The glycosylation status of the murine hepatitis coronavirus M protein affects the interferogenic capacity of the virus in vitro and its ability to replicate in the liver but not the brain (original) (raw)
Abstract
The coronavirus M protein, the most abundant coronaviral envelope component, is invariably glycosylated, which provides the virion with a diffuse, hydrophilic cover on its outer surface. Remarkably, while the group 1 and group 3 coronaviruses all have M proteins with N-linked sugars, the M proteins of the group 2 coronaviruses [e.g., mouse hepatitis virus (MHV)] are O-glycosylated. The conservation of the N- and O-glycosylation motifs suggests that each of these types of carbohydrate modifications is beneficial to their respective virus. Since glycosylation of the M protein is not required for virus assembly, the oligosaccharides are likely to be involved in the virus–host interaction. In order to investigate the role of the M protein glycosylation in the host, two genetically modified MHVs were generated by using targeted RNA recombination. The recombinant viruses carried M proteins that were either N-glycosylated or not glycosylated at all, and these were compared with the parental, O-glycosylated, virus. The M protein glycosylation state did not influence the tissue culture growth characteristics of the recombinant viruses. However, it affected their interferogenic capacity as measured using fixed, virus-infected cells. Viruses containing M proteins with N-linked sugars induced type I interferons to higher levels than viruses carrying M proteins with O-linked sugars. MHV with unglycosylated M proteins appeared to be a poor interferon inducer. In mice, the recombinant viruses differed in their ability to replicate in the liver, but not in the brain, whereas their in vivo interferogenic capacity did not appear to be affected by their glycosylation status. Strikingly, their abilities to replicate in the liver correlated with their in vitro interferogenic capacity. This apparent correlation might be explained by the functioning of lectins, such as the mannose receptor, which are abundantly expressed in the liver but also play a role in the induction of interferon-α by dendritic cells.
Introduction
Glycosylation is a frequent modification of proteins in eukaryotic cells. Basically, two types of protein glycosylation occur. Oligosaccharide side chains are attached to proteins either by N-linkage to asparagine residues or by O-linkage to hydroxyl groups of serine and threonine residues. A great variety of functions have been attributed to the oligosaccharide side chains. Carbohydrates have been shown to be important for protein folding, structure, and stability and to be involved in intracellular sorting of proteins (e.g., during quality control) and to play essential roles in molecular recognition processes (e.g., in the generation of immune responses) Drickamer and Taylor 1998, Helenius and Aebi 2001, Rudd and Dwek 1997, Van den Steen et al 1998. Most viral glycoproteins are structural proteins that become incorporated into the membrane bilayer of enveloped viruses. Glycosylation of these envelope proteins plays an essential role in establishing their bioactive conformation and has effects on receptor binding, fusion activity, and antigenic properties of the virus Alexander and Elder 1984, Braakman and van Anken 2000, Schulze 1997.
Coronaviruses, positive-stranded RNA viruses, contain three to four proteins in their membrane envelope. The small E protein, a minor component of the virion, is not glycosylated. The spike (S) glycoprotein and the hemagglutinin-esterase protein—the latter only present in some virus strains—both carry many N-linked sugar side chains. The S protein interacts with the coronavirus receptor and mediates virus–cell and cell–cell fusion. Cotranslational N-glycosylation is needed for this protein to fold properly and to exit the endoplasmic reticulum (ER). The S protein was shown to aggregate when glycosylation was inhibited (Delmas and Laude, 1990) (our unpublished results), whereas growth of coronavirus in the presence of the N-glycosylation inhibitor tunicamycin resulted in the production of spikeless, noninfectious virions, which were devoid of S protein Holmes et al 1981, Rottier et al 1981, Stern and Sefton 1982.
The membrane (M) protein is the most abundant envelope component and is almost invariably glycosylated. Intriguingly, whereas the group 1 and group 3 coronaviruses—with transmissible gastroenteritis virus (TGEV) and infectious bronchitis virus (IBV) as important representatives, respectively—all contain M proteins with only N-linked sugars, the M proteins of group 2 coronaviruses such as mouse hepatitis virus (MHV) are only O-glycosylated (reviewed by Rottier, 1995). The M protein is a type III membrane protein. The N- and O-glycosylation motifs are located within the variable part of the amino-terminal ectodomain. The O-glycosylation motif consists of a cluster of hydroxylamino acids and a nearby proline residue, positioned at the extreme amino terminus of the M protein (de Haan et al., 1998b).
The M protein plays a central role in coronavirus assembly. Together with the minor E protein it is responsible for the assembly of the coronavirus envelope Baudoux et al 1998a, Godeke et al 2000, Vennema et al 1996. In addition, the M protein directs the incorporation of the S protein Nguyen and Hogue 1997, Opstelten et al 1995 and the nucleocapsid (Narayanan et al., 2000) into the budding particle. Glycosylation of the M protein is not required for virus assembly as we demonstrated by using a viruslike particle (VLP) assembly system (de Haan et al., 1998a). Furthermore, M protein glycosylation was also shown not to be critical for interaction with the S protein (de Haan et al., 1999). These observations are consistent with studies that used tunicamycin Rossen et al 1998, Stern and Sefton 1982 and monensin (Niemann et al., 1982) to inhibit glycosylation in infected cells.
The distinct conservation of N- and O-glycosylation in the M proteins of the different groups of coronaviruses suggests that the presence and the particular type of carbohydrates are somehow beneficial to the virus. As glycosylation is not required for virus assembly, we hypothesized that the carbohydrates are involved in virus–host interaction. In order to investigate the role of M protein glycosylation, we generated genetically modified MHVs by using targeted RNA recombination. The recombinant viruses prepared carried M proteins that were O-glycosylated, N-glycosylated, or not glycosylated at all. Preliminary observations revealed no differences in the growth properties of the viruses but suggested that they might differ in their interferogenic properties (de Haan et al., 2002). This prompted us to carry out a more in-depth study of the viruses, particularly with respect to interferon induction in relation to virulence and to hepatic replication and pathogenesis in mice. The results surprisingly show that in vitro interferon induction correlated with replication in and damage to the liver.
Results
Construction of M protein glycosylation mutants
To examine the role of glycosylation of the ectodomain of the M protein, mutations expected to either ablate or alter oligosaccharide addition were directly introduced into the genome of MHV strain A59 by targeted RNA recombination Masters 1999, Masters et al 1994. In the first mutant, the functional acceptor site as well as an alternative acceptor site of O-glycosylation were eliminated by changing threonine residues 4 and 5 to alanines (de Haan et al., 1998b). In the second mutant, a consensus N-linked glycosylation site was created in the amino terminus of the M protein by insertion of two asparagine residues in place of serine residue 2, in addition to the substitutions of the threonine residues introduced to make the first mutant. Thus, the protein expressed from this latter mutant M gene would be expected to be N-glycosylated only. These mutations were first generated in the transcription vector pFV1 (Fischer et al., 1997) (Fig. 1A) and subsequently were introduced into MHV through homologous RNA recombination with the N gene deletion mutant Alb4, which was counterselected on the basis of its thermolabile phenotype Masters 1999, Masters et al 1994. Two independent candidate glycosylation-null mutants, Alb244 and Alb246, and two independent candidate N-glycosylation mutants, Alb248 and Alb250, were isolated. The genotypes of these were confirmed by direct genomic RNA sequencing of the 5′ end of the M gene (Fig. 1C) as well as by sequencing of the region that is deleted in the Alb4 virus genome (not shown). As isogenic wild-type controls for subsequent experiments we used Alb138 and Alb139, two independent wild-type recombinants obtained previously from Alb4 using pFV1-derived donor RNA.
Fig. 1.
