Target Specificity of the Endonuclease from the Xenopus laevis Non-Long Terminal Repeat Retrotransposon, Tx1L (original) (raw)
Abstract
Elements of the Tx1L family are non-long terminal repeat retrotransposons (NLRs) that are dispersed in the genome of Xenopus laevis. Essentially all genomic copies of Tx1L are found inserted at a specific site within another family of transposable elements (Tx1D). This suggests that Tx1L is a site-specific retrotransposon. Like many (but not all) other NLRs, the Xenopus element encodes an apparent endonuclease that is related in sequence to the apurinic-apyrimidinic endonucleases that participate in DNA repair. This enzyme is thought to introduce the single-strand break in target DNA that initiates transposition by the target-primed reverse transcription (TPRT) mechanism. To explore the issue of target specificity more fully, we expressed the polypeptide encoded by the endonuclease domain of open reading frame 2 from Tx1L (Tx1L EN) and characterized its cleavage capabilities. This endonuclease makes a specific nick in the bottom strand precisely at one end of the presumed Tx1L target duplication. Because this activity leaves a 5′-phosphate and 3′-hydroxyl at the nick, it has the location and chemistry required to initiate new insertion events by TPRT. Tx1L EN does not make a specific cut at a preferred target site for Tx1D elements, ruling out the alternative possibility that the composite Tx1L-Tx1D element moves as a unit under the control of functions encoded by Tx1L. Further characterization revealed that the endonuclease remains active for many hours at room temperature and that it is capable of enzymatic turnover. Scanning substitution mutagenesis located the recognition site for Tx1L EN within 10 bp surrounding the primary nick site. Implications of these features for natural transposition events are discussed.
Transposons are ubiquitous mobile genetic elements found in the genomes of most, if not all, organisms. They can be grouped into two main categories based on sequence organization and mode (or presumed mode) of transposition (3). The first group of transposons consists of the cut-and-paste elements, which move strictly through DNA intermediates. Examples of this type of transposon include the bacterial insertion sequences, the eukaryotic Tc1/Mariner elements, maize Ac/Ds elements, and Drosophila P elements (8, 20, 24, 29, 32). The second group, the retrotransposons, transpose through an RNA intermediate.
The retrotransposons can be further subdivided into two subgroups that differ in sequence organization and mechanisms of retrotransposition. The retrovirus-like long terminal repeat (LTR) retrotransposons reverse transcribe their RNA genome in the cytoplasm, producing a double-stranded DNA copy with terminal direct repeats. This species is transported to the cell nucleus, where it is integrated into chromosomal DNA courtesy of an element-encoded integrase. Examples of this type of retrotransposon are Ty_1_ and Ty_3_ of Saccharomyces cerevisiae, Copia and 412 from Drosophila, and Tf1 from Schizosaccharomyces pombe (3).
Although they also rely on reverse transcription, the non-LTR retrotransposons (NLRs) transpose through a fundamentally different mechanism than the retrovirus-like elements (7). This process, target-primed reverse transcription or TPRT, is diagrammed in Fig. 1 (23). Element RNA is packaged in a cytoplasmic ribonucleoprotein particle (RNP) that includes element-encoded proteins (14, 15, 19, 33, 38). This RNP moves to the cell nucleus, where it finds and nicks one strand of its DNA target (9, 10, 37). The free 3′-OH at the nick site is used by reverse transcriptase to prime first-strand cDNA synthesis with element RNA as the template (23). This step links the element DNA to the target as an inherent feature of reverse transcription, so there is no need for a separate integrase function. Ultimately the second target strand is cut, the second cDNA strand is synthesized, and the integration junctions are sealed, but the orchestration of these later steps remains obscure. Other examples of NLR retrotransposons are L1 elements in mammals (e.g., L1Hs in humans), the Drosophila I factor, and the R1 and R2 elements in insects (3, 6, 17).
FIG. 1.

TPRT model. Several of the steps in this reaction remain hypothetical. DNA strands are depicted as black lines; the target sequence that is duplicated upon insertion is shown by thick black lines; the element RNA is shown as a gray line. Half arrowheads represent 3′ ends. (a) The element RNA, with associated element-encoded proteins, is transported to the nucleus, and the target sequenced is located. (b) The element-encoded endonuclease nicks the bottom strand at the left end of the target. (c) The exposed 3′-OH at the nick is used to prime first-strand cDNA synthesis, using the element RNA as the template. (d) The second strand is cleaved, perhaps by the same endonuclease. (e) The exposed 3′-OH primes synthesis of second-strand cDNA. Element RNA is degraded by an RNase H activity or is displaced during second-strand synthesis. (f) After completion of the second strand, both junctions are sealed, presumably by cellular repair enzymes. Modified from reference 23.
Typical NLR elements have two open reading frames (ORFs), although the arthropod R2 elements have only one. The product of the first ORF (ORF1p) has affinity for single-stranded nucleic acid and binds to element RNA (14, 15, 19, 25, 33). The product of the second ORF (ORF2p) has homology to reverse transcriptase, and this activity has been demonstrated for L1Hs, R2Bm, Jockey, CRE1, and Tx1L (4, 11, 16, 23, 26). The second ORF also contains an endonuclease domain (9, 10, 37), which is responsible for generating the target nick that initiates TPRT (23, 38). By mutating crucial residues in the endonuclease of L1Hs, Feng et al. (9) demonstrated that its activity is required for active transposition in cultured cells.
To date, two types of NLR endonucleases have been characterized (9, 37). The endonucleases of the arthropod R2 elements represent one type and are thought to be similar to type IIS restriction endonucleases with separate DNA cleavage and DNA-binding domains (39). The endonuclease domain of the L1Hs element (L1Hs EN) is representative of the second type in that it has weak homology to apurinic-apyrimidinic (AP) endonuclease and DNase I (9). Within this second category, only the endonuclease of the L1Tc element from a trypanosome has been shown to cut apurinic sites (31); most others probably lack genuine AP endonuclease activity (5). All AP endonucleases borne by NLR elements are more closely related to each other than to any other AP endonuclease or DNase I-like sequences. To emphasize their role in transposition, we will refer to these element-encoded endonucleases as ADR (AP- and DNase I-like retrotransposon) endonucleases.
