Forming functional fat: a growing understanding of adipocyte differentiation (original) (raw)
. Author manuscript; available in PMC: 2020 Apr 21.
Published in final edited form as: Nat Rev Mol Cell Biol. 2011 Sep 28;12(11):722–734. doi: 10.1038/nrm3198
PREFACE
Adipose tissue, which is primarily composed of adipocytes, is crucial for maintaining energy and metabolic homeostasis. Adipogenesis is thought to occur in two stages: commitment of mesenchymal stem cells to a preadipocyte fate and terminal differentiation. Cell shape and extracellular matrix remodelling have recently been described to regulate preadipocyte commitment and competency by modulating WNT and RHO GTPase signalling cascades. Adipogenic stimuli induce terminal differentiation in committed preadipocytes through the epigenomic activation of PPARγ. Coordination of PPARγ with C/EBP transcription factors maintains adipocyte gene expression. A better understanding of these mechanisms may identify therapeutic targets for the growing worldwide epidemic of metabolic disease.
INTRODUCTION
Adipose tissue is a complex organ that regulates and coordinates energy homeostasis1. It is primarily composed of adipocytes surrounded by fibroblasts, fibroblastic preadipocytic cells, endothelial cells, nerves and immune cells2. Although adipose tissue was originally thought to just be an energy storage site, studies in the past years have revealed that it carries out many key endocrine functions1. Indeed, dysfunction of the adipose compartment is central to the pathology associated with metabolic diseases such as obesity, type 2 diabetes, cancer cachexia and lipodystrophies1.
There are two main types of adipose tissue, white and brown; these are primarily composed of white or brown adipocytes, respectively2. White adipose tissue (WAT), which is characterized by adipocytes containing large unilocular lipid droplets, is an active endocrine organ that regulates diverse activities such as insulin sensitivity, lipid metabolism and satiety1. WAT the main type of adipose tissue found in adult humans and is distributed throughout the body in subcutaneous regions, surrounding visceral organs and in the face. Interestingly, despite their histological similarities, subcutaneous and visceral adipose tissue are thought to have distinct depot-specific metabolic functions3, possibly owing to different exposures to paracrine and endocrine signals4 or to distinct developmental programmes5,6. Indeed, accumulation of visceral fat during the development of obesity is correlated with pathologic inflammation and insulin resistance7, whereas subcutaneous adipose tissue is thought to offer improved glucose tolerance8.
By contrast, brown adipose tissue (BAT) mainly participates in thermogenesis and is located in discrete pockets in the paravertebral, supraclavicular and periadrenal regions9. BAT is histologically distinct from WAT; it is composed of multiloculated adipocytes that contain large numbers of mitochondria, accounting for their ‘brown’ colouring upon visualization9. For many years, BAT was thought to be absent in human adults, but recent fluorodeoxyglucose positron emission tomography studies of normal humans has identified regions of high glucose uptake that represent metabolically active tissue10–12.
Despite the functional, developmental and location differences between white and brown adipocytes, the two cell types share many common differentiation features. All adipocytes, along with osteoblasts, myocytes and chondrocytes, differentiate from mesenchymal stems cells (MSCs)13 (Figure 1) in a process known as adipogenesis. Adipogenesis can be divided into two main phases: commitment and terminal differentiation. This Review focuses on the integration of underappreciated and novel mechanical and molecular cues governing the conversion of MSCs to committed preadipocytes and the epigenomic transition state that is required for the activation of the master adipogenic regulator peroxisome proliferator-activated receptor-γ (PPARγ). We also discuss the signalling cascades that lead to terminal differentiation and the conservation of these pathways between species. The Review focuses on white fat, but a discussion of brown fat is given where appropriate.
Figure 1: Cues influencing white adipogenic progression.

Differentiation of multipotent mesenchymal stem cells (MSCs) to mature adipocytes involves a complex integration of cytoarchitecture, signalling pathways, and transcriptional regulators. The first step of adipogenesis is the transition of embryonic stem cells to MSCs (not shown.) MSCs then transition to committed white preadipocytes (mediated by factors such as cell shape, confluency or matrix stiffness). Alternatively, MSCs can be stimulated to differentiate into myoblasts, chondrocytes or osteoblasts. Committed white preadipocytes can become mature white adipocytes upon addition of adipogenic stimuli, such as glucocorticoids, insulin and cyclin AMP. Factors emphasized in this Review are shown along the adipogenic progression at the stage where they act on precursor cells. Factors that have been shown to play a part during different phases of adipogenesis are listed multiple times.
IN VIVO DEVELOPMENT OF FAT
White adipose compartments begins to develop in late gestation14,15 and the rate of adipogenesis rapidly surges in response to increased nutrient availability, leading to a marked postnatal expansion of adipose compartments15. During this period, precursor cells undergo the morphological and functional conversion from spindly fibroblasts into round lipid-laden mature fat cells. By contrast, mammalian subscapular fat, which is mostly BAT, expands primarily in utero, possibly to maintain body temperature upon birth9.
Whether adipogenesis occurs in the adult adipose tissue remains controversial. In animal studies, white adipocyte numbers increase through puberty but are relatively steady in the mature fat pad16. Similar findings have been obtained in humans17; however, within human adult WAT, adipocytes seem to undergo approximately a 10% annual turnover17. Thus, adipogenesis takes place in adults to maintain the adipose compartment. Additionally, adipogenesis probably has a role in the pathology of obesity; when animals are kept on a high-fat diet, adipocyte cell size initially increases, followed by an increase in fat cell number upon prolonged over-nutrition18–20. Human studies correlating fat mass with cell number have yielded conflicting results17,21,22, but this may be attributed to differences in study protocols and basal characteristics of the studied populations. Moreover, short-term overfeeding studies in adults demonstrate increases in adipocyte cell numbers21. By contrast, preclinical and human studies have shown that weight loss is associated with decreased adipocyte cell size, but has no effect on adipocyte cell numbers17,19. Further work must be carried out to determine whether increased adipogenesis has a crucial role in the development of obesity in humans.
Interestingly, lineage studies (Box 1) have revealed that BAT is more closely related to skeletal muscle than WAT, as both muscle and BAT have progenitors that express the early muscle marker MYF5 but WAT does not23. An inducible Pax7-Cre model shows that BAT and muscle share a common Pax7-expressing progenitor as late as embryonic day 10.524. Knockdown of the zinc finger transcriptional co-regulator PR domain-containing protein 16 (Prdm16) in primary brown preadipocytes leads to increased myocyte gene expression and promotes skeletal muscle morphology23. This surprising finding means that in early development there is a divergence between white and brown precursor cells (Figure 2). In addition, upon prolonged cold exposure or in response to β-adrenergic signalling, WAT can display characteristics of BAT, such as expression of uncoupling protein 1 (UCP1)25, which is associated with improved metabolic profiles in response to high fat diet26,27. The ‘brown’-like adipocytes within WAT are developmentally distinct from brown adipocytes found in BAT23; thus, the plasticity of WAT in response to these stimuli may be due to transdifferentiation of mature white adipocytes into brown adipocytes28 or the result of de novo adipocyte formation.
Box 1: Lineage tracing studies.
Mouse models have been crucial for understanding the relationship between undifferentiated cells in a developing embryo and the mature differentiated cell types that develop from progenitor populations. Lineage tracing studies genetically and permanently identify cell populations that may express a precursor gene only for a short period of time by taking advantage of recombination systems such as Cre-lox, a system to knock out alleles, which have been engineered to contain loxP sequences that direct Cre recombinase-mediated recombination162. For example, mice expressing the Rosa26R3-YFP, a loxP flanked reporter gene, and expressing Cre recombinase from the Myf5 promoter were used to permanently indicate all cells that at one point during development expressed MYF5, an early marker of skeletal muscle development23. All tissues in adult mice containing progenitors that currently or transiently expressed MYF5 would be marked with YFP expressed from the Rosa26 promoter, which is active in all cells. Skeletal muscle and brown fat were both positive for YFP expression in this model, whereas white adipose tissue was not, suggesting that skeletal muscle and brown fat are more closely related in lineage than white and brown fat23. However, the in vivo precursor population that can differentiate into muscle or brown fat, but not white fate, has yet to be discovered.
Lineage tracing based on expression of Pparg in mice has provided insights into in vivo white adipogenesis, particularly with the discovery of committed preadipocyte precursors neighbouring blood vessels within the mature fat pad14,127. This lineage tracing method coupled with timed feeding of animals with nucleotide analogues127 can be used to assess the kinetics of adipogenesis in vivo.
Figure 2: Relationship between white and brown adipogenesis.
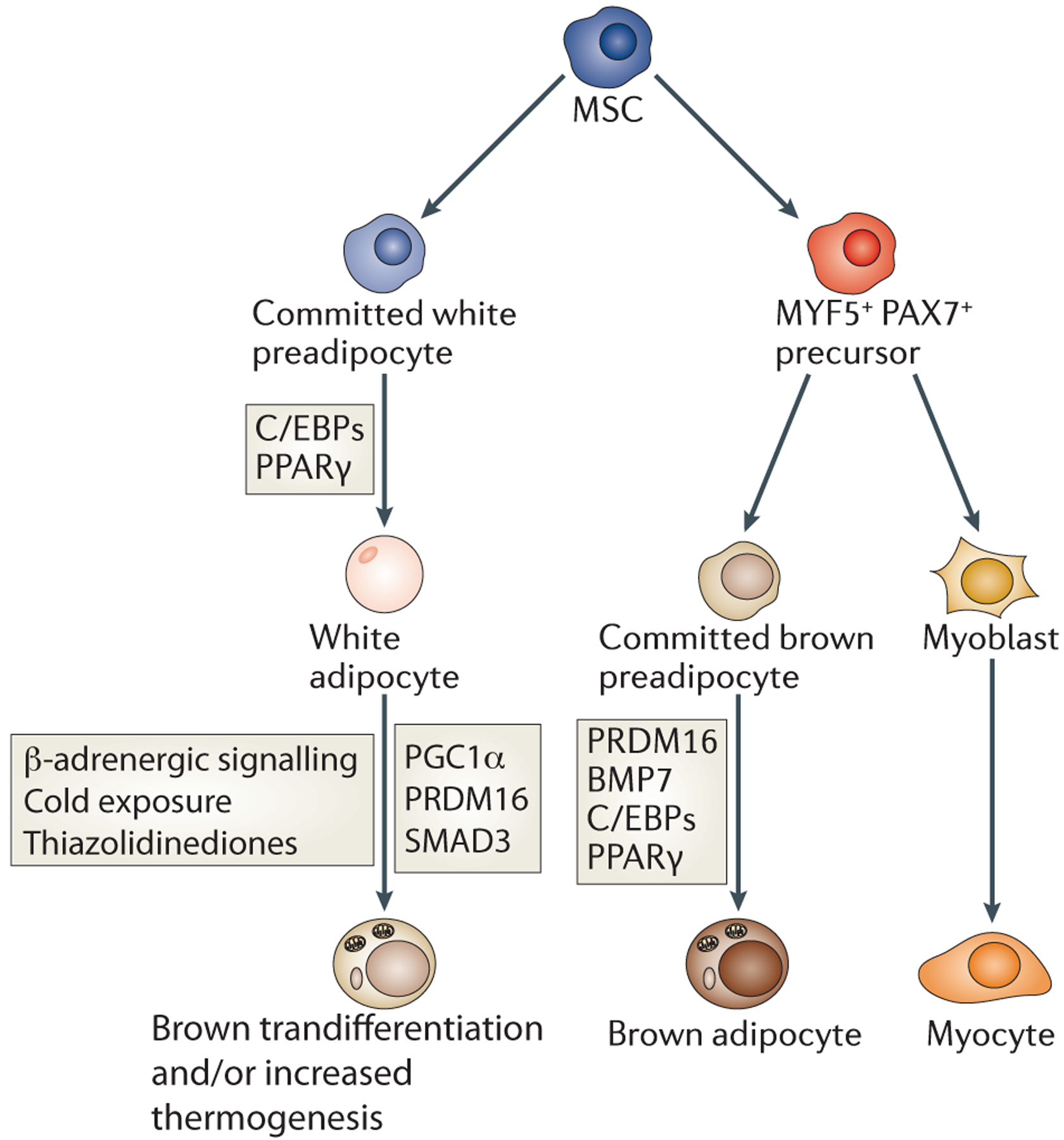
Historically, white and brown adipocytes were thought to derive from the same precursor cell. However, brown adipocytes instead share a common MYF5/PAX7-positive precursor with muscle cells (Box 1). The MYF5/PAX7-positive precursor is driven to brown adipocyte terminal differentiation by PPARγ and C/EBPs cooperating with the transcriptional co-regulator PRDM16. By contrast, PRDM16 does not affect white adipogenesis. White adipocytes can also be stimulated to display characteristics of brown adipocytes by cold exposure, β-adrenergic signalling, and thiazolidinediones, which appear to function via the indicated factors.