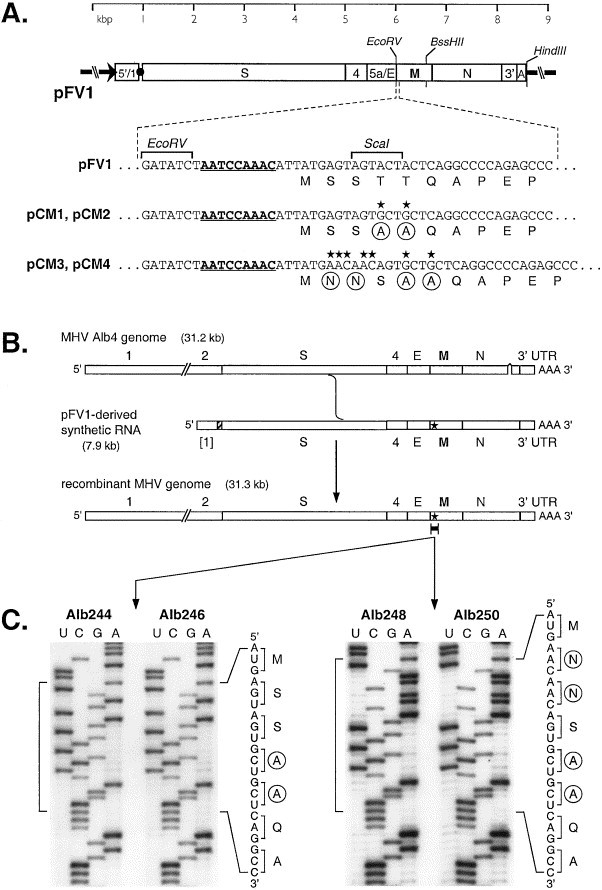
Construction of MHV M protein glycosylation mutants. (A) Schematic of transcription vector pFV1 (Fischer et al., 1997), which contains a cDNA copy of the 5′ 467 nt of the MHV genome fused to the 3′ 7406 nt of the MHV genome. The T7 RNA polymerase promoter is denoted by an arrow. The _Eco_RV and _Bss_HII sites, used for mutant plasmid construction, and the _Hin_dIII site, used for transcription vector truncation, are indicated. The expanded segments of sequence show the wild-type region encompassing the start of the M gene in pFV1, and, below this, the corresponding sequences of mutant vectors pCM1 to pCM4. Nucleotide point mutations or insertions are marked by stars, and the resulting changed amino acids are circled. The transcriptional regulatory sequence preceding the M gene is underlined. (B) Strategy for targeted recombination between recipient virus Alb4 and synthetic donor RNA transcribed from pFV1 or a mutant derivative of pFV1. The star indicates mutations transduced from the donor RNA into the recombinant genome. (C) Portions of sequence of purified genomic RNA isolated from virions of recombinants Alb244 and Alb246, which were derived from pCM1 and pCM2, respectively, and Alb248 and Alb250, which were derived from pCM3 and pCM4, respectively. Sequence was obtained using a primer complementary to nt 113 to 131 of the M protein coding region, and lanes labeled U, C, G, and A were from reactions terminated with ddATP, ddGTP, ddCTP, and ddTTP, respectively.
Analysis of viral proteins
To verify whether the viral genomic mutations resulted in the desired phenotypes, we analyzed the glycosylation status of the M protein in assembled virions. To this end, cells infected with the different MHV recombinants were labeled with 35S-labeled amino acids and the culture media were collected and processed for immunoprecipitation with an anti-MHV serum, followed by sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS–PAGE) through a 15% polyacrylamide gel. In some cases, tunicamycin was added during the infection to the culture media to inhibit N-linked glycosylation. As shown in Fig. 2, when no tunicamycin was added, S, N, and M proteins could be precipitated from the culture media, indicating that virus particles had been assembled and released from the cells infected with the MHV recombinants. The S protein appeared as the mature 180 kDa glycoprotein (S/gp 180). A fraction of the S protein had been cleaved into two subunits, both with a molecular weight of approximately 90 kDa (S/gp90). The M proteins of the different viruses showed clear differences in their electrophoretic mobilities. The M proteins obtained with Alb138 or Alb139 (i.e., the “wild-type” recombinants) appeared as the typical set of O-glycosylated forms described before Krijnse Locker et al 1992, Tooze et al 1988. The predominant forms are the M protein species containing galactose and sialic acid (M3 and M4), but the unglycosylated form (M0; the fastest migrating form) and the slightly slower migrating form containing only _N_-acetyl-galactosamine (M1) were also visible. In contrast, the M protein released from cells infected with Alb244 or Alb246 migrated in the gel as a single band with approximately the same electrophoretic mobility as the unglycosylated M protein species released from Alb138- or Alb139-infected cells. The M proteins released from cells infected with Alb248 or Alb250 appeared as a smear in the gel, quite similar to the M proteins from group I coronaviruses, which carry N-linked sugars Rossen et al 1998, Vennema et al 1990. The appearance of the M proteins as a smear in the gel is probably the result of extensive, heterogeneous modifications of the sugar side chains. To verify that the M proteins released from Alb248- and Alb250-infected cells contained no O-linked sugars in addition to N-linked side chains, we treated the cells with tunicamycin, which blocks N-linked, but not O-linked, glycosylation. Since tunicamycin also affects protein synthesis (Rossen et al., 1998) a longer exposure of this part of the gel is shown in Fig. 2. After treatment with tunicamycin the N and M proteins, but not the S protein, could be precipitated from the culture media. This is in agreement with experiments, mentioned previously, which demonstrated that noninfectious virions, devoid of S protein, are assembled in the presence of tunicamycin. Although the glycosylation state of the M proteins released from cells infected with Alb138 was not affected by treatment with tunicamycin, confirming that O-glycosylation was not perturbed, the electrophoretic mobility of the M proteins released from Alb248- or Alb250-infected cells was drastically changed by the presence of the drug. These M proteins now migrated as a single band with the same electrophoretic mobility as unglycosylated M protein, indicating that in the absence of tunicamycin these M proteins become N-glycosylated, not O-glycosylated. These results demonstrated that the recombinant viruses have the predicted M protein glycosylation phenotypes. Furthermore, each pair of independently obtained recombinant viruses behaved identically. Alb138 and Alb139 contain M proteins with O-linked sugars (O+N−) and Alb244 and Alb246 carry M proteins without any attached sugars (O−N−), whereas Alb248 and Alb250 contain M proteins with N-linked sugars only (O−N+).
Fig. 2.
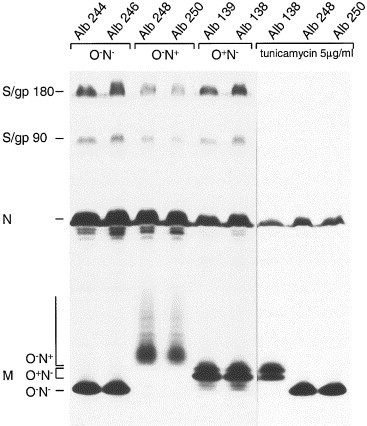
Analysis of the synthesis of recombinant MHV proteins. LR7 cells were infected with recombinant viruses as indicated on the top of the gel. Cells were labeled for 3 h with 35S-labeled amino acids starting 5 h postinfection. When indicated, tunicamycin (5 μg/ml) was added to the culture media from 2 h postinfection. At the end of the labeling period, culture media were collected and prepared for immunoprecipitation with the anti-MHV serum k134 The immunoprecipitates were analyzed by SDS–15%PAGE. The positions of the different proteins are indicated on the left.
