the presence of replicating virus in infected tissues. This would suggest that a competitive ELISA format based upon the NP as the target antigen might provide increased sensitivity for early detection of infection. Towards this goal, the ability of SARS convalescent serum to compete for binding of mAbs to the nucleoprotein or to Spike on whole SARS-CoV was tested via ELISA. The serum samples were gamma-irrradiated prior to use. Antibodies in the serum of patient S3 clearly inhibit binding of both the nucleoprotein mAb F26G15 and the Spike protein specific mAb F26G6 (Fig. 3). In contrast, antibody in pooled normal human sera does not compete for binding by either mAb. While not a statistical analysis these data show that these mAbs are useful for the development of serological competition assays which can be subjected to validation tests. The sera from several other SARS-CoV infected patients demonstrate a similar ability to inhibit binding by the F26G15 (nucleoprotein specific mAb) and F26G6 (spike specific mAb). However, in some samples there is inhibition of only the binding to nucleoprotein without inhibition of the mAb to the Spike protein suggesting that Spike antibody responses take longer to develop in infected humans (data not shown). The predominance of mAbs to the Spike protein in mice immunised with intact viral particles led us to test for biological activity in virus neutralisation assays.
Fig. 3.
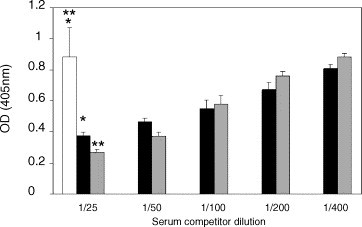
Competition ELISA measuring the binding of murine mAbs to highly purified SARS-CoV in the presence of human patient serum. Dilutions (as indicated) of a normal human serum control (white bar), or serum from convalescent SARS patient S3 were applied to wells coated with highly purified whole SARS-CoV. mAb F26G6 (Spike specific; black bars) or F26G15 (nucleoprotein specific; grey bars) were then added to the reactions. The results depicted for the pooled normal human serum (NHS) represent the mean of three replicate tests performed in the presence of mAb F26G6 combined with three replicate tests performed in the presence of mAb F26G15. Results are representative of identical assays performed in duplicate with gamma-irradiated patient sera (2 Mrad) (*_P_=0.01, **_P_=0.004, Student’s _t_-test).
3.2. Biological characterisation of mAb binding to SARS-CoV
The SARS-CoV Spike protein is the target of neutralising antibodies. Neutralisation positive mAbs bind both to linear epitopes in the Spike protein and to unknown conformational epitope(s) either in the Spike protein or in other proteins. A total of six mAbs were identified that could neutralise SARS-CoV infectivity: F26G3, G7, G9, G10, G18, and G19 (Table 1). Significantly, two of these mAbs, F26 G18 and 19, were positively identified as being specific for Spike, as determined by Western immunoblotting. The specific targets of the remaining neutralising mAbs remain to be elucidated. No cross-neutralisation was observed for the animal coronavirus TGEV. This shows that these mAbs are specific for SARS-CoV epitopes and do not cross-neutralise via TGEV epitopes. The remaining mAbs were unable to prevent viral growth when they were applied to the neutralisation assays. These data suggest that vaccines capable of engendering neutralising antibody responses to the Spike protein may be effective in blocking infection with SARS-CoV.
The four Western immunoblot negative, virus-neutralising mAbs were tested for their ability to bind native SARS-CoV in infected cells by immunofluorescence assay. Non-neutralising mAb F26G6 was used as a positive control, since this mAb recognises Spike protein in immunohistochemical staining of infected Vero cells (data not shown). Immunofluorescence analysis reveals that the neutralising mAbs F26G3, G7, G9, and G10 specifically recognise SARS-CoV infected but not uninfected Vero cells (Fig. 4). Irrelevant, isotype matched mAbs, produced in an identical fashion, do not react with SARS-CoV infected Vero cells.
Fig. 4.