Despite advances in tracing adipocyte development in vivo, studies of factors that regulate adipogenesis have frequently been limited to in vitro models (in particular mouse 3T3-L1 cells) because of the inadequacy of current models to study adipocyte differentiation in vivo (Box 2).
Box 2: The case for developing tools to study in vivo adipogenesis.
Much of what is known about factors required for adipogenesis is based on studies of adipogenic cell lines such as mouse 3T3-L1 cells83. The differentiation of these cells into adipocytes can be achieved in vitro by adipogenic stimuli, including glucocorticoids, cyclic AMP agonists and insulin163–165. Other in vitro models of adipogenesis, such as mouse 3T3-F442A cells and human SGBS cells, have many similar properties with 3T3-L1 cells84,166. Although these in vitro models share many similarities with primary adipocytes, including triglyceride storage, insulin sensitivity, expression of adipocyte genes (such as GLUT4 and FABP4 (also known as AP2)) and factor secretion167,168, there are some important differences. For example, triglycerides are stored in many droplets in the cell lines, whereas white adipocytes characteristically contain a single large droplet83. Also, some adipogenic cell lines, including 3T3-L1 and 3T3-F442A cells, express the adipocyte secreted factor leptin at much lower levels than primary adipocytes167.
Ideal mouse models have yet to be developed for studying adipogenesis. Most studies use the Fabp4 promoter-enhancer169 to drive adipocyte-specific expression of factors of interest or the Cre recombinase. However, this model system has major limitations for studying adipogenesis. First, FABP4 is expressed during terminal differentiation124,170 in a PPARγ-dependent manner128. Thus, PPARγlox/lox/FABP4-Cre mice (in which PPARγ has been conditionally deleted in adipocytes) develop WAT151, despite significant evidence that PPARγ is required for in vivo adipogenesis125 and is a marker for preadipocytes within the adipose compartment14. Second, the 5.4 kb upstream proximal promoter of Fabp4 used in these models can drive transgene expression ectopically in macrophages171, during embryonic development172 and in the brain173, raising questions about the specific involvement of adipocytes in the phenotypes of mouse models developed using Fabp4 transgenes. Adiponectin-Cre models have also been developed and seem to be more adipocyte-specific than FABP4174; however, adiponectin is generally expressed in mature adipocytes and not precursor cells. The study of adipogenesis in vivo will require the development of models in which factors can be expressed or deleted in multipotent cells and committed preadipocytes. Prx1-Cre is activated mid-gestation and has recently been suggested as one such tool to study osteogenic and adipogenic cell-fate decisions in vivo175.
ADIPOGENIC COMPETENCY AND COMMITMENT
Preadipocytes are defined as cells that are restricted to becoming adipocytes, but do not spontaneously undergo terminal differentiation in the absence of exogenous adipogenic stimuli. Here we distinguish between adipogenic competency and commitment as follows: adipogenic competency refers to the ability of cells to undergo adipocytic differentiation upon the addition of defined stimuli, whereas adipogenic commitment refers to the cell fate decision of a multipotent cell type to undergo adipocyte conversion.
Identification of preadipocytes in vivo.
Defining the characteristics distinguishing the morphologically similar adipogenic and non-adipogenic fibroblasts had proven difficult until recent years. Technological advances in flow cytometry, transgenic animals and identification of stem cell surface markers in other tissues have made it possible to isolate the subpopulation of fibroblasts in the stromal vascular fraction (SVF) of WAT that has adipogenic potential14,29. Such stem cell surface markers can be used to sort cell populations in the SVF and test them for adipogenic potential. Lineage tracing studies based on the expression of PPARγ, the master regulator of adipogenesis (see below), have identified dividing adipogenic precursor cells within the SVF that dominate the adipose compartment during the first month of postnatal development (Box 1)14. When transplanted into wild-type mice, these PPARγ-positive precursor cells could differentiate into adipocytes14. Both sorted SVF and genetically marked preadipocytes are perivascular within the developing adipose tissue and mature adipose organ14,29. Interestingly, transplantation of isolated adipogenic precursors (Lin−CD29+CD34+Sca1+CD24+ cells) rescued the diabetic phenotype in lipodystrophic A-Zip mice29, serving as a proof of principle for using primary preadipocytes to ameliorate adipose-related metabolic disease.
Brown preadipocytes that are Sca+ have been isolated from BAT, subcutaneous WAT and muscle30. However, further studies are required to determine whether they also express low, but detectable, levels of PPARγ. The isolation of these adipogenic fibroblast populations from WAT and BAT will allow a more complete understanding of how the pathways described throughout this Review are regulated during the development of adipose tissue.
WNT signalling.
Wingless-type MMTV integration site family members (WNTs) are secreted glycoproteins that have key roles during development. Canonical WNT signalling is activated following the binding of WNT ligands to the heterodimeric cell surface receptors low density lipoprotein receptor-related protein 5 (LRP5) and LRP6 and Frizzled. This induces the family of TCF transcription factors to recruit a β-catenin-dependent co-activator complex to activate target gene transcription31. In the context of adipogenesis, these include cyclin D1 and the nuclear receptor COUP-TFII32, although the role of COUP-TFII in adipogenesis is controversial33,34.
Canonical WNT signalling has been shown to inhibit adipogenesis31 (Figure 3): addition of a canonical WNT ligand to committed preadipocytes inhibits adipogenesis35, and mouse embryonic fibroblasts (MEFs) lacking the canonical WNT receptor LRP6 display increased adipocyte differentiation36. Moreover, mice expressing WNT10b, the main WNT ligand expressed by preadipocytes, in adipocytes show decreases in white and brown fat tissue mass37. WNT10b promotes osteogenesis in MSCs38, indicating that canonical WNT signalling also regulates brown adipogenesis and MSC cell fate. Repression of WNT10b and other canonical ligands by the histone methyltransferase EZH2 in white primary preadipocytes is required for adipocyte differentiation39.
Figure 3: WNT signalling in adipogenesis.

In the presence of canonical WNT ligands, such as WNT10B, β-catenin translocates into the nucleus, where it recruits a co-activator complex to TCF transcription factors and activates WNT target genes. In committed preadipocytes this pathway promotes cell survival, but inhibits adipogenesis. However, the WNT targets that inhibit adipogenesis are not completely understood, it is known that activation of this pathway in MSCs promotes osteogenesis. WNT5B, a non-canonical WNT ligand, promotes adipogenesis by inhibiting β-catenin nuclear localization to these targets. The non-canonical ligand WNT5A also signals to inhibit adipogenesis and promote osteogenesis. This is achieved through the activation of the histone methyltransferase SET domain bifurcated 1 (SETDB1) following the assembly of a SETDB1–NLK–CHD7 complex, inhibiting target gene transcription. It remains unclear which receptors are critical to non-canonical WNT signalling in adipogenesis.
However, there is also evidence that the canonical WNT pathway is essential for survival of adipocyte precursors. WNT10b increases in confluent cultures of 3T3-L1 cells35, and WNT1 can protect preadipocytes from apoptosis during serum starvation by regulating the expression of insulin growth factor 1 (IGF1) and IGF240 (which are known to protect preadipocytes from cell death upon serum starvation41).
WNT ligands can also signal through β-catenin-independent pathways, known as non-canonical signalling, by signalling through alternate cell surface receptors and activating different intracellular pathways. The non-canonical WNT ligand WNT5A activates the histone methyltransferase SET domain bifurcated 1 (SETDB1)42. SETDB1 forms a complex with chromodomain helicase DNA-binding protein (CHD7) and Nemo-like kinase (NLK) to inhibit the ability of PPARγ to transcriptionally activate its downstream metabolic target genes in the MSC cell line ST2 and 3T3-L1 cells42,43. Activation of this non-canonical WNT pathway can also promote osteogenesis in MSCs42,44, indicating that this pathway is crucial for lineage determination in these multipotent cells. However, the non-canonical WNT ligand WNT5B, which is associated with type 2 diabetes in Japanese populations45, promotes adipocyte differentiation by inhibiting the canonical WNT pathway upon addition of adipogenic stimuli to committed preadipocytes by preventing nuclear translocation of β-catenin46.
Activation of the canonical and non-canonical pathway in preadipocytes can be modulated by regulating expression of WNT ligands. For example, expression of the canonical ligand WNT10b can be repressed upon addition of cAMP agonists, which act as adipogenic stimuli to committed preadipocytes47. Interestingly, many of the cell structure pathways described below regulate adipogenesis in part by controlling the expression of pro- and anti-adipogenic WNT ligands.
TGFβ superfamily signalling.
Transforming growth factor-β (TGFβ) superfamily ligands are secreted morphogens, some which are crucial for MSC lineage decisions and adipogenic competency of committed preadipocytes cell lines48. The exact role of TGFβ, the canonical member of the superfamily, during adipogenesis has remained unclear. TGFβ expression positively correlates with obesity in humans and animal models48, but paradoxically inhibits in vitro adipogenesis of 3T3-F442A cells by signalling through SMAD349. However, SMAD3-null mice are resistant to diet-induced obesity, and MEFs from these mice have diminished adipogenic potential50. The discrepancy between the in vitro and in vivo studies may be due to differences between ectopic activation TGFβ–SMAD3 signalling and the nuanced regulation of endogenous TGFβ and SMAD3. Alternatively, TGFβ–SMAD3 signalling may promote adipogenesis in multipotent progenitor cells during early WAT expansion but inhibit adipogenesis in committed preadipocytes populations. Interestingly, SMAD3-null mice also display increases in BAT markers in WAT compartments when kept on a regular diet, suggesting this pathway may be involved in the transdifferentiation of WAT to BAT upon cold exposure50.
Several bone morphogenetic proteins (BMPs), which are members of the TGFβ superfamily, have also been implicated in adipogenesis48, and single nucleotide polymorphisms neighbouring a BMP receptor, BMPR1A, are associated with obesity in humans51. Overall, BMPs promote adipogenesis by activating various SMADs and by signalling through the p38 kinase pathway52,53. BMP2 stimulates adipogenesis when provided in conjunction with a PPARγ agonist53,54, although it can also promote osteogenesis in committed preadipocytes when given with retinoic acid55. BMP2 activates SMAD1 and increases nuclear translocation of the transcriptional activator Schnurri2 (Shn2) to directly stimulate PPARγ expression during early adipocyte differentiation56. Shn2-null mice have decreased WAT, and Shn2-null MEFs cannot differentiate in the presence of BMP2, although BAT remains unaffected56. Similarly, BMP4 has been shown to increase the capacity of C3H10T1/2 cells to respond to adipogenic stimuli and specifically promote white adipocyte differentiation. By contrast, BMP7 induces brown adipogenesis30,57. Consistent with this, BMP7-null mice had less BAT than controls, and mice overexpressing BMP7 showed increases in brown adipocyte gene expression in BAT while other metabolic organs remained unaffected57.
The composition and stiffness of the ECM can regulate adipogenesis.
The conversion of spindly fibroblasts to round adipocytes is in part characterized by major remodelling of intra- and extracellular structures58,59, and studies have shown that this can be influenced by the composition of the extracellular matrix (ECM), by ECM stiffness and by tension. For example, the differentiation of 3T3-F442A cells into adipocytes is inhibited by fibronectin, and this can be rescued by chemical inhibition of actin stress fibre formation60. Integrin α5 binds fibronectin and is expressed by preadipocytes but not mature adipocytes. Ectopic expression of integrin α5 blocks adipocyte differentiation by maintaining high levels of the active form of the RHO GTPase Rac, which must be reduced in 3T3-L1 cells for adipogenesis to proceed61 (Figure 4a).
Figure 4: Rho-GTPase Family in Adipogenesis.