Tissue culture growth phenotype
After confirmation of the glycosylation phenotypes of the different viruses, we analyzed their in vitro growth phenotypes. The recombinant viruses displayed no obvious differences with respect to plaque size or growth in tissue culture. Preliminary results indicated that they also exhibited similar one-step growth characteristics in LR7 cells (de Haan et al., 2002). These results were confirmed using independent recombinants, which showed that all recombinants behaved identically and caused extensive syncytia and cytopathic effects indistinguishably (data not shown). Passage 3 stocks of each mutant virus had titers in the range of 1.4 to 2.2 × 108 PFU/ml, which was similar to the titers obtained with wild-type recombinant virus. In addition, we compared the infectivity of the viruses to several other cell lines, including bone-marrow-derived macrophages, macrophage cells lines, dendritic cells derived from the spleen, and primary hepatocytes. No differences were observed; all cell types were infected to a similar extent and exhibited similar cytopathic effects (data not shown). These results indicate that the glycosylation state of the coronavirus M protein does not influence the tissue culture growth characteristics.
Induction of type I interferon by formaldehyde-fixed, coronavirus-infected cells
The induction of type I interferons (IFNs) by most RNA viruses is initiated by virus-derived double-stranded RNA. However, several enveloped viruses were shown to stimulate interferon-α (IFNα) expression in peripheral blood mononuclear (PBM) cells via an alternative pathway in which virus glycoproteins are involved Ito 1994, Milone and Fitzgerald-Bocarsly 1998. Also TGEV virions and TGEV VLPs, as well as fixed, TGEV-infected cells, are efficient inducers of IFNα (Baudoux et al., 1998b). Cell cultures infected with several other coronaviruses, including MHV, have also been demonstrated to have interferogenic capacity (Baudoux et al., 1998a). For TGEV it was demonstrated that the N-linked sugars on the M protein play an important role in the induction of IFNα (Laude et al., 1992). The availability of the recombinant MHVs allowed us to investigate the influence of the M protein glycosylation state on the interferogenic capacity of these viruses. For this purpose we infected LMR cells, which are porcine cells susceptible to MHV due to their constitutive expression of the MHV receptor (Rossen et al., 1996), with the different recombinant MHVs. LMR cells were chosen since they can also be infected with TGEV, which served as a positive control. LMR monolayers were infected with low (0.1) and high (10) multiplicity of infection (m.o.i.). After fixation at 9.5 h postinfection the cell monolayers were incubated overnight with porcine PBM cells. Serial 2-fold dilutions of the culture media were subsequently assayed for antiviral activity (i.e., IFNα activity) on LMR cells using vesicular stomatitis virus (VSV) as a challenge virus. The IFN produced by the porcine PBM cells has been characterized as type I IFN, as it was neutralized for more than 90% by an antitype I IFN antiserum Charley and Laude 1988, La Bonnardiere et al 1986. Fig. 3shows the results from two typical experiments. LMR cells infected with Alb248 and Alb250 (O−N+) reproducibly induced the highest type I IFN activity, after infection at both low and high m.o.i. The interferogenic capacity of these monolayers was almost as high as that of the cells infected with TGEV, the positive control. The interferogenic activity of Alb138 and Alb139 (O+N−) was reproducibly lower (5- to 30-fold) than that of Alb248 and 250 (O−N+), whereas Alb244- and Alb246- (O−N−) infected cells displayed an even lower interferogenic capacity (30- to 200-fold), especially at m.o.i. 0.1. Each pair of independent recombinant viruses behaved almost identically in the IFN assay, further demonstrating the reproducibility of the assay.
Fig. 3.
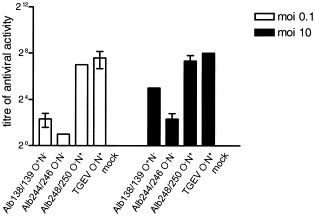
Induction of type I interferon by formaldehyde-fixed, coronavirus-infected cells. LMR cells were infected in duplicate with each recombinant virus or TGEV at a multiplicity of 0.1 or 10 PFU per cell. At 9.5 h postinfection cells were fixed with 3% formaldehyde. PBM cells were induced to produce IFNα by overnight incubation at 37°C on the TGEV- or MHV-infected, fixed cells. Supernatants were collected and serial twofold dilutions were assayed for IFN in an infection inhibition assay on LMR cells using VSV as a challenge virus. For each type of recombinant the mean value is shown, and the standard deviation is indicated. The IFN produced by the porcine PBM cells has been characterized as type I IFN, as it was neutralized by more than 90% by an antitype I IFN-antiserum Charley and Laude 1988, La Bonnardiere et al 1986.
The extent of induction of IFN by TGEV was found to parallel the level of expression of M antigen by the fixed cells (Baudoux et al., 1998b). It was therefore necessary to confirm that the effects we observed were not due simply to differences in M protein expression levels. Unfortunately, the amino acid changes in the ectodomain of the M protein are known to affect the antigenicity of this domain of the M protein (de Haan et al., 1998b), which would make interpretation of M protein antigen levels exposed by the cells troublesome. As an alternative control the fixed monolayers, used to induce IFN synthesis by the PBM cells, were immunocytochemically studied for MHV infection. The different recombinant MHVs appeared to have infected the LMR cells to the same extent (data not shown). Additionally, MHV-infected LMR cells were labeled with 35S-labeled amino acids, lysed and processed for immunoprecipitation using the anti-MHV serum that contains antibodies to domains other than the ectodomain, including the endodomain Rottier et al 1981, Rottier et al 1990. Prior to gel electrophoresis the immunoprecipitates were treated with PNGase F. PNGase F removes N-linked sugars from proteins and facilitates the quantitation of N-linked proteins, which otherwise would appear as a smear in the gel due to the extensive heterogeneous modifications. Quantitation of the M protein expression levels, using a phosphorimager, indicated that the N-glycosylated, O-glycosylated, and unglycosylated M proteins were all expressed at similar levels (data not shown). These two controls, together with the knowledge that neither the ectodomain of the M protein nor its glycosylation is important for M protein intracellular transport de Haan et al 1998a, de Haan et al 1998b, Mayer et al 1988, Rottier et al 1990, led us to conclude that the differences in the induction of IFN can not be the result of different levels of M protein exposure at the cell surface. The most likely explanation for the differences found are the different glycosylation states of the M proteins. Thus, coronaviruses containing M proteins with N-linked sugars induced type I IFN to higher levels than viruses carrying O-glycosylated M proteins. MHVs in which the M proteins are unglycosylated appeared to be poor IFN inducers.
Virulence of recombinant viruses
MHV-A59 is a dual-tropic virus producing hepatitis and encephalitis. As a first step to characterize the properties of the recombinant MHV-A59 viruses in their natural host, we determined the virulence of the recombinant viruses. Mice were inoculated intracranially with four 10-fold serial dilutions of Alb138, Alb244, and Alb248. Infected mice were observed for survival and a 50% lethal dose (LD50) was calculated for each virus. All recombinant viruses displayed the same LD50 value of 3.8 × 103 or virulence. Thus, the M protein glycosylation state does not appear to influence MHV-A59 virulence as assayed by intracranial inoculation.
Replication in the brain
Since no difference in virulence was observed after intracranial inoculation, the recombinant viruses were characterized in more detail by analyzing their in vivo replication. Thus mice were inoculated intracranially with viruses Alb138, Alb244, and Alb248 at a dose of 1 × 103 PFU. No significant differences in viral replication were observed (de Haan et al., 2002). In view of the artificial nature of intracranial inoculation, the experiment was repeated, but this time 1.4 × 105 PFU were inoculated intranasally, the presumed natural route of infection. Once again, however, the different recombinant viruses appeared to replicate to the same extent in the brain (Fig. 4). Apparently, the M protein glycosylation state is not an important factor for migration to and replication in the brain of MHV-A59.
Fig. 4.