Interestingly, both nonspecific and site-specific NLR elements exist. It is presumed that target site selection is a property of the element-encoded endonuclease, and this has been supported for the relatively nonspecific L1Hs (9) and for the highly specific insect elements, R2Bm (23, 37) and R1Bm (10).
The Tx1L family consists of 6.9-kb sequences that are present in about 150 dispersed copies in the Xenopus laevis genome (13). They have sequence features very similar to those of transposable elements of the NLR family, including ORFs that encode an RNA-binding protein (ORF1p) (33) and a reverse transcriptase (ORF2p) (4) (Fig. 2B). A curious feature of the Tx1L elements is that they are always found inserted in specific sequences within a family of apparent cut-and-paste transposons, Tx1D (Fig. 2A). There are approximately 1,500 Tx1D elements in the genome, each of which has 19-bp inverted terminal repeats and is flanked by a 4-bp target duplication (12). About 10% of the Tx1D's are interrupted by a Tx1L insertion (13). All Tx1L elements are found at a specific site within an internal tandem repeat (PTR-1) of Tx1D, and each insertion is flanked by a 23-bp duplication of PTR-1 sequence.
FIG. 2.

Tx1 element structures. (A) Tx1D elements are composed of left common flank (LCF), several 400-bp PTR-1 repeats, several 400-bp PTR-2 repeats, and right common flank (RCF) (12). Short inverted terminal repeats are indicated with triangles, and the target site duplication, TTAA, that surrounds each element has also been included. The composite Tx1C elements differ from Tx1D by the insertion of a 6.9-kb Tx1L sequence into one of the PTR-1 repeats. (B) Expanded view of the structure of Tx1L. The 775-amino-acid (775aa) ORF1 and the 1,304-amino-acid (1304aa) ORF2 are depicted by thick arrows. Untranslated regions (UTR) are shown as thin lines. The flanking 23-bp target site duplication is shown as small black arrowheads. Locations of salient features of the two ORFs are indicated. Sequence from reference 13.
These structural features support the presumption that Tx1L is an independent retrotransposon with site specificity for the Tx1D target. In the absence of direct evidence for this interpretation, we also considered the possibility that the composite element, Tx1D-Tx1L (also called Tx1C), is the mobile unit (13). This hypothesis was motivated in part by the absence of an obvious ORF in the Tx1D sequence and led to the suggestion, albeit unprecedented, that the Tx1L ORF products might act at the Tx1D ends to mobilize both the simple cut-and-paste and the composite elements. A further possibility is that the Tx1L proteins participate in both types of reaction, cut-and-paste and retrotransposition.
In their compilation of NLR sequences, Feng et al. (9) noted the homology of the N terminus of ORF2p of Tx1L to the endonuclease domains of L1Hs and related elements (see also reference 31). Given the apparent role of this endonuclease in determining the transposition target, this has made it possible for us to test the alternatives described above. If Tx1L is an independent NLR, its endonuclease (Tx1L EN) should recognize the target sequence within Tx1D, while recognition of a chromosomal target for Tx1D would indicate a role in catalyzing mobility of the composite elements. The experiments described here clearly show that the Tx1L target, and not the Tx1D target, is cleaved. This cleavage occurs at precisely the expected location for TPRT of the Tx1L element. Nucleotide sequences flanking the nick are demonstrated to be important for target recognition by the endonuclease.
MATERIALS AND METHODS
Cloning of Tx1L EN.
The first 717 bp of ORF2, corresponding to the first 239 amino acids, were amplified by PCR from the clone pBORF2b, which contains the complete ORF2 sequence of the Tx1L element from lambda clone B10 (13), using the DNA oligonucleotides GTAATACGACTCACTATAGGGC and GGCGGATCCTTAGTGGTGATGGTGATGGTGAGATCCTCTGATTGACATTCTCAGGGATAC as primers. The first primer was complementary to vector sequences just upstream of the N terminus of ORF2. The second primer was complementary to codons 232 through 239 of ORF2 and also included nucleotides encoding an RGSHHHHHH tag on the C terminus of Tx1L EN, as well as a _Bam_HI restriction site for cloning purposes. The amplified DNA fragment was treated with _Nco_I, which cuts at the junction of vector sequence and the first ATG codon of Tx1L ORF2, and _Bam_HI, and the resulting fragment was ligated into the _Nco_I and _Bam_HI sites of pET16b (Novagen) by T4 DNA ligase (New England Biolabs), using conditions recommended by the supplier. The ligation products were electroporated into Escherichia coli XL1-Blue (Stratagene) by using a Gene Pulser (Bio-Rad). Transformed bacteria were plated on ampicillin-containing Luria-Bertani (LB) plates (1). The inserts of candidate clones were sequenced by the University of Utah Core Sequencing Facility, using oligonucleotide primers specific for the pET16b vector. The clone chosen for use in these experiments has been given the name pE1EN.