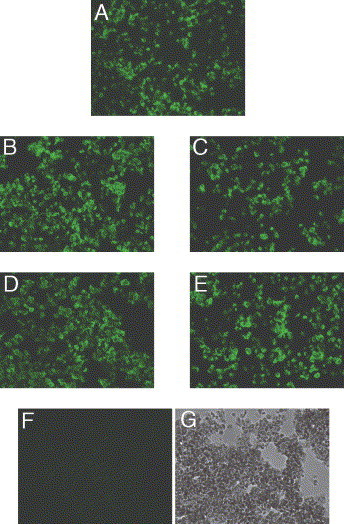
Immunofluorescence staining of SARS-CoV-infected Vero cells with Western immunoblot negative, neutralising mAbs to SARS-CoV. (A) F26G6, non-neutralising mAb specific for the Spike protein. (B) F26G3, neutralising mAb. (C) F26G7, neutralising mAb. (D) F26G9, neutralising mAb. (E) F26G10, neutralising mAb. (F) Anthrax toxin mAb, F25G1. (G) Anthrax toxin mAb F25G1 in bright field.
The SARS-neutralising mAbs bind epitopes with higher conformational requirements than the non-neutralising mAbs as they are less tolerant to denaturation of the epitopes. The method of antigen preparation and quality of the antigen greatly affect the interpretation of mAb binding results obtained from immuno-dotblot assays. Importantly, none of the mAbs react with mock-infected lysates as assayed in Immuno-dotblots (data not shown). This observation suggests that the majority of the neutralising mAbs likely target surface exposed, protein epitopes on the native viral particle. One such putative protective antigen has been identified as the Spike protein by Western immunoblot analysis with neutralising mAbs F26G18 and F26G19.
Antigen quality and conformation affects the binding of the anti-SARS-CoV mAbs in ELISA. While purified virus is clearly the optimal antigen tested in this series of experiments, the lower quality SARS-CoV-infected Vero cell lysates are, however, much easier to prepare for diagnostic assays. Therefore, the limits of mAb binding to SARS-CoV infected Vero cell lysates were further examined via ELISA. The majority of these mAbs exhibit decreased binding when the antigen is heat denatured (Table 2). Heat denaturation has very little effect on the binding of non-neutralising mAb F26G16, which also maintains a high level of binding in ELISA using infected Vero cell lysates. F26G16 does, however, show higher background on the irrelevant antigen, BSA, and has inconsistent reactivity in Western immunoblots with heat denatured viral lysate (Fig. 1, Table 1). The combination of lower quality antigen in infected Vero cell lysates, along with heat denaturation of the antigens, has a stronger negative effect on binding by the neutralising mAbs compared to non-neutralising mAbs (Table 2). Indeed, regardless of Western immunoblot reactivity, the non-neutralising clones retain a greater ability to bind heat denatured antigen compared to the neutralising mAbs (lower mean percent reduction in OD per group P<0.001, Student’s _t_-test, Table 2). This supports the earlier observation in ELISA on purified SARS-CoV (Fig. 1) that shows that the neutralising mAbs have higher requirements for native epitope conformation compared to non-neutralising mAbs.
Table 2.
Effect of heat denaturation on mAb binding to SARS-CoV
| Bio-activity | mAb | Western reactivity | Antigen | Reduction in O.D. | Mean percent reductiond | ||
|---|---|---|---|---|---|---|---|
| Viral lysatea | Denatured viral lysateb | Foldc | Percent | ||||
| Non-neutralising | F26G2 | – | 0.743 | 0.424 | 1.7 | 43 | 51 |
| F26G4 | – | 0.751 | 0.363 | 2.1 | 52 | ||
| F26G5 | – | 1.224 | 0.383 | 3.2 | 69 | ||
| F26G6 | Spike | 1.600 | 0.6 | 2.7 | 62 | ||
| F26G8 | Spike | 1.408 | 0.497 | 2.8 | 29 | ||
| F26G12 | – | 0.533 | 0.338 | 2.9 | 37 | ||
| F26G13 | – | 1.048 | 0.481 | 2.2 | 54 | ||
| F26G14 | – | 1.448 | 0.633 | 2.3 | 56 | ||
| F26G15 | N | 1.134 | 0.604 | 1.9 | 47 | ||
| F26G16 | – | 2.037 | 1.534 | 1.3 | 25 | ||
| F26G17 | – | 1.986 | 0.560 | 3.5 | 73 | ||
| Neutralising | F26G3 | – | 1.253 | 0.276 | 4.5 | 78 | 78* |
| F26G7 | – | 1.917 | 0.382 | 5.0 | 80 | ||
| F26G9 | – | 1.345 | 0.278 | 4.8 | 79 | ||
| F26G10 | – | 1.259 | 0.290 | 4.3 | 77 | ||
| F26G18 | Spike | 1.807 | 0.501 | 3.6 | 72 | ||
| F26G19 | Spike | 1.505 | 0.253 | 6.0 | 83 |
The higher conformational requirement by neutralising mAbs may help to explain some discrepancies observed in the immuno-dotblot methods. The immuno-dotblot is, overall, a less sensitive method, and the results are more difficult to quantify, compared to ELISA. For example, mAb F26G18 binds to the SARS-CoV Spike protein in Western immunoblot and neutralises SARS-CoV infection in vitro_._ While binding in Western immunoblot generally suggests the epitope is linear in nature, nonetheless, heat treatments clearly affect binding of some mAbs to the whole virion. The immuno-dotblot data reveal that with the lower quality antigen of the infected Vero cell lysate, under the conditions of heat-plus detergent, or heat-plus detergent and reducing agent, mAb F26G18 cannot bind (Table 1).