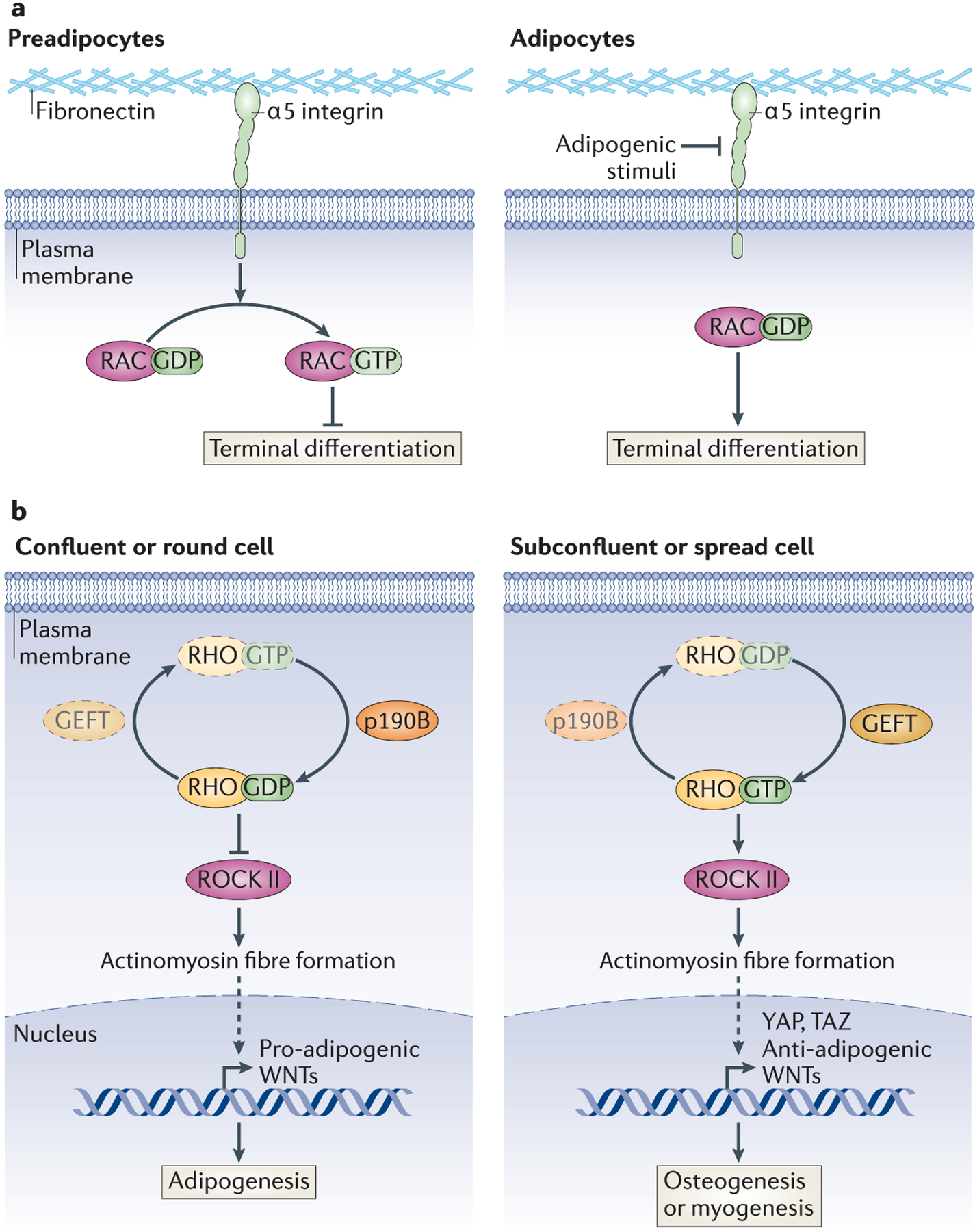
a | RAC-GTP inhibits adipogenesis. Integrins transduce extracellular structural signals into intracellular signalling cascades. Integrin α5, a fibronectin-binding protein, prevents the progression of preadipocytes to mature adipocytes in the absence of adipogenic stimuli by promoting the activation of RAC. Integrin α5 is repressed by adipogenic stimuli, leading to inactivation of RAC and terminal differentiation. b | Regulation of RHO determines MSC lineage fate. The shape of MSCs determines their ability to differentiate into adipocytes or alternate lineages by regulating RHO activity. Factors that favour the inactive form of RHO (RHO-GDP), such as p190-B RHOGAP, promote the adipogenic programme in these precursor cells, by inhibiting ROCK II activation of the actinomyosin cytoskeleton, which leads to the expression of pro-adipogenic WNTs. Conversely, factors that promote Rho-GTP lead to osteogenic or myogenic differentiation programmes, and this is mediated through the expression of anti-adipogenic WNTs and YAP and TAZ. Whether shape directly regulates p190-B RHOGAP or GEFT is unknown. Dashed arrows indicate an indirect interaction.
The stiffness of the ECM has also been shown to have a role in specifying lineage commitment in human MSCs62–64. Indeed, human MSCs grown on polyacrylamide gels with low stiffness are more likely to become adipocytes than cells seeded on stiffer matrices64. Similarly, primary mouse preadipocytes embedded in stiffer matrices that also have a higher concentration of collagen I show reduced rates of differentiation into adipocytes than when grown in softer gels65. Together, these studies suggest that force imposed on the cell by the ECM is a key gatekeeper of adipogenic competency.
ECM stiffness causes tissue tension, and, consistently, tension, which leads to enhanced actin and myosin fibre formation and cell stretching, mediates cell fate decisions in MSCs66,67. Although myogenic cell lines can transdifferentiate into adipocytes when treated with adipogenic stimuli68 (such as glucocorticoids and insulin) or by ectopically expressing PPARγ69, stretched myoblasts cannot undergo adipogenesis, correlating with increased expression of anti-adipogenic WNT ligands (see above)66. Furthermore, MSCs exposed to mechanical strain show increases in nuclear β-catenin, consistent with WNT activation, and cannot differentiate70. Lung embryonic MSCs can differentiate into adipocytes if not stretched, but when stretched will become smooth muscle cells, in part owing to altered expression patterns of different tension-induced protein isoforms (transcription co-factors that may regulate the activity of nuclear receptors such as PPARγ)67.
Cells can regulate the stiffness and composition of their environment by digesting the surrounding ECM with matrix metalloproteinases (MMPs), a family of secreted or membrane-bound zinc-dependent peptidases that cleave ECM components or other secreted factors71. Humans have 23 MMPs, which have a range of targets and functions, as well as a distinct family of four endogenous tissue inhibitors of MMPs (TIMPs)71. Complete inhibition of MMP activity blocks the differentiation of committed preadipocytes and impairs adipose tissue development in vivo72–75, suggesting that the balance between MMPs and TIMPs can determine adipogenic potential. In support of this hypothesis, polymorphisms neighbouring MMP14 (also known as MT1-MMP), a membrane-tethered collagenase known for its role in tumour metastasis76, are associated with human obesity77 and mice lacking MMP14, have reduced adipose tissue65. Interestingly, although preadipocytes lacking MMP14 can differentiate under two-dimensional growth conditions, they do not differentiate when embedded in three-dimensional collagen gels65, which better mimic physiological tissue development. This defect in three-dimensional differentiation is rescued by lowering the concentration of collagen surrounding the cells65. Whether other MMPs also regulate adipogenesis has yet to be described.
The MMP inhibitor TIMP3 is repressed during adipogenesis, and its ectopic expression inhibits 3T3-L1 adipocyte differentiation by blocking the expression of transcription factors involved in the early stages of terminal differentiation78. Although the mechanism of TIMP3-mediated repression of adipogenesis is unknown, TIMP3 strongly binds ECM proteins and may act as a pericellular regulator of MMP activity79. By contrast, TIMP1 is a secreted factor and does not seem to affect adipocyte differentiation, although it may have a role in regulating lipid droplet and blood vessel formation in mature adipose tissue80,81.
One of the key remaining questions is how stiffness and tension are detected by preadipocytes. Many scaffolds are currently being developed for ex vivo adipose tissue development and cell culture82. These new tools may help to elucidate the molecules that detect matrix stiffness and how they modulate signalling pathways regulating adipogenic commitment.
Cell–cell contact and cell shape influence adipogenesis.
Cellular confluency — when all cells in culture are all physically in contact with one another — is a requirement for many in vitro models of adipogenesis83,84. Remarkably, seeding MSCs in a spread configuration can direct lineage commitment towards osteoblasts, whereas confluent MSCs become adipocytes85. Because confluent preadipocytes can no longer proceed through the cell cycle, this cell contact was primarily thought to inhibit cellular proliferation in preparation for adipogenesis86.
However, in 3T3-L1 cells, preadipocytes undergo a period of mitotic clonal expansion upon addition of adipogenic stimuli87, suggesting that cell cycle progression in early adipogenesis does not exclude their ability to terminally differentiate. Thus, confluency could be mediating adipogenic commitment by inducing other cellular changes, such as modifying cell structure. Indeed, cells embedded within a methylcellulose gel, where they no longer divide, can also undergo adipogenesis83,86, but this is due to changes in cell shape88 that mimic the phenotype of confluent cells. Furthermore, inhibition of actinomyosin fibre formation commits pre-confluent human MSCs to adipogenesis instead of osteogenesis89. Single human MSCs plated on small surfaces that lead to a rounded morphology express pro-adipogenic WNT genes (see above) and differentiate into adipocytes upon dual adipogenic–osteogenic stimulation89,90. By contrast, single cells grown on larger features retain a spindly fibroblastic morphology that favours osteogenesis89,90.
These changes in cell shape regulate RHO GTPase–RHO-associated kinase (ROCK) signalling89 (Figure 4b). The inactive form, RHO-GDP, is the predominant species in confluent or rounded human MSCs and promotes adipogenesis; consistent with this, ectopic addition of constitutively active RHO, RHO-GTP, inhibits adipocyte differentiation89. RHO-GTP in spread cells activates ROCK; this, in turn, promotes actinomyosin fibre formation, which inhibits adipogenesis89,90. Knockdown and genetic studies have shown that ROCK II, but not ROCK I, is the kinase downstream of RHO regulating cell structure in 3T3-L1 cells and MEFs91. A recent study has suggested that active RHO inhibits adipogenesis by promoting the expression of the transcription factors YAP and TAZ; knockdown of these factors promotes adipogenesis even in MSCs grown under otherwise osteogenic conditions92, although how these factors are regulated by RHO or how they may be inhibiting adipogenesis has not been elucidated.
Not surprisingly, factors that control RHO activity (GTPase-activating proteins (GAPs) and guanine nucleotide exchange factors (GEFs)) have also been found to participate in MSC lineage commitment to adipogenesis. Mice lacking p190-B RHOGAP have decreased adiposity, and MEFs from these mice have a decrease in adipogenic capacity that is rescued with the addition of a general ROCK inhibitor93. Furthermore, p190-B RHOGAP-deficient MEFs undergo increased myogenic differentiation93, suggesting that RHO may not only determine MSC cell fate between the osteoblasts and adipocytes, but also distinguish adipogenic commitment from myogenesis. GEFT, a RHO-specific GEF that stabilizes active RHO-GTP, can also inhibit adipogenesis and promote myogenesis in cell culture models94. However, whether cell shape regulates RHOactivity in MSCs through this GAP or GEF remains unclear.
Transcriptional factors regulating adipogenic competency.
Identifying specific transcription factors that define the preadipocyte population provides additional insight into the signals required to transition from a multipotent MSC to an adipocyte progenitor cell. Expression profiling of known transcriptional regulators between adipogenic and non-adipogenic fibroblasts has shown that zinc finger protein 423 (ZFP423) is found almost exclusively in adipogenic cells95 although how it is regulated has not been identified. ZFP423 is necessary for 3T3-L1 adipogenesis and can promote white adipogenesis in non-adipogenic NIH-3T3 cells95. Furthermore, BAT development was impaired in ZFP423-null mice95, suggesting it is also involved in brown adipogenesis. However, the effect of ZFP423 on in vivo white fat development remains unclear, as ZFP423-null mice do not survive after birth, when most WAT expansion occurs96. Interestingly, ZFP423 has a SMAD-binding domain that is required for BMP4-dependent adipogenesis. However, ZFP423 mutants lacking this domain can still promoted adipocyte differentiation in the presence of glucocorticoids, cAMP agonists and insulin95, suggesting ZFP423 may promote adipogenesis in both a BMP-dependent and BMP-independent manner.
Other stem cells populations, including embryonic stem cells, require cooperation between multiple transcription factors to maintain their precursor state97; it is likely that adipogenic precursors also subscribe to this paradigm and that other factors cooperate with ZFP423 to determine adipogenic competency. One such factor may be TCF7L198, which is downstream of the canonical WNT pathway. Unlike other TCFs, which function as transcriptional activators, TCF7L1 is primarily a repressor of the canonical WNT pathway and has been shown to regulate embryonic stem cell pluripotency, skin stem cell differentiation and neurological development99. Like ZFP423, TCF7L1 is present in adipogenic but not non-adipogenic fibroblasts, where ectopic expression promotes adipogenic competency98. TCF7L1 appears to function by repressing cell structure-related genes and inhibiting myosin fibre formation upon addition of adipogenic stimuli98.