Expression and purification of Tx1L EN.
pE1EN DNA was transformed into competent E. coli BL21(DE3)-pLysS cells (Novagen) and plated on carbenicillin-containing LB plates. A single colony was used to inoculate a 5-ml culture (LB with 100 μg of ampicillin or carbenicillin per ml and 20 μg of chloramphenicol per ml) which was grown to an optical density at 600 nm of 0.6 at 37°C. The 5-ml culture was used to seed a 500-ml culture in the same medium, which was grown to an optical density at 600 nm of 0.6 at 37°C prior being induced with 1 mM isopropyl-β-d-thiogalactopyranoside (IPTG) and incubated for a further 2 h at room temperature. The bacteria from the 500-ml culture were collected by centrifugation (2,200 × g for 14 min). The cell pellet from each 100 ml of culture was washed with 30 ml of cold H2O and frozen in liquid nitrogen. To initiate purification, each frozen pellet was resuspended in 0.5 ml of binding buffer (10 mM β-mercaptoethanol [BME], 0.1% Triton X-100, 50 mM sodium phosphate [pH 8.0], 300 mM NaCl, 10% glycerol, 10 mM imidazole). The resuspended bacteria were combined and disrupted with a Branson Sonifier 450, using three sets of 16 pulses at 50% power and 50% duty cycle. The sonicated bacterial lysate was spun in an Eppendorf centrifuge (14,000 rpm) for 30 min. The resulting supernatant was removed, forced through a 0.2-μm-pore-size syringe filter, and then loaded onto a 0.5-ml Talon resin column (Clontech) equilibrated in binding buffer. The column was washed twice with 1.5 ml of binding buffer, followed by two washes with 10 mM BME–0.1% Triton X-100–50 mM sodium phosphate (pH 8.0)–300 mM NaCl–10% glycerol and two washes with 10 mM BME–0.1% Triton X-100–50 mM sodium phosphate (pH 8.0)–700 mM NaCl–30% glycerol. Two final washes with 10 mM BME–0.1% Triton X-100–50 mM sodium phosphate (pH 8.0)–300 mM NaCl–10% glycerol–30 mM imidazole were followed by elution of the bound protein with 1.5 ml of elution buffer (10 mM BME, 0.1% Triton X-100, 50 mM sodium phosphate [pH 8.0], 300 mM NaCl, 10% glycerol, 250 mM imidazole). The eluted protein was frozen in liquid nitrogen in 20- to 40-μl aliquots. The same process was performed in parallel for E. coli BL21(DE3)-pLysS containing the pET16b vector (control eluate) and again for a mock purification without any bacterial extract (blank eluate). The concentration of Tx1L EN was determined by analysis of a Coomassie blue-stained gel after sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) in comparison with known amounts of bovine serum albumin on the same gel as a mass standard. The concentration of Tx1L EN used for most of the experiments described here was 2.2 pmol/μl.
Endonuclease assays.
Complementary oligonucleotides corresponding to the Tx1L target (70 bp) and the Tx1D target (71 bp) were made by Integrated DNA Technologies. Sequencing primers for the Tx1L target (CGCATACAAACAGTCCCGTGG and CCCCGCAAAAATGCAGTCAATG) and the Tx1D target (GCAATAATACACAGAATCCC and GTTCAAAAGG TCAGATTTATTAT) were made by the DNA-Peptide Core Facility at the University of Utah. The complementary 42-bp Tx1L target oligonucleotides used in the experiments shown in Fig. 7 and 8 were made by the Core Facility (see Fig. 8 for sequences). All oligonucleotides were gel purified.
FIG. 7.

Time course of nicking by Tx1L EN. Bottom strand-labeled (noted as an asterisk) target DNA was incubated with the endonuclease (ENDO) or control (CTRL) eluate just as in Fig. 4 but for longer periods of time. Incubation times in minutes are given above the lanes, and corresponding sequencing lanes are shown at the left. In addition to the major nick site (bold arrowhead), the location of a minor site is indicated (thin arrow) both in the gel and on the substrate sequence.
FIG. 8.

Enzymatic turnover. Tx1L EN was incubated with the Tx1L target DNA at an enzyme/target ratio of 1:38. Conditions were as in previous experiments except that a 42-bp target duplex was used. Incubation times in hours are given above the lanes.
Fifty picomoles of top-strand oligonucleotide and 50 pmol of bottom-strand oligonucleotide were end labeled with 32P in separate 10-μl reactions using 450 μCi of [γ-32P]ATP (Amersham) and T4 polynucleotide kinase (New England Biolabs). The kinase reaction was stopped by heat inactivation; 40 μl of 10 mM Tris-HCl (pH 8.0)–50 mM NaCl–1 mM EDTA was added prior to loading the reaction mixture onto 0.8-ml-bed-volume Sephadex G-25 (medium grade) spin columns equilibrated with the same buffer (1). Equal amounts of cold top and bottom strands were also run over spin columns. Double-stranded substrates labeled on one strand were generated by combining in 50 μl 25 pmol of labeled strand and 25 pmol of cold complementary strand. Annealing was accomplished by boiling for 1 min followed by slow cooling to room temperature. Salt was removed from the annealed oligonucleotides on a G-25 spin column equilibrated in 10 mM Tris-HCl (pH 8.0).
Each standard 40-μl endonuclease reaction contained 1 μl (0.5 pmol) of labeled template DNA and 2 μl of eluate in reaction buffer (50 mM HEPES [pH 7.0], 1.5 mM MgCl2, 0.1% Triton X-100, 100 μg of bovine serum albumin per μl, 1 mM dithiothreitol, 10% glycerol, 5 ng sheared salmon sperm DNA per μl. Blank eluate was used to bring the total eluate volume to 2 μl when necessary. The reaction mix was incubated at 22°C before the reaction was terminated with 4.5 μl of stop buffer (2% SDS, 50 mM EDTA). Terminated reactions were run over H2O-equilibrated Sephadex G-25 spin columns into 15 μl of 95% formamide stop-buffer (GibcoBRL) and either 4.5 μl of 10× Taq buffer (GibcoBRL) or 4.5 μl of 10× PFU buffer (Stratagene), so that the reactions would be in the same buffer as the sequencing ladder being used to map the positions of the nick sites. The samples were boiled for 2 min and quickly cooled in an ice-water bath, and the equal counts from each sample were loaded onto a 6% polyacrylamide sequencing gel. The sequencing ladders were generated using a cycle sequencing kit (GibcoBRL), the sequencing primers listed above, and either the long oligonucleotides or the relevant parent plasmids as templates.