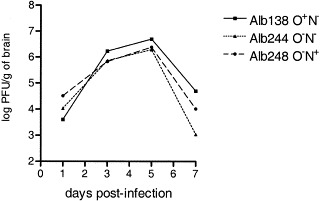
Replication of MHV recombinants in the brain. C57BI/6 mice were inoculated with the indicated recombinant intranasally at a dose of 1.4 × 105 PFU. Mice were sacrificed on days 1, 3, 5, and 7 postinfection and virus titers in the brains were determined by plaque-assay on L2 cell monolayers.
Replication in the liver
Finally, we investigated replication of the different viruses in liver. When mice were inoculated intranasally with a low dose (1 × 103 PFU), virus could never be detected in the brain, but was detected in the liver, in approximately half of the animals. The recombinant viruses appeared to replicate in the liver to a different extent, MHV-Alb248 (O−N+) being able to replicate to the highest titers (data not shown). These results, however, did not differ significantly and therefore prompted us to study viral replication in the liver in a more controllable way by intraperitoneal inoculation. Mice were inoculated with 1 × 105 PFU, and replication in the liver was determined at day 4 postinfection. As Fig. 5Areveals, Alb248 (O−N+) appeared to replicate to a much higher extent than Alb138 (O+N+), which in turn, replicated slightly better than Alb244 (O−N−). Statistical analysis done by Student’s t test revealed the significance of the differences between the titers (Fig. 5A). Similar results were obtained with the other, independently generated recombinant viruses, which confirms the observed phenotypes. The results could further be confirmed by immunohistochemical analysis of the livers (Fig. 5B). To this end, liver samples were fixed in phosphate-buffered formalin, embedded in paraffin, sectioned, and stained with hematoxylin. Viral antigen was detected with the polyclonal anti-MHV serum. A number of small lesions were seen in the livers of mice infected with Alb138 or Alb244. Lesions appeared slightly larger and showed larger necrotic centers after infection with Alb138. In animals infected with Alb248, hepatocellular degeneration and necrosis were so extreme that lesions were no longer separated. These results, reproduced in independent experiments, clearly demonstrated that MHV expressing M proteins with N-linked sugars replicated to a higher extent in the liver than MHV carrying O-linked sugars. MHV containing M proteins without any sugars replicated to the lowest extent.
Fig. 5.
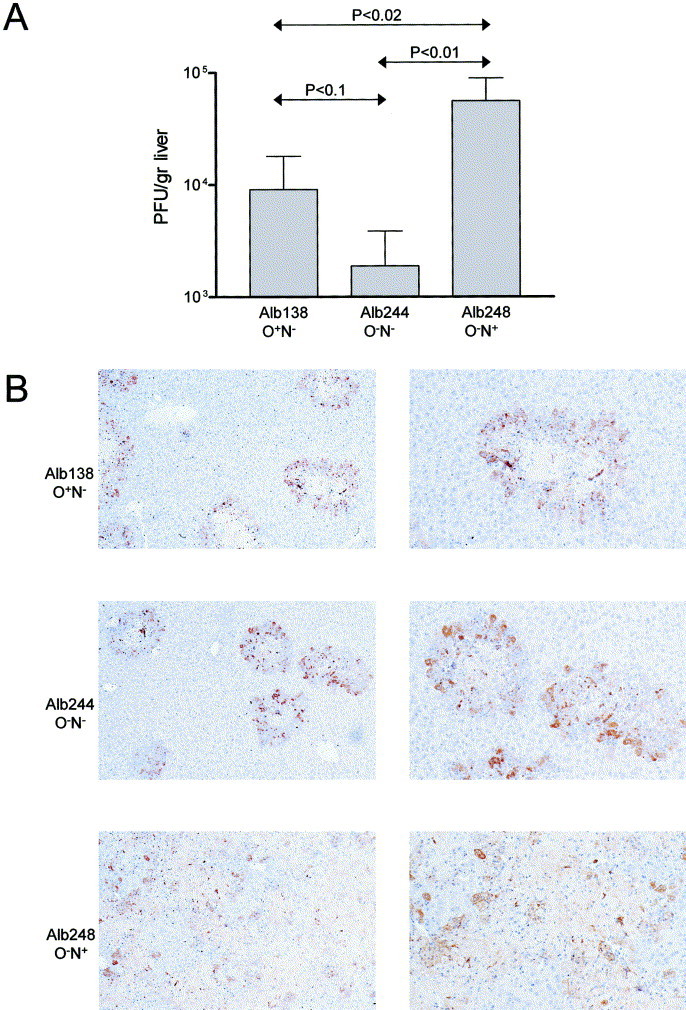
Replication of MHV recombinants in the liver. C57BI/6 mice were inoculated with the indicated recombinant viruses by injection into the intraperitoneal cavity. Mice were sacrificed and the livers were removed at day 4 postinfection. (A) Virus titers were determined by plaque-assay on LR7 cell monolayers following homogenization of the organs. P values determined by Student’s t test are indicated. (B) For immunohistochemical analysis, the liver samples were fixed in phosphate-buffered formalin, embedded in paraffin, sectioned and stained with hematoxylin. Viral antigen was detected with the anti-MHV serum k134, using peroxidase conjugated swine immunoglobulins to rabbit immunoglobulins as secondary antibodies. Peroxidase was visualized using diaminobenzidine. Magnification: ×25 (left side) and ×50 (right side).
Sensitivity to IFNα in vitro and induction of IFNα in the liver
Strikingly, the different extents to which the viruses replicated in the liver appeared to correlate with the different interferogenic capacities of the fixed, virus-infected cells (compare Fig. 3 with Fig. 5A). To study this apparent correlation in more detail, the induction of IFNα in vivo was analyzed. Using a IFN bioassay, we were not able to detect differences in IFN levels in mouse sera early after infection with the MHV recombinants (data not shown). Also, when we measured the IFNα levels in livers of mice that had been infected with recombinant viruses carrying N-glycosylated or unglycosylated M proteins using a mouse IFNα-specific ELISA, very similar IFN levels were observed both at day 1 and at day 3 postinfection (Fig. 6). Apparently, although the glycosylation status of the coronavirus M protein modulates the interferogenic capacity of the viruses in the in vitro assay, the induction of IFNα in vivo does not appear to be affected. Finally, we analyzed whether the differences in viral replication in the liver might be explained by different sensitivities of the recombinant viruses to IFNα. Therefore, the antiviral activity of IFNα against the three types of viruses was analyzed on LR7 cells, taking VSV along as a control. The recombinant viruses displayed a similar sensitivity to IFNα (data not shown), that was much less than that observed for VSV and that cannot explain the differences in viral replication in the liver.
Fig. 6.
Induction of IFNα in liver. C57BI/6 mice were inoculated with the indicated recombinant viruses by injection into the intraperitoneal cavity. Mice were sacrificed and the livers were removed at day 1 and 3 postinfection. IFNα levels were determined using a mouse IFNα ELISA kit and standard deviations are indicated.
Discussion
The coronaviral membrane basically consists of a dense matrix of laterally interacting M proteins, in which the other, much less abundant, envelope proteins are incorporated (de Haan et al., 2000). The oligosaccharides exposed on the coronavirus M protein thus constitute a diffuse, hydrophilic cover on the outer surface of the virion. Oligosaccharide types and their attachment sites on polypeptides are ultimately defined by the viral genome. Due to the lack of editing functions in the RNA-dependent RNA polymerase, replication of the coronaviral genome is prone to mutation at a high frequency. Nevertheless, a remarkably well-conserved dichotomy exists among coronaviruses with respect to the type of M protein glycosylation, which prompted us to search for the presumed function of these sugars. To this end, we used targeted recombination to generate mutant MHVs carrying M proteins that were O-glycosylated, N-glycosylated, or not glycosylated at all. Although no differences were observed between these recombinant viruses when grown in cell culture, in mice glycosylation appeared to influence the ability of the recombinant viruses to replicate in the liver but not in the brain. The glycosylation state of the recombinant viruses also affected the interferogenic capacity of fixed, virus-infected cells in a way that correlated with the extent of viral replication in the liver and, hence, with the degree of hepatic pathology. However, when the interferogenic capacity of the recombinant viruses in the liver was determined, no differences were observed.