TERMINAL DIFFERENTIATION
Once preadipocytes have committed to the adipogenesis programme, a transcriptional cascade that induces the expression of metabolic genes and adipokines associated with the adipocyte phenotype, such as FABP4, GLUT4, leptin and adiponectin, is activated; this is known the terminal differentiation stage100–102. Many of the molecular mechanisms regulating this stage of differentiation have been determined by exploiting known targets of the adipogenic stimuli used to stimulate adipogenesis in confluent committed preadipocytes, in particular glucocorticoid activation of glucocorticoid receptor (GR) and cAMP agonist activation of both protein kinase A (PKA)-dependent and PKA-independent pathways103–108, although the importance of these pathways in vivo has yet to be determined. While many transcription factors have a role in adipogenesis100,102,109, it is the expression of PPARγ, C/CAAT enhancer binding protein-α (C/EBPα), C/EBPβ and C/EBPδ, as well as the epigenomic coordination between these factors, that are the primary drivers of adipocyte gene induction during terminal differentiation110,111.
C/EBP activation by adipogenic stimuli.
C/EBP proteins are widely expressed transcription factors that have a role in the development of many diverse cell types. Genome-wide studies have shown that C/EBPβ in committed preadipocytes is present at low levels before the addition of adipogenic stimuli and is bound to quiescent ‘adipogenic hotspots’. These are regions of the genome that display marks of active enhancers and recruit other adipogenic transcription factors (C/EBPδ, signal transducer and activator of transcription 5A (STAT5A), GR and RXR) after the addition of the adipogenic cocktail112. C/EBPβ is required for the binding of the other ‘hotspot’ transcription factors, except for C/EBPδ105,112.
In addition to acting as a marker for adipogenic enhancers in preadipocytes, C/EBPβ expression and function are extensively regulated upon addition of adipogenic stimuli (Figure 5a). C/EBPβ expression is markedly induced by cAMP agonists within hours of stimulation103, and this is mediated by the transcriptional activator cAMP response element-binding protein (CREB), which is phosphorylated in response to cAMP agonists, allowing it to act as a direct activator of C/EBPβ during early adipogenesis113. A cascade involving Janus kinase 2 (JAK2) and STAT3 also directly promote C/EBPβ expression in committed preadipocytes114,115, although the initial stimulus has not been determined. Krüppel-like factor 4 (KLF4) also promotes adipogenesis by directly activating C/EBPβ transcription in early terminal differentiation116. In addition, the transcription factor KROX20 (also known as EGR2) is present during the first 6 hours of differentiation and can promote C/EBPβ expression, but this seems to be an indirect effect, suggesting that KROX20 may induce another, as-yet-unidentified transcriptional activator of C/EBPβ117. Interestingly, depletion of C/EBPβ increases the expression of both KLF4 and KROX20 in 3T3-L1 cells, suggesting a negative feedback loop during early adipogenesis116. Of note, cAMP also promotes the expression of EPACs (exchange proteins directly activated by cAMP)107,108, although whether this pathway directly induces C/EBPβ and δ activity has not been shown.
Figure 5: Activation of C/EBPs and PPARγ during terminal differentiation.
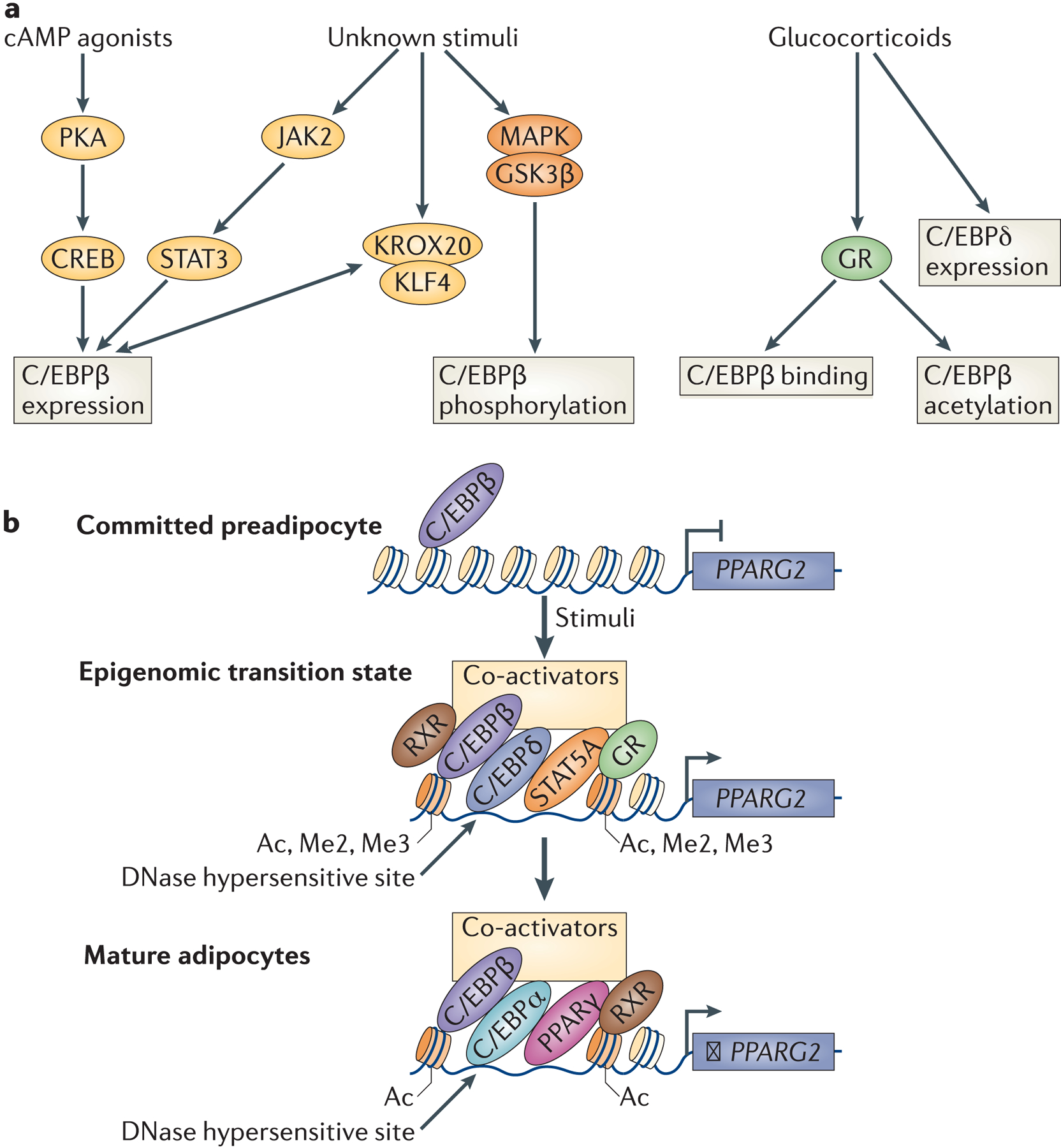
a | C/EBP activation by adipogenic stimuli. Glucocorticoids and cAMP agonists are common components of the adipogenic stimuli used to promote adipogenesis in both MSCs and committed preadipocytes. Experiments adding these compounds individually have elucidated some of the mechanisms through which C/EBPs, especially C/EBPβ, are induced during adipogenesis. C/EBPβ and C/EBPδ expression is induced upon addition of these adipogenic stimuli. C/EBPβ activity and binding are also regulated independently of its levels by glucocorticoids. In addition, C/EBPβ expression and phosphorylation are regulated by unknown components of the adipogenic cocktail, which may include insulin, growth hormone or BMPs. b | Recruitment of the transcription activation complex to PPARγ. Schematic of the recruitment of transcription factors to the PPARγ locus during adipogenesis. In preadipocytes, PPARγ enhancer regions are occupied by C/EBPβ and C/EBPδ, but are not accessible. Upon addition of adipogenic stimuli, levels of these transcription factors increase and lead to the recruitment of a transcriptional activation complex, including the transcription factors GR, STAT5a and RXR and a co-activator complex. These ‘hotspots’ are also marked by an increase in DNAse I hypersensitivity and activating histone marks. Once PPARγ is robustly activated in differentiation, it can auto-regulate its expression in cooperation with C/EBPα and C/EBPβ.
C/EBPβ is also regulated in adipogenesis by post-translational modifications. C/EBPβ phosphorylation by mitogen-activated protein kinases and glycogen synthase kinase 3β is required for the ability of C/EBPβ to bind DNA during differentiation118,119. Glucocorticoids, probably in part through GR120, have also been reported to have a role in regulating C/EBPβ transcriptional activity by directing the acetylation of C/EBPβ and interfering with the interaction of C/EBPβ with the transcriptional co-repressor histone deacetylase 1 (HDAC1)121,122.
Much less is known about C/EBPδ regulation, which is also induced rapidly by glucocorticoids upon addition of adipogenic stimuli103. C/EBPδ is likely just as important to adipose development as C/EBPβ since both C/EBPβ- and C/EBPδ-null mice have mild adipose tissue developmental defects, MEFs and mice lacking both proteins have marked primary defects in adipogenesis and adipose tissue development123. By contrast, C/EBPα is expressed later in terminal differentiation and is a direct target of C/EBPβ124.
Epigenomics provides insights into early PPARγ activation.
Terminal differentiation cannot occur in the absence of the nuclear receptor PPARγ125; consistently, thiazolidinediones (TZDs), which target PPARγ to treat diabetes, can promote adipogenesis in adipocyte progenitor cells both in vitro and in vivo126,127. There are two PPARγ protein isoforms: PPARγ1 is expressed at highest levels by adipocytes but also by other cell types, including preadipocytes and other MSC-derived cell types; PPARγ2 is adipocyte specific128,129. However, adipogenic conversion can be equally mediated by either isoform102. The endogenous PPARγ ligand has yet to be found; nevertheless, cAMP agonists contribute to the production of an endogenous PPARγ ligand within the first 48 hours of terminal differentiation in 3T3-L1 cells106.
Recent studies using in vitro adipogenic models have revealed that the PPARg locus is dynamically and extensively regulated upon addition of adipogenic stimuli by C/EBPs and GR during an epigenomic transition state105,112,130 (Figure 5b). Genome-wide mapping of histone marks associated with active transcription, such as His3 Lys9 acetylation (H3K9ac) or H3K27ac, have shown that the PPARg locus has multiple functional enhancers located as far at −122 kb upstream of the PPARg1 transcription start site105,130.
Given the dynamic changes in histone acetylation during adipogenesis, regulators of these marks have been implicated in adipocyte differentiation, although their role seems to be complicated. Exposure of preadipocytes to inhibitors of HDACs has led to conflicting results131–134, indicating HDAC subtype-specific effects and potentially off-target effects of the chemical agents. Moreover, studies in which the class I HDAC, HDAC1, was manipulated have also yielded conflicting results131,135. The class II HDAC9 inhibits adipogenesis, although this does not require its deacetylase activity134, and inhibitors of class II HDACs reportedly block adipogenesis136. Moreover, both sirtuin 1 and sirtuin 2, which are class III HDACs, are downregulated during adipogenesis and also inhibit white adipocyte differentiation, potentially by regulating the acetylation of transcription factors rather than histone acetylation137,138. Whether these factors are important for regulating acetylation at the PPARg gene remains unclear.
Regulation of histone methylation has also been implicated in adipogenesis. In addition to the SETDB142,43, the histone methytransferase SETD8 has been shown to be required for committed preadipocyte differentiation by adding the activating histone mark His4 Lys20 monomethylation (H4K20me) at the promoters of PPARg and of PPARγ target genes upon addition of adipogenic stimuli43. MLL3, a His4 Lys3 methyltransferase, is also required for adipogenesis, and MLL3-null mice have small WAT but normal BAT139. Reciprocally, the histone demethylase jumonji domain-containing 2C (JMJD2C) inhibits adipogenesis in vitro, although this effect may be due to HDAC binding and not demethylase activity140.