The endonuclease reactions used to demonstrate enzymatic turnover were similar to those described above except that the salmon sperm DNA was replaced with 83 pmol of unlabeled target (the 70-bp Tx1L target oligonucleotide). Instead of the final spin columns, the reactions were ethanol precipitated and resuspended in 50% formamide.
For the ligation reaction, three standard endonuclease reactions were incubated for 30 min, combined, extracted with phenol-chloroform-isoamyl alcohol (25:24:1), ethanol precipitated, and dissolved in 10 mM Tris-HCl (pH 8.0)–1 mM EDTA. One-fourth of the dissolved DNA was treated with approximately 1,000 U of T4 ligase (New England Biolabs) in a 100-μl reaction, and one-fourth of the resuspended DNA was mock ligated (no T4 ligase). All quantitations of cutting efficiency were performed with ImageQuant software on a Molecular Dynamics model 400E PhosphorImager.
RESULTS
Enrichment and activity of Tx1L EN.
The endonuclease domain of Tx1L, represented by the first 239 amino acids of ORF2, was expressed in E. coli from a pET16b construct. A C-terminal His6 tag was added to enable enrichment of the resulting protein by passing the bacterial lysate over a cobalt chelate column and eluting Tx1L EN with imidazole (Fig. 3). This same purification procedure was performed on bacteria carrying only the pET16b vector. In addition to the expected polypeptide (predicted molecular mass of 28.4 kDa), a few faint contaminating bands in the Tx1L EN preparation were present in the control eluate. We estimate that the Tx1L EN was approximately 70% pure.
FIG. 3.
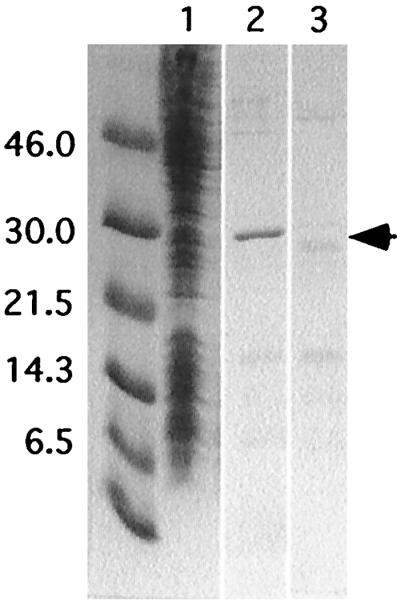
Expression and enrichment of Tx1L EN. Tx1L EN was expressed in bacteria from the pET16b vector and purified on a cobalt chelate column. Various fractions were analyzed by PAGE and stained with Coomassie blue. Lane 1, Tx1L EN-expressing bacterial extract before the nickel column; lane 2, imidazole eluate from the cobalt column; lane 3, control eluate from bacteria carrying the vector only. The unlabeled lane has molecular weight standards of the sizes (in kilodaltons) indicated at the left. The expected location of Tx1L EN is indicated with an arrow.
The activity of Tx1L EN was tested initially on a 70-bp oligonucleotide substrate that represented the Tx1L insertion site within Tx1D and encompassed the 23-bp target sequence that is duplicated around the element (boxed in Fig. 4). This substrate was end labeled with 32P on either the top strand or the bottom strand and exposed to increasing amounts of Tx1L EN eluate. At the end of the incubation, equal counts from each reaction were loaded onto a denaturing polyacrylamide gel for analysis. The positions of the nicks introduced by the enzyme were then precisely mapped by comparing the positions of the bands to a set of dideoxynucleotide sequencing reactions generated from a primer having the same 5′ end as the corresponding labeled strand.
FIG. 4.

Target cleavage by Tx1L EN. The double-stranded DNA oligonucleotide shown at the bottom was the substrate for cleavage by the endonuclease. It is composed of 70 bp of PTR-1 sequence from a Tx1D element. The 23 bp found duplicated around all Tx1L insertions are boxed. This DNA was end labeled on the top or bottom strand and treated with three different amounts (0.5, 1.5, and 5 μl) of Tx1L EN (ENDO) or control (CTRL) eluate. The products were analyzed by electrophoresis next to lanes with sequencing reactions initiated from the corresponding primers (indicated as arrows above and below the sequence at the bottom). Locations of the most prominent nicks on each strand are indicated by a bold arrowhead (bottom strand) and fine arrow (top strand) in both sections of the figure. The cut in the bottom strand corresponds to the location expected based on the TPRT mechanism.
As seen in Fig. 4, Tx1L EN creates only one major nick in the Tx1L substrate. This nick is on the bottom strand at the left extremity of the 23-bp target duplication, at precisely the position required to initiate transposition of Tx1L into this site by TPRT. No nicks of comparable intensity were made in the top strand.
To confirm that Tx1L EN is responsible for the observed nicking activity, protein extract from bacteria containing the pET16b vector was subjected to the same enrichment procedure as the Tx1L EN-containing eluate. This control eluate, which contains many of the contaminants present in the Tx1L EN eluate (Fig. 3), was devoid of the nicking activity seen in the endonuclease eluate (Fig. 4). Furthermore, boiling the endonuclease eluate prior to addition to the reaction destroyed the nicking activity, as did treatment with proteinase K (data not shown).
Target of Tx1L EN.
The activity shown in Fig. 4 indicates strongly that Tx1L ORF2p can catalyze the transposition of the Tx1L element into the PTR-1 sequence of Tx1D. As stated in the introduction, we were also interested in determining whether this protein could participate in the mobilization of Tx1D and composite elements. To resolve this question, we tested the activity of Tx1L EN on a genomic sequence that serves as a target for Tx1D. We showed previously that this site is polymorphic for the presence or absence of a Tx1D element (12).