Glycosylation of the coronavirus M protein is not important for virus assembly or replication per se. The M protein glycosylation state did not influence the tissue culture growth characteristics of the recombinant viruses in several cell types, nor did it affect replication in the mouse brain (de Haan et al., 2002; this study). These results confirm and extend previous studies, using either glycosylation inhibitors or a viruslike particle assembly system, which demonstrated that glycosylation of the M protein does not play an appreciable role in virion assembly (de Haan et al., 1998a). In addition, both for MHV (strain RI) and TGEV, mutants have been described that contain unglycosylated M proteins. Their M proteins have disrupted O- or N-glycosylation motifs, respectively. The TGEV mutant was demonstrated to replicate efficiently in vitro (Laude et al., 1992), whereas MHV-RI appeared to differ from other MHV strains in its inability to use an isoform of the MHV receptor (mmCGM2 or CEACAM1b) (Compton, 1994). Since the recombinant viruses generated in this study were able to use this MHV receptor isoform to infect the same cells (data not shown), the result obtained with MHV-RI is not due to the lack of O-linked sugars on its M protein but can probably be attributed to mutations in its S protein. Recently, MHV-2 was demonstrated to carry both N-linked and O-linked sugars at its M protein, indicating that both types of glycosylation can occur side by side on the same M protein (Yamada et al., 2000), as we had observed as well (de Haan et al., 1998a).
Glycosylation of the coronavirus M protein is a strongly conserved feature. As a consequence, we expected an important role for the carbohydrates during coronavirus infection of the natural host, even though glycosylation of the M protein was not required for efficient growth in vitro. Indeed, the recombinant viruses differed in their ability to replicate in the liver and to induce hepatitis, although they replicated to the same extent in the brain. Apparently, the difference in their hepatitis inducing abilities does not result from the recombinant viruses having different replication kinetics. Previously, Navas et al. (2001) demonstrated that the extent of replication and the degree of hepatitis in the liver induced by different MHV strains were determined largely by the S protein. Since the S protein is the viral attachment protein and mediates virus–cell fusion this is not surprising. However, we now show that, in addition to the S protein, mutations in the M protein that alter the glycosylation state of the protein affect, in a still unknown way, the outcome of infection in the liver. The strong hepatotropism of MHV-2, although mainly determined by the S protein, may therefore be supported by the attachment of sugars to the M protein both via O- and N-linkages Navas et al 2001, Yamada et al 2000.
Type I IFNs are an important component of the first line of defense against virus infections. They are produced by cells in response to stimulation with several agents, including viruses, bacteria, and synthetic nucleic acids. The induction of type I interferons (IFNs) by most RNA viruses is initiated by virus-derived double-stranded RNA. In addition, many enveloped viruses are able to stimulate, via their glycoproteins, type I IFN production by dendritic cells Fitzgerald-Bocarsly 1993, Ito 1994, Milone and Fitzgerald-Bocarsly 1998, Siegal et al 1999. We now demonstrate for MHV that the glycosylation state of the M protein plays an important role in the induction of type I IFNs in vitro. MHVs in which the M proteins were unglycosylated appeared to be poor IFN inducers compared to MHVs containing glycosylated M proteins. Attachment of oligosaccharide side chains to the M protein via N-linkage resulted in a higher interferogenic capacity than when attached via O-linkage. These results confirm and extend previous studies that demonstrated that several coronaviruses, including MHV, BCoV and TGEV, were able to induce IFNα by their glycoproteins Baudoux et al 1998a, Baudoux et al 1998b. For TGEV, the oligosaccharides N-linked to the M protein were demonstrated to be important for efficient IFNα induction. Mutations in the M protein ectodomain that impaired N-glycosylation decreased the interferogenic activity (Laude et al., 1992), whereas antibodies directed to the TGEV M ectodomain blocked IFNα induction (Charley and Laude, 1988). The TGEV M protein by itself is not able to induce IFN Baudoux et al 1998a, Baudoux et al 1998b. It needs to be coexpressed with its assembly partner E, which suggests that the induction of IFNα is dependent on a specific, probably regularly organized structure of the M protein.
Strikingly, the extent of viral replication in the liver of the different recombinant viruses appeared to correlate with the different interferogenic capacities of the viruses in vitro. These observations seem paradoxical since type I IFNs play an important role in the host defence against viral infections (Muller et al., 1994), whereas several other studies have demonstrated the protective effect of type I IFNs on MHV infection in mice Aurisicchio et al 2000, Matsuyama et al 2000, Smith et al 1987, Uetsuka et al 1996. We observed no differences in the sensitivities of our recombinant viruses to IFNα when analyzed in vitro. Furthermore, we also detected no differences in IFN levels in mouse sera and in liver after inoculation of mice with the different MHV recombinants. Apparently, the different interferogenic capacities of fixed, virus-infected cells in vitro do not necessarily reflect the interferogenic capacities of the replicating viruses in vivo. The different abilities of the recombinant viruses to replicate in the liver are difficult to explain. We speculate that they might correlate with differences in the binding of the viruses to hepatic lectins and, consequently, with their hepatic targeting efficiencies. The induction and production of IFNα in peripheral blood dendritic cells by several enveloped viruses is mediated by the mannose receptor (Milone and Fitzgerald-Bocarsly, 1998). This receptor is a so-called pattern-recognition receptor (Stahl and Ezekowitz, 1998), which has been implicated in the nonspecific recognition of enveloped viruses as well as of other pathogens Milone and Fitzgerald-Bocarsly 1998, Stahl and Ezekowitz 1998. The mannose receptor is also expressed on liver sinusoidal endothelium, where it is an essential regulator of serum glycoprotein homeostasis (Lee et al., 2002). By recognizing their carbohydrates—supposed to function as tags—the receptor appears to target serum glycoproteins for degradation in the liver. Consequently, it is conceivable that the differential recognition of the differently glycosylated MHV recombinants by hepatic lectins such as the mannose receptor modulates the extent of viral replication in the liver in proportion to the in vitro interferogenic capacities of the viruses.
Materials and methods
Cells, viruses, and antibodies
The MHV-A59 mutants Alb138, Alb139, Alb244, Alb246, Alb248, and Alb250 were propagated in mouse 17 clone 1 cells, and plaque assays and purification were carried out using mouse L2 cells. The viruses were radiolabeled and analyzed for their one-step growth kinetics in LR7 cells (Kuo et al., 2000). For the IFNα induction and bioassay LLC-PK1 cells stably expressing the MHV receptor gene (LMR cells; Rossen et al., 1996) were used. PBM cells were obtained from heparinized blood by Ficoll density centrifugation as described before (Charley and Laude, 1988). The VSV strain San Juan was used as a challenge virus in the IFNα bioassay. The rabbit polyclonal MHV strain A59 antiserum k134 has been described earlier (Rottier et al., 1981).