4. Discussion
This paper describes the development of murine mAbs which recognise SARS-CoV antigens in ELISA, immuno-dotblot, Western immunoblot, on the surface of infected cells, and in neutralisation assays. These data are consistent with the appearance of coronavirus antigens on the surface of the infected cell during replication (Talbot et al., 1984), although the fixation process may allow for reactivity of these mAbs with internal antigens as well. The conformational sensitivity of most of the SARS-CoV neutralising mAbs is consistent with properties of neutralising mAbs raised against other enveloped viruses, which generally require more native conformation for binding (Wilson et al., 2000, Zwick et al., 2001). The immuno-dotblot assays contradict the classification of several putative epitopes as being linear as is suggested by positive Western immunoblot reaction (for example with neutralising mAb F26G18). Indeed, it appears that the strict traditional classification of epitopes as being linear or conformational must account for a broader spectrum of conformational requirements, especially when dealing with antigens of variable quality in different immunological tests. However, in general the neutralising mAbs can be considered to have a higher requirement for correct epitope conformation compared to non-neutralising mAbs in ELISA. It will be important to verify, under optimised conditions (Opstelten et al., 1995) the use of viral lysates designed for maximal recovery of coronavirus proteins, and to this end the production of high quality recombinant protein antigens will provide useful insights. Unfortunately, preparation of highly purified viral antigen requires enormous efforts under bio-containment conditions, which emphasises the need for a quality recombinant antigen assay. Collectively, these data demonstrate that development of mAb-based diagnostic tools for the detection of SARS-CoV infection is well within reach.
The Spike protein of the SARS-CoV is a target of neutralising mAbs. Epitopes on the Spike protein provide neutralising targets on SARS-CoV in vitro, and this is consistent with the Spike being the target of neutralising antibodies for other coronavirus strains (Godet et al., 1994). Moreover, these mAbs may be useful for the identification of protective epitopes for vaccine formulations (Enjuanes et al., 1995). Studies are underway to determine the identity of the antigen(s) recognised by the neutralising, Western immunnoblot negative mAbs. Preliminary analysis of the nucleotide and predicted amino acid sequences of the cloned VH and VL coding regions of these mAbs suggests that the mAbs are distinct. This observation implies that the hybridoma clones expressing the anti-SARS-CoV neutralising mAbs were derived from individually rearranged and clonally selected B cells in vivo_._ It also reveals that there is no apparent consensus sequence within the complementarity determining regions that is required for the mAbs to exhibit virus neutralisation activity. This finding makes it feasible to engineer multiple mAbs with high specificity and avidity for various SARS-CoV epitopes for preparations of defined cocktails of therapeutic mAbs. A detailed description of the immunogenetic properties of these mAbs is in preparation (Berry et al., manuscript in preparation). The NP specific mAb, F26G15, is useful in competitive ELISA with patient sera. This is important as early detection of SARS infections is key to risk management of this disease. This is the first description of neutralising mAbs from a host immunised with whole SARS-CoV and these antibodies should prove useful for the development of new diagnostic tests, studies of antigenic variation, and vaccine development in the global fight against SARS.