A 71-bp oligonucleotide corresponding to this Tx1D target was prepared, with the insertion site located centrally (boxed in Fig. 5). Its top and bottom strands were labeled independently, and the resulting duplexes were treated with Tx1L EN under conditions identical to those used for the reactions with the Tx1L target. As seen in Fig. 5, much less significant cuts were made in either strand of the Tx1D target. Because the amounts of substrate and enzyme were carefully matched between the samples shown in Fig. 4 and 5, we conclude that Tx1L EN has a strong preference for the Tx1L target sequence and, therefore, that Tx1L ORF2p very likely catalyzes the transposition of the retroelement by TPRT but does not participate in transposition of Tx1D.
FIG. 5.

Treatment of a Tx1D target with Tx1L EN. An oligonucleotide corresponding to an unoccupied Tx1D target (12) was the substrate in this experiment. The 4-bp target duplication found flanking all Tx1D elements is boxed. The experiment was performed just like that in Fig. 4. The positions of the weak but most prominent nicks on the two strands are indicated with hash marks.
Properties of the Tx1L EN reaction.
To serve as a first step in TPRT, the nick made by Tx1L EN must have not only the proper location but also the appropriate chemistry (23, 37). This was tested by subjecting a partially nicked Tx1L target to reaction with T4 DNA ligase, which requires a 3′-hydroxyl and 5′-phosphate to join DNA segments. The sample shown in Fig. 6 was initially nicked in 22% of the target molecules. Following ligase treatment, nicks remained in only 4%. Conversion of 80% of the nicked bottom strands back to full length demonstrates that the nicks created by Tx1L EN leave a free 3′-OH, as required to prime first-strand DNA synthesis during transposition. The faint bands in Fig. 6 appear to have similar intensities before and after ligase treatment, which suggests that they may not be products of Tx1L EN cutting, although no attempt has been made to quantitate them.
FIG. 6.

Chemistry of the nick. The Tx1L target oligonucleotide, labeled on the bottom strand, was incubated with Tx1L EN as in Fig. 4. One portion of the sample was treated with T4 DNA ligase, and another portion was mock ligated. After electrophoresis, the percentages of total radioactivity found in the bands corresponding to full-length and nicked substrate were determined.
Two aspects of the kinetics of the Tx1L EN reaction were examined. First, an extensive time course (Fig. 7) showed that the major bottom-strand nick remained predominant throughout the reaction. There were no prominent nicks more distant from the labeled end that might have been obscured by cleavage at the TPRT site and no nicks with unusual kinetics that might have been missed in earlier experiments. A similar time course with the top-strand labeled revealed no strong bands and no additional cut sites beyond those documented in Fig. 4 (not shown). As can be seen in Fig. 7, Tx1L EN is capable of converting the vast majority of the Tx1L target DNA to product. This differs from the experience with the endonuclease of R1Bm, which nicked only a minority of its target substrates (10). Averaging the last two time points in Fig. 7, about 22% of the substrate remained uncut, 65% was converted to the major product, and 10% appeared in the principal minor band (indicated with an arrow). As expected, there was a gradual increase in the proportion of this minor band as time progressed, because a substrate with both the major and minor cuts would appear only in the band representing the cut closer to the labeled end.
Second, we tested whether Tx1L EN was capable of enzymatic turnover. The earlier experiments were performed with a slight molar excess of enzyme over target DNA. By adding a large amount of unlabeled target molecules, we increased the target/enzyme ratio to 38:1 (84 pmol of target:2.2 pmol of enzyme). Tx1L EN converted essentially all of the labeled target to nicked product within about 8-10 h of incubation (Fig. 8). This provides clear evidence that a single enzyme molecule can nick multiple targets. This is different from what has been seen for the R2 class of endonucleases (38).
Recognition site of Tx1L EN.
Tx1L EN makes a specific nick in the Tx1L target and fails to make significant cuts in the Tx1D target. What sequence features are important for recognition by the enzyme? Initial attempts to demonstrate an electrophoretic mobility shift for a complex between the endonuclease and its substrate were unsuccessful. Therefore, this question was addressed by substitution scanning mutagenesis of the Tx1L target sequence.
A series of substrate oligonucleotides was prepared, each 42 bp in length. One corresponds precisely to a shorter version of the Tx1L target sequence described earlier. This was readily nicked by Tx1L EN (Fig. 9, lane 2). Each of the other substrates in the series contained a 6-bp substitution, GATCGA (boxed in Fig. 9), that was moved progressively through the sequence in steps of approximately 4 bp. Treatment of the mutant substrates gave an indication of which positions in the sequence are important for recognition by Tx1L EN. Two of the substrates were essentially not cut at all (Fig. 9, lanes 5 and 6), and two others were nicked much less efficiently than the wild-type sequence (lanes 4 and 7). This defines the critical sequences as lying within 10 consecutive base pairs that flank the cut site on both sides (underlined in the top sequence in Fig. 9).
FIG. 9.

Substitution scanning mutagenesis of the Tx1L target site. The initial substrate (lanes 1 and 2) was a 42-bp oligonucleotide corresponding to the 23-bp target duplication and surrounding sequences. The target site duplication segment is flanked by spaces in the sequences shown, and the position of the prominent TPRT nick is indicated with an arrowhead. Each mutant target carried a 6-bp substitution (boxed) that was placed at successive locations in the substrates, as shown. Each DNA was labeled on the bottom strand (∗) and treated with Tx1L EN as before. The sample in lane 1 was incubated without enzyme. The fraction of radioactivity in the band corresponding to the TPRT nick was determined and reported for each substrate as a percentage of that found with the nonmutant sequence. The line between the strands in the initial substrate shows the region in which substitutions caused a substantial reduction (≥65%) in the nicking activity.
DISCUSSION
Activity of Tx1L EN.