PTIP (Pax transactivation domain-interacting protein), which forms a complex with MLL3 and the related MLL4, increases H3K4Me3 at the PPARg and CEBPa promoters, and deletion of PTIP inhibits differentiation of both white and brown preadipocytes141. However, in contrast to MLL3-null mice, PTIP-null mice have defects in BAT formation, but normal WAT development141. Moreover, the transcriptional regulators ASXL1 and ASXL2, which can recruit histone methyltransferases to promoter regions, have opposite effects on adipogenesis: ASXL1 inhibits adipogenesis and recruits repressive histone marks to PPARγ target genes, whereas ASXL2 promotes adipogenesis and recruits activating histone marks to these same loci142. Thus, additional studies are clearly needed to better understand the role of histone methylation, as well as other epigenomic changes, in adipocyte differentiation.
Other factors regulating PPARγ.
PPARγ levels and activity are also regulated by circadian rhythm factors109. REV-ERBα, a circadian transcription factor, is regulated by adipogenic stimuli, and both knockdown and overexpression of this factor inhibit adipogenesis and PPARγ induction during terminal differentiation143. Nocturnin is a circadian rhythm-regulated protein that is required for adipogenesis by acting as a co-activator for PPARγ and enhancing its activity during terminal differentiation144.
Other transcription factors also regulate PPARγ expression and its ability to cooperate with C/EBP proteins in terminal differentiation. Examples of such factors include GATA-binding proteins 2 (GATA2) and GATA3. These are expressed by committed preadipocytes, in which they inhibit C/EBPβ and C/EBPα function, and therefore PPARγ activation145,146, and must be repressed for adipogenesis to proceed in the presence of adipogenic stimuli145.
Maintenance of mature adipocyte gene expression.
Genome-wide studies of mature adipocytes have shown that PPARγ and C/EBPα are present at ~ 60% of all genes induced during terminal differentiation110. Knockdown studies have revealed that PPARγ, C/EBPα and C/EBPβ are all required for sustained expression of PPARγ and C/EBP target genes in mature adipocytes, such as adiponectin, FABP4, and hormone sensitive lipase110. PPARγ and these C/EBP proteins also regulate each other in a positive feedback circuit central to terminal105,124,147.
The central role of PPARγ in maintaining mature adipocyte function has been shown in vitro and in vivo. Depletion of PPARγ in mature 3T3-L1 adipocytes with small interfering RNA leads to decreased expression of adipocyte metabolic genes148 and reduced ability to respond to insulin149 without any effect on adipocyte morphology. However, in a mouse model where PPARγ is deleted in mature adipocytes, this transcription factor was required for cell survival150,151. These experiments suggest that mature adipocytes can survive and maintain their lipid content in the presence of minimal PPARγ (such as in the knockdown experiments), but that in the complete absence of PPARγ these differentiated cells cannot be sustained (as in the genetic mouse model).
Conservation of adipocyte gene activation across species.
PPARγ and many other metabolic genes are induced during adipogenesis in models from multiple species130. Comparison of genome-wide maps of histone marks, PPARγ binding and C/EBPα between human- and mouse-derived adipogenic cell lines has further developed our understanding of the conservation of developmental programmes130,152,153. First, these studies have revealed that there is conservation of adipogenic stimulus-dependent induction of PPARγ binding and histone modification near genes that are induced during adipogenesis130. Intriguingly, although many of the same metabolic genes are induced and epigenomically modulated in human and mouse adipogenesis, the precise genomic loci that are regulated during differentiation are not well conserved despite the conservation of these sequences in the genome of the other species130,153. Moreover, genes that were expressed in mature adipocytes of both species were more likely to have roles in pathways related to the metabolic function of these cells, whereas genes expressed only in one species did not153, suggesting that comparison between expression profiles of different species can filter out species-specific artefacts and highlight the biologically important genes. These studies also suggested that conservation of a PPARγ binding site could be predicted in the syntenic region of the genome from the other species contained a C/EBPα-binding site152, supporting a role for the cooperation of these factors in maintaining mature adipocyte function in evolution.
Brown adipocyte-specific transcriptional regulators.
Brown adipocyte differentiation also requires PPARγ and C/EBP activity123,154. To date no genome-wide studies of PPARγ or C/EBP binding have been done in brown adipocytes to compare the extent of overlap between these related, but distinct cells types. In addition, many transcription factors and signalling pathways, such as ZFP423 and WNT, have been shown to have crucial roles in BAT development37,95. However, brown adipocyte differentiation and mature adipocyte function have unique requirements for transcriptional co-regulators than their white adipocyte counterparts.
The transcriptional regulator PRDM16 is critical for brown fat adipogenesis. PRDM16 exists in a complex with C/EBPβ to induce the expression of genes common to white and brown adipocytes, such as PPARγ and FABP4, as well as brown fat-specific genes such as UCP123,155. Activation of brown fat genes occurs following recruitment of PPARγ to a PRDM16 transcriptional complex156; PRDM16 also forms a repressive complex with C-terminal binding protein 1 (CTBP1) and CTBP2 to repress the expression of white adipocyte genes156. In vitro PRDM16 knockdown in primary brown preadipocytes promotes myogenesis, consistent with a role in brown adipocyte–myocyte cell fate specification23. However, although PRDM16-knockout mice do not express brown adipocyte-specific thermogenic genes, they still express adipogenic markers common to white and brown adipocytes in brown adipose depots23. This indicates that brown adipogenesis can still proceed in part in the absence of PRDM16. Thus, other regulators of brown adipogenesis must exist, possibly including other PRDM family members. Although PRDM16 has no role in white adipocyte differentiation and is not expressed in visceral WAT, it is moderately expressed in subcutaneous WAT, where it promotes the metabolically favourable thermogenic properties27.
Another key regulator of brown fat is PPARγ co-activator 1α (PGC1α), which was identified in a screen for proteins that bind PPARγ in brown fat157. Although PGC1α and the related PGC1β do not have a significant role in brown adipogenesis, they are required to maintain the expression of thermogenic genes in mature brown adipocytes158. Interestingly, PGC1α, along with thermogenic genes, is induced in WAT from mice or humans treated with thiazolidinediones (see above), contributing to the improved metabolic profile seen upon treatment with these compounds159.
CONCLUSION AND PERSPECTIVES
Recent advances in adipogenesis have provided insights into precursor competency, remodelling of the genome and the relationship of those pathways to in vivo fat pad development. Cell structure and signalling pathways, as well as epigenomic remodelling, are potential targets of novel therapeutics targeting specific elements of adipogenic commitment and terminal differentiation. Indeed, compounds targeting epigenomic pathways such as histone acetylation are already used clinically for other disorders, such as cancer,160 and may be exploited for the treatment of metabolic disease.
However, many important questions remain about adipogenesis and adipose tissue development. First, our understanding of adipocyte development in vivo is limited by the lack of candidate markers distinguishing different stages of precursor cell development. Second, although mouse models have shown that subcutaneous and visceral fat are functionally distinct5,6,8,13 and express different developmental genes5,6, it remains inconclusive whether these differences are due to adipogenic precursor potential. Third, the mechanisms that increase the ‘brown’ phenotype in white adipocytes with cold exposure have yet to be discovered but should offer an intriguing strategy for treating metabolic disease. Last, although many pathways described here and elsewhere have been shown to regulate adipogenic commitment and terminal differentiation, how these two stages of adipogenesis are physiologically integrated during development remains unknown.
The recent isolation of preadipocyte populations in vivo14,29 should help to answer many of these questions by identifying specific markers for adipocyte precursor cells, allowing more advanced lineage tracing of WAT development in both the subcutaneous and visceral depots, and providing a pure population of more physiologically relevant cells for genome-wide studies of transcription factor binding and histone modifications to further understand the relationship between epigenomic regulation and gene expression in cellular differentiation. Ultimately, a better understanding of in vivo adipogenic commitment and triggers of terminal differentiation will be crucial for manipulating MSCs and adipose derived stem cell populations in ways that have promise above and beyond treating conditions of metabolic dysfunction82,161.
ONLINE SUMMARY.
- Adipogenesis is a highly regulated process that converts fibroblast-like precursor cells into round and lipid-laden adipocytes.
- White and brown adipocyte differentiation share many key important features, such as shared requirement for the master regulator PPARγ, but have important differences.
- Identification of committed precursor cells within adipose tissue has been important for understanding of adipogenesis in vivo.
- Adipogenic stimuli activate signalling pathways that coordinate transcription factors to promote stem cell commitment to an adipogenic fate.
- Extensive epigenomic modifications underlie the commitment and stability of adipocytic differentiation.
ACKNOWLEDGEMENTS.
We thank P. Seale and members of the Lazar laboratory for insightful discussions. Work on adipogenesis in the Lazar laboratory is supported by NIH DK49780. AGC was supported by the Gilliam Fellowship from the Howard Hughes Medical Institute. We apologize for not being able to discuss and cite all worthy papers and topics as a result of space limitations.
GLOSSARY
type 2 diabetes
chronic disease that is characterized by increased blood glucose related to insulin resistance and pancreatic failure
cancer cachexia
Weight, muscle atrophy, fatigue, weakness, and loss of appetite in the setting of cancer
lipodystrophies
Reduced or abnormally redistributed adipose compartments (acquired or genetic)
unilocular
One large lipid droplet
thermogenesis
The process of producing heat
fluorodeoxyglucose positron emission tomography
Molecular imaging technique using a labelled glucose analogue
uncoupling protein 1 (UCP1)
Mitochondrial protein that dissociates oxidative phosphorylation from energy production, leading to increased thermogenesis
Mesenchymal stem cell
Multipotent progenitor that can differentiate into adipocytes, osteoblasts, myocytes or chondrocytes
subscapular
region below the scapula, the shoulder blade
epigenomic
of the epigeome, i.e., chromatin modifications including DNA methylation and histone modification that regulate gene expression and function without a corresponding alteration in DNA sequence
A-Zip mice
mouse model in which a dominant negative transcription factor that interferes with C/EBP function is expressed by adipocytes, leading to lipodystrophy
Integrins
Heterodimeric, cation-dependent cell surface receptors that attach cells to their surrounding environment, connecting ECM cues to intracellular signalling
AUTHOR BIOGRAPHIES
Ana G. Cristancho is an M.D. and Ph.D. candidate at the Perelman School of Medicine at the University of Pennsylvania, USA. She earned her B.S. at the University of Miami, USA, and recently completed her Ph.D in the laboratory of Mitchell A. Lazar. During her time in the Lazar laboratory she has focused on understanding the mechanisms that govern transcriptional repression during adipogenesis.
Mitchell A. Lazar is Sylvan Eisman Professor of Medicine and Genetics, Chief of the Division of Endocrinology, Diabetes, and Metabolism, and Director of the Institute for Diabetes, Obesity, and Metabolism at the Perelman School of Medicine of the University of Pennsylvania. He received his undergraduate degree in Chemistry from MIT, Massachusetts, USA, a Ph. D. in Neurosciences and an M.D. from Stanford University, and trained in Internal Medicine and Endocrinology at Brigham and Women’s Hospital and Massachusetts General Hospital, USA. His laboratory focuses on the functions of nuclear receptors to regulate the epigenome and gene expression in metabolic tissues such as adipose and liver.
REFERENCES
- 1.Galic S, Oakhill JS & Steinberg GR Adipose tissue as an endocrine organ. Mol Cell Endocrinol 316, 129–39 (2010). [DOI] [PubMed] [Google Scholar]
- 2.Cinti S The adipose organ. Prostaglandins Leukot. Essent. Fatty Acids 73, 9–15 (2005). [DOI] [PubMed] [Google Scholar]
- 3.Ibrahim MM Subcutaneous and visceral adipose tissue: structural and functional differences. Obes Rev 11, 11–8 (2010). [DOI] [PubMed] [Google Scholar]
- 4.Girard J & Lafontan M Impact of visceral adipose tissue on liver metabolism and insulin resistance. Part II: Visceral adipose tissue production and liver metabolism. Diabetes Metab 34, 439–445 (2008). [DOI] [PubMed] [Google Scholar]
- 5.Gesta S et al. Evidence for a role of developmental genes in the origin of obesity and body fat distribution. Proc Natl Acad Sci U S A 103, 6676–81 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Yamamoto Y et al. Adipose depots possess unique developmental gene signatures. Obesity (Silver Spring) 18, 872–8 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hamdy O, Porramatikul S & Al-Ozairi E Metabolic obesity: the paradox between visceral and subcutaneous fat. Curr Diabetes Rev 2, 367–373 (2006). [DOI] [PubMed] [Google Scholar]
- 8.Tran TT, Yamamoto Y, Gesta S & Kahn CR Beneficial effects of subcutaneous fat transplantation on metabolism. Cell Metab 7, 410–20 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Frontini A & Cinti S Distribution and development of brown adipocytes in the murine and human adipose organ. Cell Metab 11, 253–6 (2010). [DOI] [PubMed] [Google Scholar]
- 10.van Marken Lichtenbelt WD et al. Cold-activated brown adipose tissue in healthy men. N Engl J Med 360, 1500–8 (2009). [DOI] [PubMed] [Google Scholar]
- 11.Virtanen KA et al. Functional brown adipose tissue in healthy adults. N. Engl. J. Med 360, 1518–1525 (2009). [DOI] [PubMed] [Google Scholar]
- 12.Cypess AM et al. Identification and importance of brown adipose tissue in adult humans. N. Engl. J. Med 360, 1509–1517 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gesta S, Tseng YH & Kahn CR Developmental origin of fat: tracking obesity to its source. Cell 131, 242–56 (2007). [DOI] [PubMed] [Google Scholar]
- 14.Tang W et al. White fat progenitor cells reside in the adipose vasculature. Science 322, 583–6 (2008).Identifies preadipocytes that reside within the adipose compartment using a novel lineage tracing mouse model.