Acknowledgements
The authors would like to thank Ms. Nicole Beausoleil, Mr. Daryl Dick, Mr. Darrell Johnstone, Ms. Kathy Frost, Ms. Hilary Holland, and Mr. Richard Nickel for expert technical assistance. MG is supported by a Health Canada OCS Postdoctoral Fellowship. Thanks to Dr. John Copps (NCFAD) for expert veterinarian services and for assisting in the design of the emergency Animal Use Document. Thanks also to Dr. Susy Carman (University of Guelph, Canada) and Dr. Lorne Babiuk (VIDO, Canada) for providing animal coronavirus strains. Funding for this work was provided by Health Canada and the Canadian Food Inspection Agency. This work is dedicated to the memory of Lloyd D. Berry.
References
- Beaty, B.J., Calisher, C.H., Shope, R., 1989. Arboviruses. In: Schmidt, N.J., Emmons, R.W. (Eds.), Diagnostic Procedures for Viral, Rickettsial and Chlamydial infections, 6th ed. American Public Health Association, Washington, DC, pp. 797–856.
- Brian, D.A., Hogue, B., Lapps, W., Potts, B., Kapke, P, 1983. In: Proceedings of the Fourth International Symposium on Neonatal Diarrhea. S.D. Acres, Saskatoon, Canada.
- Collins A.R, Knobler R.L, Powell H, Buchmeier M.J. Monoclonal antibodies to murine hepatitis virus-4 (strain JHM) define the viral glycoprotein responsible for attachment and cell–cell fusion. Virology. 1982;119:358–371. doi: 10.1016/0042-6822(82)90095-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Enjuanes L, Mendez A, Ballesteros M. Tropism and immunoprotection in transmissible gastroenteritis coronaviruses. Dev. Biol. Stand. 1995;84:145–152. [PubMed] [Google Scholar]
- Fields, B.N., Knipe, D.M., Howley, P.M., Griffin, D., 2001. Fields Virology, 4th ed. Lippincott Williams & Wilkins, Philadelphia.
- Gerna G.M, Battaglia M, Cereda P, Passarani N. Reactivity of human coronavirus OC43 and neonatal calf diarrhoea coronavirus membrane-associated antigens. J. Gen. Virol. 1982;60:385–390. doi: 10.1099/0022-1317-60-2-385. [DOI] [PubMed] [Google Scholar]
- Godet M, Grosclaude J, Delmas B, Laude H. Major receptor-binding and neutralization determinants are located within the same domain of the transmissible gastroenteritis virus (coronavirus) Spike protein. J. Virol. 1994;68:8008–8016. doi: 10.1128/jvi.68.12.8008-8016.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jimenez G, Correa I, Melgosa M.P, Bullido M.J, Enjuanes L. Critical epitopes in transmissible gastroenteritis virus neutralization. J. Virol. 1986;60:131–139. doi: 10.1128/jvi.60.1.131-139.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kohler G, Milstein C. Continuous cultures of fused cells secreting antibody of predefined specificity. Nature. 1975;256:495–497. doi: 10.1038/256495a0. [DOI] [PubMed] [Google Scholar]
- Krokhin O, Li Y, Andonov A, Feldmann H, Flick R, Jones S, Stroeher U, Bastien K, Dasuri K, Cheng J, Simonsen J, Perrault J, Wilkins J, Enw W, Plummer F, Standing K. Mass spectrometric characterization of proteins from the SARS virus: a preliminary report. Mol. Cell. Proteomics. 2003;2:346–356. doi: 10.1074/mcp.M300048-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laude H, Chapsal J.M, Gelfi J, Labiau S, Grosclaude J. Antigenic structure of transmissible gastroenteritis virus. I. Properties of monoclonal antibodies directed against virion proteins. J. Gen. Virol. 1986;67:119–130. doi: 10.1099/0022-1317-67-1-119. [DOI] [PubMed] [Google Scholar]
- Li G, Chen X, Xu A. Profile of specific antibodies to the SARS-associated coronavirus. N. Engl. J. Med. 2003;349:508–509. doi: 10.1056/NEJM200307313490520. [DOI] [PubMed] [Google Scholar]