The ADR endonuclease domain of Tx1L ORF2p is active when expressed independently in a truncated form. This enzyme makes a specific cut in the bottom strand of the target for site-specific insertion of Tx1L elements at precisely the location required for priming of first-strand cDNA synthesis by the TPRT mechanism. Not only is the nick located at the end of the 23-bp target duplication, but the product has the expected chemical attributes (i.e., a free 3′-OH). This fact, combined with the observation that the endonuclease does not recognize and nick the target sequence for insertion of Tx1D elements, strongly supports the idea that Tx1L is an independent non-LTR retrotransposon that has target specificity for a sequence within Tx1D. Based both on these data and on mechanistic grounds, it seems unlikely that the proteins encoded by Tx1L participate directly in Tx1D mobility.
This conclusion leaves open the question of what protein(s) catalyzes Tx1D transposition, since the elements sequenced to date have no obvious ORFs (12). Whatever the catalysts of Tx1D transposition, it seems probable that a Tx1L insertion could be carried along passively to the new target site without a requirement for the activities of its own gene products. Thus, Tx1L can integrate at new chromosomal locations either by moving independently or by piggybacking on Tx1D.
Tx1L effectively maximizes its chances for survival by (i) choosing a preexisting high-copy-number element as its target; (ii) minimizing potential damage by selecting safe insertion sites already scouted by Tx1D; and (iii) taking advantage of the possibility of being transported to new sites by Tx1D, as well as by its own independent mechanism. This strategy also has its drawbacks, in that a particular Tx1L element may be eliminated along with Tx1D, either by selection against a disadvantageous insertion or by attrition. Furthermore, if the Tx1D family should disappear from the X. laevis genome, no targets for Tx1L insertion would remain. This hazard is no greater, however, than that experienced by the abundant short interspersed repetitive sequences, which are dependent for their transposition on functions provided by long interspersed repetitive sequences or other independent retrotransposons (2, 30, 34, 35). In addition, the site specificity of Tx1L elements is capable of coevolving with the sequence of its DNA target. We have identified a second family in the X. laevis genome, called Tx2, that is comprised of homologues of the L, D, and composite (C) elements found in the Tx1 family (13). Both the target sequence within PTR-1 repeats of Tx2D and the cleavage specificity of the Tx2L endonuclease are different from those in the Tx1 family, and no hybrid elements (e.g., Tx1L in Tx2D) are observed, suggesting independent coevolution in the two families (S. Christensen, G. Pont-Kingdon, and D. Carroll, unpublished data).
Top-strand cleavage.
The bottom-strand nick made by Tx1L EN produces a 3′ end that could prime first-strand cDNA synthesis at the target site. To complete the integration process, a nick must ultimately be made in the top strand to prime second-strand synthesis. The isolated endonuclease domain does not make a strong cut in the top strand at the other end of the duplicated target sequence. We offer several possible explanations for the absence of an obvious top-strand cut. (i) Top-strand cleavage may be inefficient in our reaction conditions, perhaps because no element RNA was added (23, 38) (see below). (ii) Top-strand cleavage may not occur precisely at the end of the apparent target duplication, perhaps because we have defined it incorrectly (it is difficult to know which sequences to assign to the target and which to the element, since they may be identical). Alternatively, if the donor RNA carries on its 5′ end all or part of the target duplication from its previous integration site, this could serve as the source of those sequences at the new site. In either case, the very weak nicks seen on the top strand near the end of the target duplication (Fig. 4 and 10) could represent inefficient versions of the top-strand cut. (iii) The top-strand cut may not be made by the N-terminal endonuclease domain alone. The full-length ORF2p or a host factor may be necessary to alter the specificity of the endonuclease to recognize the top-strand site, or it may be cleaved by a completely different activity.
FIG. 10.

Target sequences of NLRs whose ADR endonuclease has been characterized. Spaces in the target sequence demarcate the target duplication. The observed bottom strand nicks made by the purified endonucleases are marked with bold arrowheads. Experimentally observed top-strand nicks that have been proposed as top-strand cleavages are marked with an arrow. The simple hash marks in the case of Tx1L represent very minor cut sites (Fig. 4). The data for L1Hs are from references 9 and 18), those for R1Bm are from reference 10, and those for Tx1L are from this work.
Cleavage sites for the three ADR endonucleases characterized to date are shown in Fig. 10. The human L1Hs EN is rather nonspecific, in agreement with the wide range of target sequences these elements are seen to occupy. Nonetheless, there is a weak consensus among observed target duplications (18), and cleavage by the endonuclease is consistent with this consensus (9).
The other site-specific ADR endonuclease that has been examined is that from the silkmoth ribosomal insertion, R1Bm (10). Like Tx1L EN, the R1Bm endonuclease cuts the bottom strand of its target precisely at the expected location, at the left end of the target sequence that will be duplicated. In this case, Feng et al. (10) also observed a nick on the top strand at the right end of the duplicated sequence; however, this was not the only nick made on the top strand of the proffered substrate, nor was it the most prominent. It is not known whether this corresponds to the mechanistically relevant top-strand cut.
The other NLR endonuclease that has been studied in detail is that from the silkmoth ribosomal insertion R2Bm (22, 23, 37, 38). In this case, the full-length protein encoded by the single ORF of the element both makes the first-strand nick and primes DNA synthesis from it. Further, the R2Bm protein makes a specific cut in the top strand of the target, and this reaction requires the presence of RNA (38). The R2Bm endonuclease is not related in sequence to the ADR family, and it appears to have two essential domains in distant portions of the polypeptide sequence (39). After the first strand nick is made by the R2Bm endonuclease, the protein remains associated with the target DNA (38). This is thought to be important for coordinating first-strand cleavage with reverse transcription, with top-strand cleavage, and with initiation of top-strand synthesis. In contrast, the Tx1L EN clearly releases after making a nick, and a single molecule of the truncated protein can process multiple DNA substrates. This is perhaps not surprising given the homology of the ADR family to DNase I and AP endonuclease, which also exhibit enzymatic turnover (9, 27, 28). In the context of the full-length ORF2p sequence, however, the endonuclease may be constrained to remain at the target; and, as speculated above, its specificity may be modified to effect top-strand cleavage.