- 15.Kirtland J & Harris PM Changes in adipose tissue of the rat due early undernutrition followed by rehabilitation. 3. Changes in cell replication studied with tritiated thymidine. Br J Nutr 43, 33–43 (1980). [DOI] [PubMed] [Google Scholar]
- 16.Hirsch J & Han PW Cellularity of rat adipose tissue: effects of growth, starvation, and obesity. J. Lipid Res 10, 77–82 (1969). [PubMed] [Google Scholar]
- 17.Spalding KL et al. Dynamics of fat cell turnover in humans. Nature 453, 783–7 (2008).Using a novel isotopic method, this paper calculates rates of adipocyte differentiation and apoptosis in humans.
- 18.Lemonnier D Effect of age, sex, and sites on the cellularity of the adipose tissue in mice and rats rendered obese by a high-fat diet. J. Clin. Invest 51, 2907–2915 (1972). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Faust IM, Johnson PR, Stern JS & Hirsch J Diet-induced adipocyte number increase in adult rats: a new model of obesity. Am. J. Physiol 235, E279–286 (1978). [DOI] [PubMed] [Google Scholar]
- 20.Klyde BJ & Hirsch J Increased cellular proliferation in adipose tissue of adult rats fed a high-fat diet. J. Lipid Res 20, 705–715 (1979). [PubMed] [Google Scholar]
- 21.Tchoukalova YD et al. Regional differences in cellular mechanisms of adipose tissue gain with overfeeding. Proc. Natl. Acad. Sci. U.S.A 107, 18226–18231 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.van Harmelen V et al. Effect of BMI and age on adipose tissue cellularity and differentiation capacity in women. Int. J. Obes. Relat. Metab. Disord 27, 889–895 (2003). [DOI] [PubMed] [Google Scholar]
- 23.Seale P et al. PRDM16 controls a brown fat/skeletal muscle switch. Nature 454, 961–967 (2008).Reveals that brown adipocytes and skeletal myocytes share a common progenitor, with transcription factor Prdm16 determining the brown adipogenic fate.
- 24.Lepper C & Fan C-M Inducible lineage tracing of Pax7-descendant cells reveals embryonic origin of adult satellite cells. Genesis 48, 424–436 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Cousin B et al. Occurrence of brown adipocytes in rat white adipose tissue: molecular and morphological characterization. J. Cell. Sci 103 (Pt 4), 931–942 (1992). [DOI] [PubMed] [Google Scholar]
- 26.Stefl B et al. Brown fat is essential for cold-induced thermogenesis but not for obesity resistance in aP2-Ucp mice. Am. J. Physiol 274, E527–533 (1998). [DOI] [PubMed] [Google Scholar]
- 27.Seale P et al. Prdm16 determines the thermogenic program of subcutaneous white adipose tissue in mice. J. Clin. Invest 121, 96–105 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Barbatelli G et al. The emergence of cold-induced brown adipocytes in mouse white fat depots is determined predominantly by white to brown adipocyte transdifferentiation. Am. J. Physiol. Endocrinol. Metab 298, E1244–1253 (2010). [DOI] [PubMed] [Google Scholar]
- 29.Rodeheffer MS, Birsoy K & Friedman JM Identification of white adipocyte progenitor cells in vivo. Cell 135, 240–9 (2008).Defines surface antigens that define preadipocytes within the adipose compartment.
- 30.Schulz TJ et al. Identification of inducible brown adipocyte progenitors residing in skeletal muscle and white fat. Proc. Natl. Acad. Sci. U.S.A 108, 143–148 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Takada I, Kouzmenko AP & Kato S Wnt and PPARgamma signaling in osteoblastogenesis and adipogenesis. Nat Rev Rheumatol 5, 442–447 (2009). [DOI] [PubMed] [Google Scholar]
- 32.Okamura M et al. COUP-TFII acts downstream of Wnt/beta-catenin signal to silence PPARgamma gene expression and repress adipogenesis. Proc Natl Acad Sci U S A 106, 5819–24 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Xu Z, Yu S, Hsu C-H, Eguchi J & Rosen ED The orphan nuclear receptor chicken ovalbumin upstream promoter-transcription factor II is a critical regulator of adipogenesis. Proc. Natl. Acad. Sci. U.S.A 105, 2421–2426 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Li L et al. The nuclear orphan receptor COUP-TFII plays an essential role in adipogenesis, glucose homeostasis, and energy metabolism. Cell Metab 9, 77–87 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ross SE et al. Inhibition of adipogenesis by Wnt signaling. Science (New York, N.Y 289, 950–3 (2000).Identification of a critical role of Wnt signalling in adipogenesis.
- 36.Kawai M et al. Wnt/Lrp/beta-catenin signaling suppresses adipogenesis by inhibiting mutual activation of PPARgamma and C/EBPalpha. Biochem Biophys Res Commun 363, 276–82 (2007). [DOI] [PubMed] [Google Scholar]
- 37.Longo KA et al. Wnt10b inhibits development of white and brown adipose tissues. J Biol Chem 279, 35503–9 (2004). [DOI] [PubMed] [Google Scholar]
- 38.Kang S et al. Wnt signaling stimulates osteoblastogenesis of mesenchymal precursors by suppressing CCAAT/enhancer-binding protein alpha and peroxisome proliferator-activated receptor gamma. J. Biol. Chem 282, 14515–14524 (2007). [DOI] [PubMed] [Google Scholar]
- 39.Wang L, Jin Q, Lee J-E, Su I.-hsin & Ge K Histone H3K27 methyltransferase Ezh2 represses Wnt genes to facilitate adipogenesis. Proc. Natl. Acad. Sci. U.S.A 107, 7317–7322 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Longo KA et al. Wnt signaling protects 3T3-L1 preadipocytes from apoptosis through induction of insulin-like growth factors. J Biol Chem 277, 38239–44 (2002). [DOI] [PubMed] [Google Scholar]
- 41.Gagnon A, Dods P, Roustan-Delatour N, Chen CS & Sorisky A Phosphatidylinositol-3,4,5-trisphosphate is required for insulin-like growth factor 1-mediated survival of 3T3-L1 preadipocytes. Endocrinology 142, 205–212 (2001). [DOI] [PubMed] [Google Scholar]
- 42.Takada I et al. A histone lysine methyltransferase activated by non-canonical Wnt signalling suppresses PPAR-gamma transactivation. Nat Cell Biol 9, 1273–85 (2007). [DOI] [PubMed] [Google Scholar]
- 43.Wakabayashi K.-ichi et al. The peroxisome proliferator-activated receptor gamma/retinoid X receptor alpha heterodimer targets the histone modification enzyme PR-Set7/Setd8 gene and regulates adipogenesis through a positive feedback loop. Mol. Cell. Biol 29, 3544–3555 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kennell JA & MacDougald OA Wnt signaling inhibits adipogenesis through beta-catenin-dependent and -independent mechanisms. J Biol Chem 280, 24004–10 (2005). [DOI] [PubMed] [Google Scholar]
- 45.Kanazawa A et al. Association of the gene encoding wingless-type mammary tumor virus integration-site family member 5B (WNT5B) with type 2 diabetes. Am. J. Hum. Genet 75, 832–843 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kanazawa A et al. Wnt5b partially inhibits canonical Wnt/beta-catenin signaling pathway and promotes adipogenesis in 3T3-L1 preadipocytes. Biochem Biophys Res Commun 330, 505–10 (2005). [DOI] [PubMed] [Google Scholar]
- 47.Fox KE et al. Regulation of cyclin D1 and Wnt10b gene expression by cAMP-responsive element-binding protein during early adipogenesis involves differential promoter methylation. J Biol Chem 283, 35096–105 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Zamani N & Brown CW Emerging roles for the transforming growth factor-{beta} superfamily in regulating adiposity and energy expenditure. Endocr. Rev 32, 387–403 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Choy L, Skillington J & Derynck R Roles of autocrine TGF-beta receptor and Smad signaling in adipocyte differentiation. J. Cell Biol 149, 667–682 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Yadav H et al. Protection from Obesity and Diabetes by Blockade of TGF-β/Smad3 Signaling. Cell Metab 14, 67–79 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Böttcher Y et al. Adipose tissue expression and genetic variants of the bone morphogenetic protein receptor 1A gene (BMPR1A) are associated with human obesity. Diabetes 58, 2119–2128 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Huang H et al. BMP signaling pathway is required for commitment of C3H10T1/2 pluripotent stem cells to the adipocyte lineage. Proc. Natl. Acad. Sci. U.S.A 106, 12670–12675 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Hata K et al. Differential roles of Smad1 and p38 kinase in regulation of peroxisome proliferator-activating receptor gamma during bone morphogenetic protein 2-induced adipogenesis. Mol. Biol. Cell 14, 545–555 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Sottile V & Seuwen K Bone morphogenetic protein-2 stimulates adipogenic differentiation of mesenchymal precursor cells in synergy with BRL 49653 (rosiglitazone). FEBS Lett 475, 201–204 (2000). [DOI] [PubMed] [Google Scholar]
- 55.Skillington J, Choy L & Derynck R Bone morphogenetic protein and retinoic acid signaling cooperate to induce osteoblast differentiation of preadipocytes. J. Cell Biol 159, 135–146 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Jin W et al. Schnurri-2 controls BMP-dependent adipogenesis via interaction with Smad proteins. Dev. Cell 10, 461–471 (2006).Characterization of Shn2 as a physiologic regulator of BMP-dependent adipose development.
- 57.Tseng Y-H et al. New role of bone morphogenetic protein 7 in brown adipogenesis and energy expenditure. Nature 454, 1000–1004 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Aratani Y & Kitagawa Y Enhanced synthesis and secretion of type IV collagen and entactin during adipose conversion of 3T3-L1 cells and production of unorthodox laminin complex. The Journal of biological chemistry 263, 16163–9 (1988). [PubMed] [Google Scholar]
- 59.Nakajima I, Yamaguchi T, Ozutsumi K & Aso H Adipose tissue extracellular matrix: newly organized by adipocytes during differentiation. Differentiation 63, 193–200 (1998). [DOI] [PubMed] [Google Scholar]
- 60.Spiegelman BM & Ginty CA Fibronectin modulation of cell shape and lipogenic gene expression in 3T3-adipocytes. Cell 35, 657–66 (1983). [DOI] [PubMed] [Google Scholar]
- 61.Liu J et al. Changes in integrin expression during adipocyte differentiation. Cell Metabolism 2, 165–177 (2005). [DOI] [PubMed] [Google Scholar]
- 62.Engler AJ, Sen S, Sweeney HL & Discher DE Matrix elasticity directs stem cell lineage specification. Cell 126, 677–89 (2006). [DOI] [PubMed] [Google Scholar]
- 63.Rowlands AS, George PA & Cooper-White JJ Directing osteogenic and myogenic differentiation of MSCs: interplay of stiffness and adhesive ligand presentation. Am J Physiol Cell Physiol 295, C1037–44 (2008). [DOI] [PubMed] [Google Scholar]
- 64.Winer JP, Janmey PA, McCormick ME & Funaki M Bone marrow-derived human mesenchymal stem cells become quiescent on soft substrates but remain responsive to chemical or mechanical stimuli. Tissue Eng Part A 15, 147–54 (2009). [DOI] [PubMed] [Google Scholar]
- 65.Chun T-H et al. A Pericellular Collagenase Directs the 3-Dimensional Development of White Adipose Tissue. Cell 125, 577–591 (2006).This paper demonstrates that the pericellular collagenase Mmp14 is required for adipocyte differentiation in three dimensions.