Plasmid constructs
Transcription vectors for the production of donor RNA for targeted recombination were constructed from the plasmid pFV1 (Fischer et al., 1997), which encodes a runoff transcript consisting of the 5′ end of the MHV genome fused to the start of the S gene and running to the 3′ end of the genome (Fig. 1A). An M gene mutant of pFV1, encoding the substitutions T4A and T5A, was made by transfer of the mutations from the previously described plasmid pTUG3-M-A4A5 (de Haan et al., 1998a). A polymerase chain reaction (PCR) product was obtained from pTUG3-M-A4A5 with primer PM303 (5′ATCTAATCCAAACATTATGAGTAGT3′), which produces half of the _Eco_RV site immediately upstream of the M coding region, and primer LK10 (5′CAACAATGCGGTGTCCGCGCCAC3′), which is complementary to the 3′ end of the M coding region. The PCR product was restricted with _Bss_HII and ligated in place of the _Eco_RV–_Bss_HII fragment of pFV1 to produce plasmids pCM1 and pCM2. An M gene mutant of pFV1 designated M-N2N3, encoding an M protein in which Ser2 was replaced by two Asn residues in addition to the substitutions T4A and T5A, was generated by PCR using primer 932 (5′CAAACATTATGAACAACAGTGCTGCTCAGG3′), which directs the desired mutations, and primer 509 (5′GTCTAACATACACGGTACCTTTC3′), corresponding to a unique _Kpn_I site in the M gene, and cloned into pTUG3 in a similar way as described for M gene mutant M-A4A5 (de Haan et al., 1998a). The mutations were transferred from pTUG3-M-N2N3 into pFV1 via a PCR product obtained with primers PM304 (5′ATCTAATCCAAACATTATGAACAACAGT3′) and LK10. The PCR product was restricted with _Bss_HII and ligated in place of the _Eco_RV–_Bss_HII fragment of pFV1 to produce plasmids pCM3 and pCM4. The sequences of all ligation junctions and all PCR-generated segments were confirmed by DNA sequencing.
Construction of MHV mutants
Incorporation of mutations into the MHV genome by targeted RNA recombination was carried out as described previously Masters 1999, Masters et al 1994. Donor RNAs transcribed in vitro from _Hin_dIII-linearized, pFV1-derived plasmids were transfected by electroporation into mouse L2 cells that had been infected with the thermolabile N gene deletion mutant Alb4. The cells were then plated onto a monolayer of mouse 17Cl1 cells. Progeny recombinants that had repaired the N gene deletion were selected as viruses able to form large plaques at the nonpermissive temperature for Alb4 (39°C). Candidates were plaque purified and screened for the presence of the cotransduced mutations in the M gene by reverse transcription–polymerase chain reaction (RT–PCR), on the basis of the loss of a _Sca_I site that occurs in codons 3 and 4 of the wild-type M gene (Fig. 1A). Mutant construction was finally confirmed by direct RNA sequencing of purified genomic RNA (Peng et al., 1995).
Metabolic labeling and immunoprecipitation
LR7 cells and LMR cells were grown and infected with each virus at a m.o.i. of 10 PFU/cell. When indicated, tunicamycin (5 μg/ml) was added to the culture media from 2 h postinfection. Before being labeled, cells were starved for 30 min in cysteine- and methionine-free minimal essential medium containing 10 mM Hepes (pH 7.2) and 5% dialyzed fetal bovine serum (FBS). The medium was then replaced by 600 μl of the same medium containing 100 μCi in vitro cell-labeling mix (Amersham). Cells were labeled from 5 to 8 h postinfection. At the end of the labeling period, culture media were collected, cleared by low-speed centrifugation, and prepared for immunoprecipitation by the addition of 1:4 volume of 5 times concentrated lysis buffer (de Haan et al., 1998a). Proteins were immunoprecipitated from cell lysates as described before (de Haan et al., 1998a). An amount of 2.5 μl of anti-MHV serum k134 was used per immunoprecipitation. Immune complexes were adsorbed to Pansorbin cells (Calbiochem) for 30 min at 4°C and were subsequently collected by centrifugation. Pellets were washed 4 times by resuspension and centrifugation using wash buffers as described previously (de Haan et al., 1998a). The final pellets were suspended in electrophoresis sample buffer. The immunoprecipitates were analyzed by sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS–PAGE) in 15% polyacrylamide gels.
IFN induction and bioassay
LMR cells grown in 16-mm dishes were infected with each virus at a m.o.i. of 0.1 or 10 PFU/cell. At 9.5 h postinfection cells were washed with PBS 3 times and then fixed overnight with 3% formaldehyde at 4°C. Cells were then washed three times with PBS–50 mM glycine and stored in the same buffer. PBM cells were induced to produce IFNα by overnight incubation at 37°C on the TGEV- or MHV-infected, fixed cells (3 × 106 PBM cells/well). Supernatants were collected and serial twofold dilutions were assayed for IFN in an infection inhibition assay on LMR cells using VSV as a challenge virus (Laude et al., 1992). To determine which fraction of the inhibitory effect was contributed by type I IFN, the bioactivity was also assayed after neutralization with a sheep antihuman type I IFN antiserum (La Bonnardiere et al., 1986), which was kindly provided by B. Charley (INRA). The serum was used at a final dilution of 1:100 as described before (Charley and Laude, 1988).
Virulence assay
An LD50 assay was carried out by inoculating 4-week-old, MHV-negative, C57BI/6 mice intracranially with four 10-fold serial dilutions (5 × 105–5 × 102) of recombinant viruses. Mice were anesthetized with Isoflurane (IsoFlo; Abbott Laboratories, North Chicago, IL). Viruses were diluted using PBS containing 0.75% bovine serum albumin. A volume of 25 μl was used for injection into the left cerebral hemisphere. Five animals were analyzed for each dose of virus. LD50 values were calculated by the Reed–Muench method based on death by 21 days postinfection (Reed and Muench, 1938).
Viral replication in brain and liver
Four-week-old C57BI/6 mice were inoculated intracranially (as described above) or intranasally. Five animals per dose of virus were analyzed. Mice were sacrificed on days 1, 3, 5, and 7 postinfection and perfused with 10 ml PBS, and the brains and liver were removed. All organs were weighed and then frozen at −80°C until they were titered for virus. Virus titers were determined by plaque-assay on L2 cell monolayers following homogenization of the organs (Hingley et al., 1994). For analysis of viral replication in the liver, 8-week-old C57BI/6 mice were inoculated by injection of 100-μl virus dilution into the intraperitoneal cavity. Mice were sacrificed and the livers were removed at day 4 postinfection. Livers were placed in 1.5 ml DMEM, weighed, and then frozen at −80°C until titered for virus. Virus titers were determined by plaque assay on LR7 cell monolayers following homogenization of the organs. For immunohistochemical analysis, the liver samples were fixed in phosphate-buffered formalin, embedded in paraffin, sectioned, and stained with hematoxylin. Viral antigen was detected with k134 at a 1:400 dilution. Peroxidase-conjugated swine immunoglobulins to rabbit immunoglobulins (Dakopatts) were used as secondary antibodies at a 1:100 dilution. Peroxidase was visualized using diaminobenzidine.
IFN induction in liver
Eight-week-old C57BI/6 mice were inoculated by injection of 100 μl of virus dilution (1.0 × 105 PFU) into the intraperitoneal cavity. Mice were sacrificed and the livers were removed at days 1 and 3 postinfection. Livers were placed in 1.5 ml DMEM, weighed, and then stored frozen at −80°C until use. Following homogenization of the organs, IFNα levels were determined using a mouse IFNα ELISA kit according to the manufacturer’s instructions (PBL).
Acknowledgements
We gratefully acknowledge Su-hun Seo for excellent technical assistance with the virulence assays, Frank van der Meer for providing the porcine blood, our colleagues at the Department of Pathology of the Faculty of Veterinary Medicine, Utrecht University, for their advice and assistance with the preparation of the liver sections, and John Jansen for photographing the liver sections. We thank B. Charley for providing the sheep antihuman type I IFN antiserum and Kay Holmes for providing the BHK cells expressing CEACAM1b. This work has been carried out in part with financial support from the Commission of the European Communities specific RTD program, “Quality of Life and Management of Living Resources,” QLK2-CT-1999-00002, “Generic coronavirus vaccine vectors for protection of farm animals against mucosal infections” to C.A.M.d.H. and P.J.M.R. It does not necessarily reflect its views and in no way anticipates the Commission’s future policy in this area. This work was also supported in part by Public Health Service Grants AI-39544 and AI-17418 from the National Institutes of Health to P.S.M. and S.R.W., respectively.