Recognition site of Tx1L EN.
The recognition site for Tx1L EN consists of sequences flanking the TPRT nick site. The minimum recognition site appears to be about 10 bp and is approximately centered around the cut site. These features correspond well to similar observations made for DNase I, although Tx1L EN has an added level of sequence specificity. Based on the crystal structure and DNA footprinting experiments, it is known that DNase I contacts approximately one helical turn of DNA and that the protein is roughly centered around the phosphodiester cut site (21, 36).
We suggest that, in the context of the full-length ORF2 protein, the endonuclease has additional determinants of its sequence specificity. First, the isolated endonuclease domain makes weak cuts in noncanonical sequences in the substrates we have studied here and in some other sequences that we have utilized (4). (If enough such secondary sites were analyzed, it might be possible to derive a consensus recognition sequence for Tx1L EN.) Second, a 10-bp recognition site is not large enough to explain the fact that Tx1L elements are found only within Tx1D elements in the X. laevis genome. On statistical grounds, multiple additional copies of a sequence of this length are expected to exist. Either the full-length ORF2p recognizes a longer sequence, or there are additional structural determinants of cleavage specificity, perhaps in the form of an organized chromatin structure or a specific local DNA geometry (5, 21).
ACKNOWLEDGMENTS
This work was supported in part by research grant NP-803 from the American Cancer Society. Assistance was also provided by the Markey Center for Protein Biophysics and the Huntsman Cancer Institute at the University of Utah.
We are grateful to Tom Eickbush and Harmit Malik for thoughtful and helpful comments and to an editor for a useful suggestion on interpreting the strategic aspects of Tx1L transposition.
REFERENCES
- 1.Ausubel F M, Brent R, Kingston R E, Moore D D, Seidman J G, Smith J A, Struhl K, editors. Current protocols in molecular biology. New York, N.Y: John Wiley & Sons, Inc.; 1998. [Google Scholar]
- 2.Boeke J D. LINEs and Alus—the polyA connection. Nat Genet. 1997;16:6–7. doi: 10.1038/ng0597-6. [DOI] [PubMed] [Google Scholar]
- 3.Capy P, Bazin C, Higuet D, Langin T. Dynamics and evolution of transposable elements. Austin, Tex: Landes Bioscience; 1998. [Google Scholar]
- 4.Christensen S. Ph.D. thesis. Salt Lake City: University of Utah; 1999. [Google Scholar]
- 5.Cost G J, Boeke J D. Targeting of human retrotransposon integration is directed by the specificity of the L1 endonuclease for regions of unusual DNA structure. Biochemistry. 1998;37:18081–18093. doi: 10.1021/bi981858s. [DOI] [PubMed] [Google Scholar]
- 6.Dombroski B A, Mathias S L, Nanthakumar E, Scott A F, Kazazian H H., Jr Isolation of an active human transposable element. Science. 1991;254:1805–1808. doi: 10.1126/science.1662412. [DOI] [PubMed] [Google Scholar]
- 7.Eickbush T H. Transposing without ends: the non-LTR retrotransposable elements. New Biol. 1992;4:430–440. [PubMed] [Google Scholar]
- 8.Engels W R. P elements in Drosophila. Curr Top Microbiol Immunol. 1996;204:103–123. doi: 10.1007/978-3-642-79795-8_5. [DOI] [PubMed] [Google Scholar]
- 9.Feng Q, Moran J V, Kazazian H H, Jr, Boeke J D. Human L1 retrotransposon encodes a conserved endonuclease required for retrotransposition. Cell. 1996;87:905–916. doi: 10.1016/s0092-8674(00)81997-2. [DOI] [PubMed] [Google Scholar]
- 10.Feng Q, Schumann G, Boeke J D. Retrotransposon R1Bm endonuclease cleaves the target sequence. Proc Natl Acad Sci USA. 1998;95:2083–2088. doi: 10.1073/pnas.95.5.2083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gabriel A, Boeke J D. Reverse transcriptase encoded by a retrotransposon from the trypanosomatid Crithidia fasciculata. Proc Natl Acad Sci USA. 1991;88:9794–9798. doi: 10.1073/pnas.88.21.9794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Garrett J E, Carroll D. Tx1: a transposable element from Xenopus laevis with some unusual properties. Mol Cell Biol. 1986;6:933–941. doi: 10.1128/mcb.6.3.933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Garrett J E, Knutzon D S, Carroll D. Composite transposable elements in the Xenopus laevis genome. Mol Cell Biol. 1989;9:3018–3027. doi: 10.1128/mcb.9.7.3018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hohjoh H, Singer M F. Cytoplasmic ribonucleoprotein complexes containing human LINE-1 protein and RNA. EMBO J. 1996;15:630–639. [PMC free article] [PubMed] [Google Scholar]