- 66.Akimoto T et al. Mechanical stretch inhibits myoblast-to-adipocyte differentiation through Wnt signaling. Biochemical and biophysical research communications 329, 381–5 (2005). [DOI] [PubMed] [Google Scholar]
- 67.Jakkaraju S, Zhe X, Pan D, Choudhury R & Schuger L TIPs are tension-responsive proteins involved in myogenic versus adipogenic differentiation. Developmental cell 9, 39–49 (2005). [DOI] [PubMed] [Google Scholar]
- 68.Teboul L et al. Thiazolidinediones and fatty acids convert myogenic cells into adipose-like cells. J. Biol. Chem 270, 28183–28187 (1995). [DOI] [PubMed] [Google Scholar]
- 69.Hu E, Tontonoz P & Spiegelman BM Transdifferentiation of myoblasts by the adipogenic transcription factors PPAR gamma and C/EBP alpha. Proc Natl Acad Sci U S A 92, 9856–60 (1995). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Sen B et al. Mechanical strain inhibits adipogenesis in mesenchymal stem cells by stimulating a durable beta-catenin signal. Endocrinology 149, 6065–6075 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Visse R & Nagase H Matrix metalloproteinases and tissue inhibitors of metalloproteinases: structure, function, and biochemistry. Circ. Res 92, 827–839 (2003). [DOI] [PubMed] [Google Scholar]
- 72.Chavey C et al. Matrix metalloproteinases are differentially expressed in adipose tissue during obesity and modulate adipocyte differentiation. The Journal of biological chemistry 278, 11888–96 (2003). [DOI] [PubMed] [Google Scholar]
- 73.Croissandeau G, Chretien M & Mbikay M Involvement of matrix metalloproteinases in the adipose conversion of 3T3-L1 preadipocytes. Biochem J 364, 739–46 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Lijnen HR et al. Matrix metalloproteinase inhibition impairs adipose tissue development in mice. Arteriosclerosis, thrombosis, and vascular biology 22, 374–9 (2002). [DOI] [PubMed] [Google Scholar]
- 75.Maquoi E, Munaut C, Colige A, Collen D & Lijnen HR Modulation of adipose tissue expression of murine matrix metalloproteinases and their tissue inhibitors with obesity. Diabetes 51, 1093–101 (2002). [DOI] [PubMed] [Google Scholar]
- 76.Itoh Y MT1-MMP: A key regulator of cell migration in tissue. IUBMB Life (International Union of Biochemistry and Molecular Biology: Life) V58, 589–596 (2006). [DOI] [PubMed] [Google Scholar]
- 77.Chun T-H et al. Genetic link between obesity and MMP14-dependent adipogenic collagen turnover. Diabetes 59, 2484–2494 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Bernot D et al. Down-regulation of tissue inhibitor of metalloproteinase-3 (TIMP-3) expression is necessary for adipocyte differentiation. J Biol Chem 285, 6508–14 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Yu WH, Yu S, Meng Q, Brew K & Woessner JF TIMP-3 binds to sulfated glycosaminoglycans of the extracellular matrix. J Biol Chem 275, 31226–32 (2000). [DOI] [PubMed] [Google Scholar]
- 80.Demeulemeester D et al. Overexpression of tissue inhibitor of matrix metalloproteinases-1 (TIMP-1) in mice does not affect adipogenesis or adipose tissue development. Thromb Haemost 95, 1019–24 (2006). [DOI] [PubMed] [Google Scholar]
- 81.Scroyen I, Jacobs F, Cosemans L, De Geest B & Lijnen HR Blood vessel density in de novo formed adipose tissue is decreased upon overexpression of TIMP-1. Obesity (Silver Spring) 18, 638–40 (2010). [DOI] [PubMed] [Google Scholar]
- 82.Tran TT & Kahn CR Transplantation of adipose tissue and stem cells: role in metabolism and disease. Nat Rev Endocrinol 6, 195–213 (2010).This review provides an overview of applications for adipose-derived stem cells, surveys methods of culturing and differentiating adipocyte precursor cells, and discusses the potential clinical use of adipose transplantation.
- 83.Green H & Meuth M An established pre-adipose cell line and its differentiation in culture. Cell 3, 127–33 (1974). [DOI] [PubMed] [Google Scholar]
- 84.Kuri-Harcuch W & Green H Adipose conversion of 3T3 cells depends on a serum factor. Proc Natl Acad Sci U S A 75, 6107–9 (1978). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Grigoriadis AE, Heersche JN & Aubin JE Differentiation of muscle, fat, cartilage, and bone from progenitor cells present in a bone-derived clonal cell population: effect of dexamethasone. J Cell Biol 106, 2139–51 (1988). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Pairault J & Green H A study of the adipose conversion of suspended 3T3 cells by using glycerophosphate dehydrogenase as differentiation marker. Proc. Natl. Acad. Sci. U.S.A 76, 5138–5142 (1979). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Tang QQ, Otto TC & Lane MD Mitotic clonal expansion: a synchronous process required for adipogenesis. Proc Natl Acad Sci U S A 100, 44–9 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Dike LE & Farmer SR Cell adhesion induces expression of growth-associated genes in suspension-arrested fibroblasts. Proc Natl Acad Sci U S A 85, 6792–6 (1988). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.McBeath R, Pirone DM, Nelson CM, Bhadriraju K & Chen CS Cell shape, cytoskeletal tension, and RhoA regulate stem cell lineage commitment. Dev. Cell 6, 483–495 (2004).Provides evidence for the role of cell shape and the Rho pathway in the regulation adipogenic/osteogenic cell fate decisions.
- 90.Kilian KA, Bugarija B, Lahn BT & Mrksich M Geometric cues for directing the differentiation of mesenchymal stem cells. Proc Natl Acad Sci U S A 107, 4872–7 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Noguchi M et al. Genetic and pharmacological inhibition of Rho-associated kinase II enhances adipogenesis. The Journal of biological chemistry 282, 29574–83 (2007). [DOI] [PubMed] [Google Scholar]
- 92.Dupont S et al. Role of YAP/TAZ in mechanotransduction. Nature 474, 179–183 (2011).This recent study from demonstrates that YAP and TAZ are transcription factors downstream of Rho-dependent mechanotransduction that regulate adipogenic commitment in MSCs.
- 93.Sordella R, Jiang W, Chen GC, Curto M & Settleman J Modulation of Rho GTPase signaling regulates a switch between adipogenesis and myogenesis. Cell 113, 147–58 (2003). [DOI] [PubMed] [Google Scholar]
- 94.Bryan BA et al. Modulation of muscle regeneration, myogenesis, and adipogenesis by the Rho family guanine nucleotide exchange factor GEFT. Mol Cell Biol 25, 11089–101 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Gupta RK et al. Transcriptional control of preadipocyte determination by Zfp423. Nature 464, 619–623 (2010).This paper demonstrates that transcription factor Zfp423 is an adipogenic competency factor.
- 96.Cheng LE, Zhang J & Reed RR The transcription factor Zfp423/OAZ is required for cerebellar development and CNS midline patterning. Dev. Biol 307, 43–52 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Takahashi K & Yamanaka S Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell 126, 663–676 (2006). [DOI] [PubMed] [Google Scholar]
- 98.Cristancho AG et al. Transcriptional Repressor TCF7L1 Promotes Adipogenic Competency in Precursor Cells Proc Natl Acad Sci U S A In Press, (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Yi F & Merrill BJ Stem cells and TCF proteins: a role for beta-catenin--independent functions. Stem Cell Rev 3, 39–48 (2007). [DOI] [PubMed] [Google Scholar]
- 100.Lefterova MI & Lazar MA New developments in adipogenesis. Trends Endocrinol. Metab 20, 107–114 (2009). [DOI] [PubMed] [Google Scholar]
- 101.Hwang C-S, Loftus TM, Mandrup S & Lane MD ADIPOCYTE DIFFERENTIATION AND LEPTIN EXPRESSION. Annu. Rev. Cell Dev. Biol 13, 231–259 (1997). [DOI] [PubMed] [Google Scholar]
- 102.Rosen ED & MacDougald OA Adipocyte differentiation from the inside out. Nature reviews 7, 885–96 (2006). [DOI] [PubMed] [Google Scholar]
- 103.Yeh WC, Cao Z, Classon M & McKnight SL Cascade regulation of terminal adipocyte differentiation by three members of the C/EBP family of leucine zipper proteins. Genes Dev 9, 168–181 (1995). [DOI] [PubMed] [Google Scholar]
- 104.Wu Z, Xie Y, Bucher NL & Farmer SR Conditional ectopic expression of C/EBP beta in NIH-3T3 cells induces PPAR gamma and stimulates adipogenesis. Genes Dev 9, 2350–63 (1995). [DOI] [PubMed] [Google Scholar]
- 105.Steger DJ et al. Propagation of adipogenic signals through an epigenomic transition state. Genes Dev 24, 1035–1044 (2010).This study identifies an epigenomic transition state in adipogenesis.
- 106.Tzameli I et al. Regulated Production of a Peroxisome Proliferator-activated Receptor-{gamma} Ligand during an Early Phase of Adipocyte Differentiation in 3T3-L1 Adipocytes. J. Biol. Chem 279, 36093–36102 (2004). [DOI] [PubMed] [Google Scholar]
- 107.Martini CN, Plaza MV & Vila M del C. PKA-dependent and independent cAMP signaling in 3T3-L1 fibroblasts differentiation. Mol. Cell. Endocrinol 298, 42–47 (2009). [DOI] [PubMed] [Google Scholar]
- 108.Petersen RK et al. Cyclic AMP (cAMP)-mediated stimulation of adipocyte differentiation requires the synergistic action of Epac- and cAMP-dependent protein kinase-dependent processes. Mol Cell Biol 28, 3804–16 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Kawai M & Rosen CJ PPARγ: a circadian transcription factor in adipogenesis and osteogenesis. Nat Rev Endocrinol 6, 629–636 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Lefterova MI et al. PPARgamma and C/EBP factors orchestrate adipocyte biology via adjacent binding on a genome-wide scale. Genes & development 22, 2941–52 (2008).These two papers describes the genome-wide binding of PPARγ in 3T3-L1 adipocytes, and identified cooperation with C/EBP proteins as a hallmark of genomic PPARγ binding in adipocytes.
- 111.Nielsen R et al. Genome-wide profiling of PPARgamma:RXR and RNA polymerase II occupancy reveals temporal activation of distinct metabolic pathways and changes in RXR dimer composition during adipogenesis. Genes & development 22, 2953–67 (2008).These two papers describes the genome-wide binding of PPARγ in 3T3-L1 adipocytes, and identified cooperation with C/EBP proteins as a hallmark of genomic PPARγ binding in adipocytes.
- 112.Siersbæk R et al. Extensive chromatin remodelling and establishment of transcription factor “hotspots” during early adipogenesis. EMBO J 30, 1459–1472 (2011).Utilizes DNAse hypersensitivity followed by deep sequencing to identify open chromatin regions during early adipogenesis and in mature adipocytes.