- Li W, Moore M.J, Vasilieva N, Sui J, Wong S.K, Berne M.A, Somasundaran M, Sullivan J.L, Luzuriga K, Greenough T.C, Choe H, Farzan M. Angiotensin converting enzyme 2 is a functional receptor for the SARS coronavirus. Nature. 2003;426:450–454. doi: 10.1038/nature02145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marra M, Jones S.J.M, Astell C.R, Holt R.A, Brooks-Wilson A, Butterfield T.S.N, Khattra J, Asano J.K, Barber S.A, Chan S.Y, Cloutier A, Coughlin S.M, Freeman D, Girn N, Griffith O.L, Leach S.R, Mayo M, McDonald H, Montgomery S.B, Pandoh P.K, Petrescu A.S, Robertson A.G, Schein J.E, Siddiqui A, Smailus D.E, Stott J.M, Yang G.S, Plummer F, Andonov A, Artsob H, Bastien N, Bernard K, Booth T, Bowness D, Czub M, Drebot M, Fernando L, Flick R, Garbutt M, Gray M, Grolla A, Jones S, Feldmann H, Meyers A, Kabani A, Li Y, Normand S, Stroher U, Tipples G, Tyler S, Vogrig R, Ward D, Watson B, Brunham R.C, Krajden M, Petric M, Skowronski D.M, Upton C, Roper R.L. The genome sequence of the SARS-associated coronavirus. Science. 2003;300:1399–1404. doi: 10.1126/science.1085953. [DOI] [PubMed] [Google Scholar]
- Opstelten D.-J, Martin J.B, Raamsman K.W, Horzinek M.C, Rottier P.J. Envelope glycoprotein interactions in coronavirus assembly. J. Cell Biol. 1995;131:339–349. doi: 10.1083/jcb.131.2.339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rota P.A, Oberste M.S, Monroe S, Nix W, Campagnoli R, Icenogle J, Penaranda S, Bankamp B, Maher K, Chen M, Tong S, Tamin A, Lowe L, Frace M, DeRisi J, Chen Q, Wang D, Erdman D, Peret T, Burns C, Ksiazek T, Rollin P, Sanchez A, Liffick S, Holloway B, Limor J, McCaustland K, Olsen-Rasmussen M, Fouchier R, Gunther S, Osterhaus A, Drosten C, Pallansch M, Anderson L, Bellini W. Characterization of a novel coronavirus associated with severe acute respiratory syndrome. Science. 2003;300:1394–1399. doi: 10.1126/science.1085952. [DOI] [PubMed] [Google Scholar]
- Stadler K, Masignani V, Eickmann M, Becker S, Abrignani S, Klenk H.-D, Rappuoli R. SARS-beginning to understand a new virus. Nat. Rev. Microbiol. 2003;1:209–218. doi: 10.1038/nrmicro775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Talbot P.J, Salmi A.A, Knobler R.L, Buchmeier M.J. Topographical mapping of epitopes on the glycoproteins of murine hepatitis virus-4 (strain JHM): correlation with biological activities. Virology. 1984;132:250–260. doi: 10.1016/0042-6822(84)90032-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wege H, Domes R, Wege H. Hybridoma antibodies to the murine coronavirus JHM: characterization of epitopes on the peplomer protein (E2) J. Gen. Virol. 1984;65:1931–1942. doi: 10.1099/0022-1317-65-11-1931. [DOI] [PubMed] [Google Scholar]
- Wilson J.A, Hevey M, Bakken R, Guest S, Bray M, Schmaljohn A.L, Hart M.K. Epitopes involved in antibody-mediated protection from Ebola-virus. Science. 2000;287:1664–1666. doi: 10.1126/science.287.5458.1664. [DOI] [PubMed] [Google Scholar]
- Xiao X, Chakraborti S, Dimitrov A.S, Gramatikoff K, Dimitrov D. The SARS-CoV S glycoprotein: expression and functional characterization. Biochem. Biophys. Res. Commun. 2003;312:1159–1164. doi: 10.1016/j.bbrc.2003.11.054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zwick M.B, Bonnycastle L.L, Menendez A, Irving M, Barbas C.F, III, Parren P, Burton D.R, Scott J.K. Identification and characterization of a peptide that specifically binds the human broadly neutralizing anti-human immunodeficiency virus type 1 antibody b12. J. Virol. 2001;75:6692–6699. doi: 10.1128/JVI.75.14.6692-6699.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]