- 15.Hohjoh H, Singer M F. Sequence-specific single-strand RNA binding protein encoded by the human LINE-1 retrotransposon. EMBO J. 1997;16:6034–6043. doi: 10.1093/emboj/16.19.6034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ivanov V A, Melnikov A A, Siunov A V, Fodor I I, Ilyin Y V. Authentic reverse transcriptase is coded by jockey, a mobile Drosophila element related to mammalian LINEs. EMBO J. 1991;10:2489–2495. doi: 10.1002/j.1460-2075.1991.tb07788.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Jakubczak J L, Burke W D, Eickbush T H. Retrotransposable elements R1 and R2 interrupt the rRNA genes of most insects. Proc Natl Acad Sci USA. 1991;88:3295–3299. doi: 10.1073/pnas.88.8.3295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Jurka J. Sequence patterns indicate an enzymatic involvement in integration of mammalian retroposons. Proc Natl Acad Sci USA. 1997;94:1872–1877. doi: 10.1073/pnas.94.5.1872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kolosha V O, Martin S L. In vitro properties of the first ORF protein from mouse LINE-1 support its role in ribonucleoprotein particle formation during retrotransposition. Proc Natl Acad Sci USA. 1997;94:10155–10160. doi: 10.1073/pnas.94.19.10155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kunze R. The maize transposable element activator (Ac) Curr Top Microbiol Immunol. 1996;204:161–194. doi: 10.1007/978-3-642-79795-8_8. [DOI] [PubMed] [Google Scholar]
- 21.Lahm A, Suck D. DNase I-induced DNA conformation. 2 Å structure of a DNase I-octamer complex. J Mol Biol. 1991;222:645–667. doi: 10.1016/0022-2836(91)90502-w. [DOI] [PubMed] [Google Scholar]
- 22.Luan D D, Eickbush T H. Downstream 28S gene sequences on the RNA template affect the choice of primer and the accuracy of initiation by the R2 reverse transcriptase. Mol Cell Biol. 1996;16:4726–4734. doi: 10.1128/mcb.16.9.4726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Luan D D, Korman M H, Jakubczak J L, Eickbush T H. Reverse transcription of R2Bm RNA is primed by a nick at the chromosomal target site: a mechanism for non-LTR retrotransposition. Cell. 1993;72:595–605. doi: 10.1016/0092-8674(93)90078-5. [DOI] [PubMed] [Google Scholar]
- 24.Mahillon J, Chandler M. Insertion sequences. Microbiol Mol Biol Rev. 1998;62:725–774. doi: 10.1128/mmbr.62.3.725-774.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mathews D H, Banerjee A R, Luan D D, Eickbush T H, Turner D H. Secondary structure model of the RNA recognized by the reverse transcriptase from the R2 retrotransposable element. RNA. 1997;3:1–16. [PMC free article] [PubMed] [Google Scholar]
- 26.Mathias S L, Scott A F, Kazazian H H, Jr, Boeke J D, Gabriel A. Reverse transcriptase encoded by a human transposable element. Science. 1991;254:1808–1810. doi: 10.1126/science.1722352. [DOI] [PubMed] [Google Scholar]
- 27.Mol C D, Kuo C-F, Thayer M M, Cunningham R P, Tainer J A. Structure and function of the multifunctional DNA-repair enzyme exonuclease III. Nature. 1995;374:381–386. doi: 10.1038/374381a0. [DOI] [PubMed] [Google Scholar]
- 28.Oefner C, Suck D. Crystallographic refinement and structure of DNase I at 2Å resolution. J Mol Biol. 1986;192:605–632. doi: 10.1016/0022-2836(86)90280-9. [DOI] [PubMed] [Google Scholar]
- 29.Ohtsubo F, Sekine Y. Bacterial insertion sequences. Curr Top Microbiol Immunol. 1996;204:1–26. doi: 10.1007/978-3-642-79795-8_1. [DOI] [PubMed] [Google Scholar]
- 30.Okada N, Hamada M, Ogiwara I, Ohshima K. SINEs and LINEs share common 3′ sequences: a review. Gene. 1997;205:229–243. doi: 10.1016/s0378-1119(97)00409-5. [DOI] [PubMed] [Google Scholar]
- 31.Olivares M, Alonso C, Lopez M C. The open reading frame 1 of the L1Tc retrotransposon of Trypanosoma cruzi codes for a protein with apurinic-apyrimidinic nuclease activity. J Biol Chem. 1997;272:25224–25228. doi: 10.1074/jbc.272.40.25224. [DOI] [PubMed] [Google Scholar]
- 32.Plasterk R H. The Tc1/mariner transposon family. Curr Top Microbiol Immunol. 1996;204:125–143. doi: 10.1007/978-3-642-79795-8_6. [DOI] [PubMed] [Google Scholar]
- 33.Pont-Kingdon G, Chi E, Christensen S, Carroll D. Ribonucleoprotein formation by the ORF1 protein of the non-LTR retrotransposon Tx1L in Xenopus oocytes. Nucleic Acids Res. 1997;25:3088–3094. doi: 10.1093/nar/25.15.3088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Smit A F A. The origin of interspersed repeats in the human genome. Curr Opin Genet Dev. 1996;6:743–748. doi: 10.1016/s0959-437x(96)80030-x. [DOI] [PubMed] [Google Scholar]
- 35.Tchénio T, Segal-Bendirdjian E, Heidmann T. Generation of processed pseudogenes in murine cells. EMBO J. 1993;12:1487–1497. doi: 10.1002/j.1460-2075.1993.tb05792.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Weston S A, Lahm A, Suck D. X-ray structure of the DNase I-d(GGTATACC)2 complex at 2.3 Å resolution. J Mol Biol. 1992;226:1237–1256. doi: 10.1016/0022-2836(92)91064-v. [DOI] [PubMed] [Google Scholar]
- 37.Xiong Y, Eickbush T H. Functional expression of a sequence-specific endonuclease encoded by the retrotransposon R2Bm. Cell. 1988;55:235–246. doi: 10.1016/0092-8674(88)90046-3. [DOI] [PubMed] [Google Scholar]
- 38.Yang J, Eickbush T H. RNA-induced changes in the activity of the endonuclease encoded by the R2 retrotransposable element. Mol Cell Biol. 1998;18:3455–3465. doi: 10.1128/mcb.18.6.3455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Yang J, Malik H S, Eickbush T H. Identification of the endonuclease domain encoded by R2 and other site-specific, non-long terminal repeat retrotransposable elements. Proc Natl Acad Sci USA. 1999;96:7847–7852. doi: 10.1073/pnas.96.14.7847. [DOI] [PMC free article] [PubMed] [Google Scholar]