- 113.Zhang JW, Klemm DJ, Vinson C & Lane MD Role of CREB in transcriptional regulation of CCAAT/enhancer-binding protein beta gene during adipogenesis. J Biol Chem 279, 4471–8 (2004). [DOI] [PubMed] [Google Scholar]
- 114.Wang D et al. Signal transducer and activator of transcription 3 (STAT3) regulates adipocyte differentiation via peroxisome-proliferator-activated receptor gamma (PPARgamma). Biol. Cell 102, 1–12 (2010). [DOI] [PubMed] [Google Scholar]
- 115.Zhang K, Guo W, Yang Y & Wu J JAK2/STAT3 pathway is involved in the early stage of adipogenesis through regulating C/EBPβ transcription. J. Cell. Biochem 112, 488–497 (2011). [DOI] [PubMed] [Google Scholar]
- 116.Birsoy K, Chen Z & Friedman J Transcriptional regulation of adipogenesis by KLF4. Cell Metab 7, 339–347 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Chen Z, Torrens JI, Anand A, Spiegelman BM & Friedman JM Krox20 stimulates adipogenesis via C/EBPbeta-dependent and -independent mechanisms. Cell Metab 1, 93–106 (2005). [DOI] [PubMed] [Google Scholar]
- 118.Park B-H, Qiang L & Farmer SR Phosphorylation of C/EBPbeta at a consensus extracellular signal-regulated kinase/glycogen synthase kinase 3 site is required for the induction of adiponectin gene expression during the differentiation of mouse fibroblasts into adipocytes. Mol. Cell. Biol 24, 8671–8680 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Tang Q-Q et al. Sequential phosphorylation of CCAAT enhancer-binding protein beta by MAPK and glycogen synthase kinase 3beta is required for adipogenesis. Proc. Natl. Acad. Sci. U.S.A 102, 9766–9771 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Asada M et al. DNA binding-dependent glucocorticoid receptor activity promotes adipogenesis via Krüppel-like factor 15 gene expression. Lab. Invest 91, 203–215 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Wiper-Bergeron N, Wu D, Pope L, Schild-Poulter C & Hache RJ Stimulation of preadipocyte differentiation by steroid through targeting of an HDAC1 complex. EMBO J 22, 2135–45 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Wiper-Bergeron N, Salem HA, Tomlinson JJ, Wu D & Hache RJ Glucocorticoid-stimulated preadipocyte differentiation is mediated through acetylation of C/EBPbeta by GCN5. Proc Natl Acad Sci U S A 104, 2703–8 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Tanaka T, Yoshida N, Kishimoto T & Akira S Defective adipocyte differentiation in mice lacking the C/EBPbeta and/or C/EBPdelta gene. EMBO J 16, 7432–43 (1997). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Tang QQ, Zhang JW & Daniel Lane M Sequential gene promoter interactions of C/EBPbeta, C/EBPalpha, and PPARgamma during adipogenesis. Biochemical and biophysical research communications 319, 235–9 (2004). [DOI] [PubMed] [Google Scholar]
- 125.Rosen ED et al. PPAR gamma is required for the differentiation of adipose tissue in vivo and in vitro. Mol. Cell 4, 611–617 (1999). [DOI] [PubMed] [Google Scholar]
- 126.Lehmann JM et al. An antidiabetic thiazolidinedione is a high affinity ligand for peroxisome proliferator-activated receptor gamma (PPAR gamma). J. Biol. Chem 270, 12953–12956 (1995). [DOI] [PubMed] [Google Scholar]
- 127.Tang W, Zeve D, Seo J, Jo A-Y & Graff JM Thiazolidinediones regulate adipose lineage dynamics. Cell Metab 14, 116–122 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Tontonoz P, Hu E, Graves RA, Budavari AI & Spiegelman BM mPPAR gamma 2: tissue-specific regulator of an adipocyte enhancer. Genes Dev 8, 1224–1234 (1994). [DOI] [PubMed] [Google Scholar]
- 129.Chawla A, Schwarz EJ, Dimaculangan DD & Lazar MA Peroxisome proliferator-activated receptor (PPAR) gamma: adipose-predominant expression and induction early in adipocyte differentiation. Endocrinology 135, 798–800 (1994). [DOI] [PubMed] [Google Scholar]
- 130.Mikkelsen TS et al. Comparative epigenomic analysis of murine and human adipogenesis. Cell 143, 156–169 (2010).Provides an extensive collection of genome-wide profiles of transcription factor binding and histone modifications in human and murine adipogenesis.
- 131.Haberland M, Carrer M, Mokalled MH, Montgomery RL & Olson EN Redundant control of adipogenesis by histone deacetylases 1 and 2. J Biol Chem 285, 14663–70 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Kim S-N, Choi H-Y & Kim YK Regulation of adipocyte differentiation by histone deacetylase inhibitors. Arch. Pharm. Res 32, 535–541 (2009). [DOI] [PubMed] [Google Scholar]
- 133.Lagace DC & Nachtigal MW Inhibition of histone deacetylase activity by valproic acid blocks adipogenesis. J. Biol. Chem 279, 18851–18860 (2004). [DOI] [PubMed] [Google Scholar]
- 134.Chatterjee TK et al. Histone deacetylase 9 is a negative regulator of adipogenic differentiation. J. Biol. Chem 286, 27836–27847 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Yoo EJ, Chung J-J, Choe SS, Kim KH & Kim JB Down-regulation of histone deacetylases stimulates adipocyte differentiation. J. Biol. Chem 281, 6608–6615 (2006). [DOI] [PubMed] [Google Scholar]
- 136.Nebbioso A et al. HDACs class II-selective inhibition alters nuclear receptor-dependent differentiation. J. Mol. Endocrinol 45, 219–228 (2010). [DOI] [PubMed] [Google Scholar]
- 137.Picard F et al. Sirt1 promotes fat mobilization in white adipocytes by repressing PPAR-gamma. Nature 429, 771–776 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Jing E, Gesta S & Kahn CR SIRT2 regulates adipocyte differentiation through FoxO1 acetylation/deacetylation. Cell Metab 6, 105–114 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Lee J et al. Targeted inactivation of MLL3 histone H3-Lys-4 methyltransferase activity in the mouse reveals vital roles for MLL3 in adipogenesis. Proc. Natl. Acad. Sci. U.S.A 105, 19229–19234 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Lizcano F, Romero C & Vargas D Regulation of adipogenesis by nuclear receptor PPARγ is modulated by the histone demethylase JMJD2C. Genet. Mol. Biol 34, 19–24 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Cho Y-W et al. Histone methylation regulator PTIP is required for PPARgamma and C/EBPalpha expression and adipogenesis. Cell Metab 10, 27–39 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Park U-H, Yoon SK, Park T, Kim E-J & Um S-J Additional sex comb-like (ASXL) proteins 1 and 2 play opposite roles in adipogenesis via reciprocal regulation of peroxisome proliferator-activated receptor {gamma}. J. Biol. Chem 286, 1354–1363 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Wang J & Lazar MA Bifunctional role of Rev-erbalpha in adipocyte differentiation. Mol. Cell. Biol 28, 2213–2220 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Kawai M et al. A circadian-regulated gene, Nocturnin, promotes adipogenesis by stimulating PPAR-gamma nuclear translocation. Proc. Natl. Acad. Sci. U.S.A 107, 10508–10513 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Tong Q et al. Function of GATA transcription factors in preadipocyte-adipocyte transition. Science 290, 134–138 (2000). [DOI] [PubMed] [Google Scholar]
- 146.Tong Q, Tsai J, Tan G, Dalgin G & Hotamisligil GS Interaction between GATA and the C/EBP family of transcription factors is critical in GATA-mediated suppression of adipocyte differentiation. Mol. Cell. Biol 25, 706–715 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Wu Z et al. Cross-regulation of C/EBP alpha and PPAR gamma controls the transcriptional pathway of adipogenesis and insulin sensitivity. Mol. Cell 3, 151–158 (1999). [DOI] [PubMed] [Google Scholar]
- 148.Schupp M et al. Re-expression of GATA2 cooperates with peroxisome proliferator-activated receptor-gamma depletion to revert the adipocyte phenotype. The Journal of biological chemistry 284, 9458–64 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Liao W et al. Suppression of PPAR-gamma attenuates insulin-stimulated glucose uptake by affecting both GLUT1 and GLUT4 in 3T3-L1 adipocytes. Am. J. Physiol. Endocrinol. Metab 293, E219–227 (2007). [DOI] [PubMed] [Google Scholar]
- 150.Imai T et al. Peroxisome proliferator-activated receptor gamma is required in mature white and brown adipocytes for their survival in the mouse. Proc. Natl. Acad. Sci. U.S.A 101, 4543–4547 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.He W et al. Adipose-specific peroxisome proliferator-activated receptor γ knockout causes insulin resistance in fat and liver but not in muscle. Proceedings of the National Academy of Sciences 100, 15712–15717 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Schmidt SF et al. Cross species comparison of C/EBPα and PPARγ profiles in mouse and human adipocytes reveals interdependent retention of binding sites. BMC Genomics 12, 152 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Soccio RE et al. Species-Specific Strategies Underlying Conserved Functions of Metabolic Transcription Factors. Mol Endocrinol (2011).doi: 10.1210/me.2010-0454 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Nedergaard J, Petrovic N, Lindgren EM, Jacobsson A & Cannon B PPARgamma in the control of brown adipocyte differentiation. Biochim. Biophys. Acta 1740, 293–304 (2005). [DOI] [PubMed] [Google Scholar]
- 155.Kajimura S et al. Initiation of myoblast to brown fat switch by a PRDM16-C/EBP-beta transcriptional complex. Nature 460, 1154–8 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Kajimura S et al. Regulation of the brown and white fat gene programs through a PRDM16/CtBP transcriptional complex. Genes Dev 22, 1397–1409 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Puigserver P et al. A cold-inducible coactivator of nuclear receptors linked to adaptive thermogenesis. Cell 92, 829–839 (1998). [DOI] [PubMed] [Google Scholar]
- 158.Uldry M et al. Complementary action of the PGC-1 coactivators in mitochondrial biogenesis and brown fat differentiation. Cell Metab 3, 333–341 (2006). [DOI] [PubMed] [Google Scholar]
- 159.Tontonoz P & Spiegelman BM Fat and beyond: the diverse biology of PPARgamma. Annu Rev Biochem 77, 289–312 (2008). [DOI] [PubMed] [Google Scholar]
- 160.Müller S & Krämer OH Inhibitors of HDACs--effective drugs against cancer? Curr Cancer Drug Targets 10, 210–228 (2010). [DOI] [PubMed] [Google Scholar]
- 161.Sugii S et al. Human and mouse adipose-derived cells support feeder-independent induction of pluripotent stem cells. Proc Natl Acad Sci U S A 107, 3558–63 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Sauer B Inducible gene targeting in mice using the Cre/lox system. Methods 14, 381–392 (1998). [DOI] [PubMed] [Google Scholar]
- 163.Chang TH & Polakis SE Differentiation of 3T3-L1 fibroblasts to adipocytes. Effect of insulin and indomethacin on the levels of insulin receptors. J Biol Chem 253, 4693–6 (1978). [PubMed] [Google Scholar]
- 164.Costa M, Manen CA & Russell DH In vivo activation of cAMP-dependent protein kinase by aminophylline and 1-methyl, 3-isobutylxanthine. Biochem Biophys Res Commun 65, 75–81 (1975). [DOI] [PubMed] [Google Scholar]
- 165.Elks ML, Manganiello VC & Vaughan M Hormone-sensitive particulate cAMP phosphodiesterase activity in 3T3-L1 adipocytes. Regulation of responsiveness by dexamethasone. J Biol Chem 258, 8582–7 (1983). [PubMed] [Google Scholar]
- 166.Fischer-Posovszky P, Newell FS, Wabitsch M & Tornqvist HE Human SGBS cells - a unique tool for studies of human fat cell biology. Obes Facts 1, 184–9 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Mandrup S, Loftus TM, MacDougald OA, Kuhajda FP & Lane MD Obese gene expression at in vivo levels by fat pads derived from s.c. implanted 3T3-F442A preadipocytes. Proc. Natl. Acad. Sci. U.S.A 94, 4300–4305 (1997). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.MacDougald OA & Lane MD Transcriptional regulation of gene expression during adipocyte differentiation. Annu Rev Biochem 64, 345–73 (1995). [DOI] [PubMed] [Google Scholar]
- 169.Ross SR et al. A fat-specific enhancer is the primary determinant of gene expression for adipocyte P2 in vivo. Proc. Natl. Acad. Sci. U.S.A 87, 9590–9594 (1990). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Hunt CR, Ro JH, Dobson DE, Min HY & Spiegelman BM Adipocyte P2 gene: developmental expression and homology of 5’-flanking sequences among fat cell-specific genes. Proc. Natl. Acad. Sci. U.S.A 83, 3786–3790 (1986). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Makowski L et al. Lack of macrophage fatty-acid-binding protein aP2 protects mice deficient in apolipoprotein E against atherosclerosis. Nat. Med 7, 699–705 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Urs S, Harrington A, Liaw L & Small D Selective expression of an aP2/Fatty Acid Binding Protein 4-Cre transgene in non-adipogenic tissues during embryonic development. Transgenic Res 15, 647–653 (2006). [DOI] [PubMed] [Google Scholar]
- 173.Martens K, Bottelbergs A & Baes M Ectopic recombination in the central and peripheral nervous system by aP2/FABP4-Cre mice: implications for metabolism research. FEBS Lett 584, 1054–1058 (2010). [DOI] [PubMed] [Google Scholar]
- 174.Wang ZV, Deng Y, Wang QA, Sun K & Scherer PE Identification and characterization of a promoter cassette conferring adipocyte-specific gene expression. Endocrinology 151, 2933–2939 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Calo E et al. Rb regulates fate choice and lineage commitment in vivo. Nature 466, 1110–1114 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]