References
- Alexander S., Elder J.H. Carbohydrate dramatically influences immune reactivity of antisera to viral glycoprotein antigens. Science. 1984;226:1328–1330. doi: 10.1126/science.6505693. [DOI] [PubMed] [Google Scholar]
- Aurisicchio L., Delmastro P., Salucci V., Paz O.G., Rovere P., Ciliberto G., La Monica N., Palombo F. Liver-specific alpha 2 interferon gene expression results in protection from induced hepatitis. J. Virol. 2000;74:4816–4823. doi: 10.1128/jvi.74.10.4816-4823.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baudoux P., Besnardeau L., Carrat C., Rottier P., Charley B., Laude H. Interferon alpha inducing property of coronavirus particles and pseudoparticles. Adv. Exp. Med. Biol. 1998;440:377–386. doi: 10.1007/978-1-4615-5331-1_49. [DOI] [PubMed] [Google Scholar]
- Baudoux P., Carrat C., Besnardeau L., Charley B., Laude H. Coronavirus pseudoparticles formed with recombinant M and E proteins induce alpha interferon synthesis by leukocytes. J. Virol. 1998;72:8636–8643. doi: 10.1128/jvi.72.11.8636-8643.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braakman I., van Anken E. Folding of viral envelope glycoproteins in the endoplasmic reticulum. Traffic. 2000;1:533–539. doi: 10.1034/j.1600-0854.2000.010702.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charley B., Laude H. Induction of alpha interferon by transmissible gastroenteritis coronavirus: role of transmembrane glycoprotein E1. J. Virol. 1988;62:8–11. doi: 10.1128/jvi.62.1.8-11.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Compton S.R. Enterotropic strains of mouse coronavirus differ in their use of murine carcinoembryonic antigen-related glycoprotein receptors. Virology. 1994;203:197–201. doi: 10.1006/viro.1994.1475. doi:10.1006/viro.1994.1475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Haan C.A.M., de Wit M., Kuo L., Montalto C., Masters P.S., Weiss S.R., Rottier P.J.M. O-glycosylation of the mouse hepatitis coronavirus membrane protein. Virus Res. 2002;82:77–81. doi: 10.1016/S0168-1702(01)00390-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Haan C.A.M., Vennema H., Rottier P.J.M. Assembly of the coronavirus envelope: homotypic interactions between the M proteins. J. Virol. 2000;74:4967–4978. doi: 10.1128/jvi.74.11.4967-4978.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Haan C.A.M., Kuo L., Masters P.S., Vennema H., Rottier P.J.M. Coronavirus particle assembly: primary structure requirements of the membrane protein. J. Virol. 1998;72:6838–6850. doi: 10.1128/jvi.72.8.6838-6850.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Haan C.A.M., Roestenberg P., de Wit M., de Vries A.A.F., Nilsson T., Vennema H., Rottier P.J.M. Structural requirements for O-glycosylation of the mouse hepatitis virus membrane protein. J. Biol. Chem. 1998;273:29905–29914. doi: 10.1074/jbc.273.45.29905. [DOI] [PubMed] [Google Scholar]
- de Haan C.A.M., Smeets M., Vernooij F., Vennema H., Rottier P.J.M. Mapping of the coronavirus membrane protein domains involved in interaction with the spike protein. J. Virol. 1999;73:7441–7452. doi: 10.1128/jvi.73.9.7441-7452.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delmas B., Laude H. Assembly of coronavirus spike protein into trimers and its role in epitope expression. J. Virol. 1990;64:5367–5375. doi: 10.1128/jvi.64.11.5367-5375.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drickamer K., Taylor M.E. Evolving views of protein glycosylation. Trends Biochem. Sci. 1998;23:321–324. doi: 10.1016/s0968-0004(98)01246-8. [DOI] [PubMed] [Google Scholar]
- Fischer F., Stegen C.F., Koetzner C.A., Masters P.S. Analysis of a recombinant mouse hepatitis virus expressing a foreign gene reveals a novel aspect of coronavirus transcription. J. Virol. 1997;71:5148–5160. doi: 10.1128/jvi.71.7.5148-5160.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fitzgerald-Bocarsly P. Human natural interferon-alpha producing cells. Pharmacol. Ther. 1993;60:39–62. doi: 10.1016/0163-7258(93)90021-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Godeke G.J., de Haan C.A.M., Rossen J.W., Vennema H., Rottier P.J.M. Assembly of spikes into coronavirus particles is mediated by the carboxy-terminal domain of the spike protein. J. Virol. 2000;74:1566–1571. doi: 10.1128/jvi.74.3.1566-1571.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Helenius A., Aebi M. Intracellular functions of N-linked glycans. Science. 2001;291:2364–2369. doi: 10.1126/science.291.5512.2364. [DOI] [PubMed] [Google Scholar]
- Hingley S.T., Gombold J.L., Lavi E., Weiss S.R. MHV-A59 fusion mutants are attenuated and display altered hepatotropism. Virology. 1994;200:1–10. doi: 10.1006/viro.1994.1156. doi:10.1006/viro.1994.1156. [DOI] [PubMed] [Google Scholar]
- Holmes K.V., Doller E.W., Sturman L.S. Tunicamycin resistant glycosylation of coronavirus glycoprotein: demonstration of a novel type of viral glycoprotein. Virology. 1981;115:334–344. doi: 10.1016/0042-6822(81)90115-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ito Y. Induction of interferon by virus glycoprtein(s) in lymphoid cells through interaction with the cellular receptors via lectin-like action: an alternative interferon induction mechanism. Arch. Virol. 1994;138:187–198. doi: 10.1007/BF01379125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krijnse Locker J., Griffiths G., Horzinek M.C., Rottier P.J.M. O-glycosylation of the coronavirus M protein. Differential localization of sialyltransferases in N- and O-linked glycosylation. J. Biol. Chem. 1992;267:14094–14101. doi: 10.1016/S0021-9258(19)49683-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuo L., Godeke G.J., Raamsman M.J., Masters P.S., Rottier P.J.M. Retargeting of coronavirus by substitution of the spike glycoprotein ectodomain: crossing the host cell species barrier. J. Virol. 2000;74:1393–1406. doi: 10.1128/jvi.74.3.1393-1406.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- La Bonnardiere C., Laude H., Berg K. Biological and antigenic relationships between virus-induced porcine and human interferons. Ann. Inst. Pasteur Virol. 1986;137E:171–180. doi: 10.1016/S0769-2617(86)80202-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laude H., Gelfi J., Lavenant L., Charley B. Single amino acid changes in the viral glycoprotein M affect induction of alpha interferon by the coronavirus transmissible gastroenteritis virus. J. Virol. 1992;66:743–749. doi: 10.1128/jvi.66.2.743-749.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee S.J., Evers S., Roeder D., Parlow A.F., Risteli J., Risteli L., Lee Y.C., Feizi T., Langen H., Nussenzweig M.C. Mannose receptor-mediated regulation of serum glycoprotein homeostasis. Science. 2002;295:1898–1901. doi: 10.1126/science.1069540. [DOI] [PubMed] [Google Scholar]
- Masters P.S. Reverse genetics of the largest RNA viruses. Adv. Virus Res. 1999;53:245–264. doi: 10.1016/S0065-3527(08)60351-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masters P.S., Koetzner C.A., Kerr C.A., Heo Y. Optimization of targeted RNA recombination and mapping of a novel nucleocapsid gene mutation in the coronavirus mouse hepatitis virus. J. Virol. 1994;68:328–337. doi: 10.1128/jvi.68.1.328-337.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuyama S., Henmi S., Ichihara N., Sone S., Kikuchi T., Ariga T., Taguchi F. Protective effects of murine recombinant interferon-beta administered by intravenous, intramuscular or subcutaneous route on mouse hepatitis virus infection. Antiviral Res. 2000;47:131–137. doi: 10.1016/s0166-3542(00)00097-8. [DOI] [PubMed] [Google Scholar]
- Mayer T., Tamura T., Falk M., Niemann H. Membrane integration and intracellular transport of the coronavirus glycoprotein E1, a class III membrane glycoprotein. J. Biol. Chem. 1988;263:14956–14963. doi: 10.1016/S0021-9258(18)68131-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milone M.C., Fitzgerald-Bocarsly P. The mannose receptor mediates induction of IFN-alpha in peripheral blood dendritic cells by enveloped RNA and DNA viruses. J. Immunol. 1998;161:2391–2399. [PubMed] [Google Scholar]
- Muller U., Steinhoff U., Reis L.F., Hemmi S., Pavlovic J., Zinkernagel R.M., Aguet M. Functional role of type I and type II interferons in antiviral defense. Science. 1994;264:1918–1921. doi: 10.1126/science.8009221. [DOI] [PubMed] [Google Scholar]
- Narayanan K., Maeda A., Maeda J., Makino S. Characterization of the coronavirus M protein and nucleocapsid interaction in infected cells. J. Virol. 2000;74:8127–8134. doi: 10.1128/jvi.74.17.8127-8134.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Navas S., Seo S.H., Chua M.M., Sarma J.D., Lavi E., Hingley S.T., Weiss S.R. Murine coronavirus spike protein determines the ability of the virus to replicate in the liver and cause hepatitis. J. Virol. 2001;75:2452–2457. doi: 10.1128/JVI.75.5.2452-2457.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen V.P., Hogue B.G. Protein interactions during coronavirus assembly. J. Virol. 1997;71:9278–9284. doi: 10.1128/jvi.71.12.9278-9284.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niemann H., Boschek B., Evans D., Rosing M., Tamura T., Klenk H.-D. Post-translational glycosylation of coronavirus glycoprotein E1: inhibition by monensin. EMBO J. 1982;1:1499–1504. doi: 10.1002/j.1460-2075.1982.tb01346.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Opstelten D.J., Raamsman M.J., Wolfs K., Horzinek M.C., Rottier P.J. Envelope glycoprotein interactions in coronavirus assembly. J. Cell Biol. 1995;131:339–349. doi: 10.1083/jcb.131.2.339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng D., Koetzner C.A., McMahon T., Zhu Y., Masters P.S. Construction of murine coronavirus mutants containing interspecies chimeric nucleocapsid proteins. J. Virol. 1995;69:5475–5484. doi: 10.1128/jvi.69.9.5475-5484.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reed L.J., Muench H. A simple method of estimating fifty percent points. Am. J. Hygeine. 1938;27:493–497. [Google Scholar]
- Rossen J.W., de Beer R., Godeke G.J., Raamsman M.J., Horzinek M.C., Vennema H., Rottier P.J.M. The viral spike protein is not involved in the polarized sorting of coronaviruses in epithelial cells. J. Virol. 1998;72:497–503. doi: 10.1128/jvi.72.1.497-503.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rossen J.W.A., Bekker C.P.J., Strous G.J.A.M., Horzinek M.C., Dveksler G.S., Holmes K.V., Rottier P.J.M. A murine and a porcine coronavirus are released from opposite surfaces of the same epithelial cells. Virology. 1996;224:345–351. doi: 10.1006/viro.1996.0540. doi:10.1006/viro.1996.0540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rottier P.J.M., Horzinek M.C., van der Zeijst B.A. Viral protein synthesis in mouse hepatitis virus strain A59-infected cells: effect of tunicamycin. J. Virol. 1981;40:350–357. doi: 10.1128/jvi.40.2.350-357.1981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rottier P.J.M. The coronavirus membrane protein. In: Siddell S.G., editor. The Coronaviridae. Plenum Press; New York: 1995. pp. 115–139. [Google Scholar]
- Rottier P.J.M., Krijnse Locker J., Horzinek M.C., Spaan W.J.M. Expression of MHV-A59 M glycoprotein: effects of deletions on membrane integration and intracellular transport. Adv. Exp. Med. Biol. 1990;276:127–135. doi: 10.1007/978-1-4684-5823-7_19. [DOI] [PubMed] [Google Scholar]
- Rudd P.M., Dwek R.A. Glycosylation: heterogeneity and the 3D structure of proteins. Crit. Rev. Biochem. Mol. Biol. 1997;32:1–100. doi: 10.3109/10409239709085144. [DOI] [PubMed] [Google Scholar]
- Schulze I.T. Effects of glycosylation on the properties and functions of influenza virus hemagglutinin. J. Infect. Dis. 1997;176(Suppl 1):S24–S28. doi: 10.1086/514170. [DOI] [PubMed] [Google Scholar]
- Siegal F.P., Kadowaki N., Shodell M., Fitzgerald-Bocarsly P.A., Shah K., Ho S., Antonenko S., Liu Y.J. The nature of the principal type 1 interferon-producing cells in human blood. Science. 1999;284:1835–1837. doi: 10.1126/science.284.5421.1835. [DOI] [PubMed] [Google Scholar]
- Smith A.L., Barthold S.W., Beck D.S. Intranasally administered alpha/beta interferon prevents extension of mouse hepatitis virus, strain JHM, into the brains of BALB/cByJ mice. Antiviral Res. 1987;8:239–245. doi: 10.1016/S0166-3542(87)80002-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stahl P.D., Ezekowitz R.A. The mannose receptor is a pattern recognition receptor involved in host defense. Curr. Opin. Immunol. 1998;10:50–55. doi: 10.1016/s0952-7915(98)80031-9. [DOI] [PubMed] [Google Scholar]
- Stern D.F., Sefton B.M. Coronavirus proteins: structure and function of the oligosaccharides of the avian infectious bronchitis virus glycoproteins. J. Virol. 1982;44:804–812. doi: 10.1128/jvi.44.3.804-812.1982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tooze S.A., Tooze J., Warren G. Site of addition of N-acetyl-galactosamine to the E1 glycoprotein of mouse hepatitis virus-A59. J. Cell Biol. 1988;106:1475–1487. doi: 10.1083/jcb.106.5.1475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uetsuka K., Nakayama H., Goto N. Protective effect of recombinant interferon (IFN)-alpha/beta on MHV-2cc-induced chronic hepatitis in athymic nude mice. Exp. Anim. 1996;45:293–297. doi: 10.1538/expanim.45.293. [DOI] [PubMed] [Google Scholar]
- Van den Steen P., Rudd P.M., Dwek R.A., Opdenakker G. Concepts and principles of O-linked glycosylation. Crit. Rev. Biochem. Mol. Biol. 1998;33:151–208. doi: 10.1080/10409239891204198. [DOI] [PubMed] [Google Scholar]
- Vennema H., Godeke G.J., Rossen J.W., Voorhout W.F., Horzinek M.C., Opstelten D.J., Rottier P.J.M. Nucleocapsid-independent assembly of coronavirus-like particles by co-expression of viral envelope protein genes. EMBO J. 1996;15:2020–2028. doi: 10.1002/j.1460-2075.1996.tb00553.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vennema H., Heijnen L., Zijderveld A., Horzinek M.C., Spaan W.J. Intracellular transport of recombinant coronavirus spike proteins: implications for virus assembly. J. Virol. 1990;64:339–346. doi: 10.1128/jvi.64.1.339-346.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamada Y.K., Yabe M., Ohtsuki T., Taguchi F. Unique N-linked glycosylation of murine coronavirus MHV-2 membrane protein at the conserved O-linked glycosylation site. Virus Res. 2000;66:149–154. doi: 10.1016/S0168-1702(99)00134-3. [DOI] [PMC free article] [PubMed] [Google Scholar]