Study of Genetic Diversity of Eukaryotic Picoplankton in Different Oceanic Regions by Small-Subunit rRNA Gene Cloning and Sequencing (original) (raw)
Abstract
Very small eukaryotic organisms (picoeukaryotes) are fundamental components of marine planktonic systems, often accounting for a significant fraction of the biomass and activity in a system. Their identity, however, has remained elusive, since the small cells lack morphological features for identification. We determined the diversity of marine picoeukaryotes by sequencing cloned 18S rRNA genes in five genetic libraries from North Atlantic, Southern Ocean, and Mediterranean Sea surface waters. Picoplankton were obtained by filter size fractionation, a step that excluded most large eukaryotes and recovered most picoeukaryotes. Genetic libraries of eukaryotic ribosomal DNA were screened by restriction fragment length polymorphism analysis, and at least one clone of each operational taxonomic unit (OTU) was partially sequenced. In general, the phylogenetic diversity in each library was rather great, and each library included many different OTUs and members of very distantly related phylogenetic groups. Of 225 eukaryotic clones, 126 were affiliated with algal classes, especially the Prasinophyceae, the Prymnesiophyceae, the Bacillariophyceae, and the Dinophyceae. A minor fraction (27 clones) was affiliated with clearly heterotrophic organisms, such as ciliates, the chrysomonad Paraphysomonas, cercomonads, and fungi. There were two relatively abundant novel lineages, novel stramenopiles (53 clones) and novel alveolates (19 clones). These lineages are very different from any organism that has been isolated, suggesting that there are previously unknown picoeukaryotes. Prasinophytes and novel stramenopile clones were very abundant in all of the libraries analyzed. These findings underscore the importance of attempts to grow the small eukaryotic plankton in pure culture.
Marine picoeukaryotes (which are between 0.2 and 2 to 3 μm in diameter) (43) are probably the most abundant eukaryotes on Earth. They are found throughout the world's oceans at concentrations between 102 and 104 cells ml−1 in the photic zone, and they constitute an essential component of microbial food webs, playing significant roles in global mineral cycles (11, 21). Marine picoeukaryotes seem to belong to very different phylogenetic groups. In fact, nearly every algal phylum has picoplanktonic representatives (47), and in the last 10 years three novel algal classes have been described for picoeukaryotic isolates (3, 15, 27). However, the extent of the diversity and the distribution and abundance of the different taxa in situ remain unknown (33). In the open oceans most picoeukaryotes are coccoid or flagellated forms with chloroplasts (phototrophic) or without chloroplasts (heterotrophic) and with few morphologically distinct features (5, 44, 47). They can hardly be discriminated, even at the class level, by conventional optical microscopy (30). Electron microscopy generally allows assignment to taxonomic classes (4), but most cells do not have enough ultrastructural features for identification at lower taxonomic levels (38). Cultivation is the best possible way to characterize a natural organism, and isolation of small picoeukaryotic strains is thus an important task. However, there is no guarantee that organisms grown in culture are dominant or important in the natural community (16, 23). Organisms belonging to different algal classes have different diagnostic marker pigments that can be identified and quantified by high-performance liquid chromatography (HPLC) (17). HPLC pigment analysis is very useful for characterizing new isolates, but it has some technical constraints when it is applied to natural assemblages, since interpretation of the complex pigment patterns of samples requires application of algorithms (20) which generally involve untestable assumptions. At best, many of the conventional characterization techniques have limited phylogenetic capacity and are cumbersome or time-consuming.
An alternative approach for characterizing the phylogenetic diversity of marine picoeukaryotes is analysis of small-subunit (SSU) rRNA genes (2, 33). During the last decade, cloning of environmental rRNA genes has provided insight into the diversity of the marine prokaryotic picoplankton and has revealed that this assemblage is dominated by novel lineages of bacteria (13) and archaea (8, 12). Similar studies focusing on marine picoeukaryotes are just beginning. Two very recent papers described the diversity of picoeukaryotes as determined by gene cloning and sequencing of ribosomal DNA (rDNA) in one surface sample from the equatorial Pacific Ocean (29) and several deep-sea samples from the Southern Ocean (24). Both studies showed that the phylogenetic diversity of the assemblages was great and that novel lineages were present. Another study analyzed the algal assemblages at two coastal sites by using the plastidic genes found in bacterial SSU rRNA libraries (39). Other molecular studies have focused on particular taxonomic groups and have used a similar gene-cloning approach (16, 28) or taxon-specific rRNA probes in fluorescent in situ hybridization experiments (22, 45). Finally, fingerprinting techniques, such as denaturing gradient gel electrophoresis (DGGE), have been used to quickly compare the compositions of planktonic eukaryotic assemblages (9, 49).
The objective of this work was to study the diversity of marine picoeukaryotes in different marine areas by gene cloning and sequencing of eukaryotic rRNA genes. Clones derived from five genetic libraries were analyzed by restriction fragment length polymorphism (RFLP) analysis, and selected clones were partially sequenced. With this approach we determined whether different picoeukaryotic groups were present in different areas of oceans and estimated their relative abundance.
MATERIALS AND METHODS
Sampling.
Samples from different marine areas (Table 1) were collected with Niskin bottles attached to a rosette and a conductivity, temperature, and depth (CTD) probe. Seawater was transferred to 25-liter plastic containers that previously had been rinsed three times with the same water. Microbial biomass was collected on 0.2-μm-pore-size Sterivex units (Durapore; Millipore) by filtering 10 to 20 liters of seawater through a prefilter and a Sterivex unit in succession with a peristaltic pump at rates of 50 to 100 ml min−1. Different prefilters were used; these prefilters included 5-μm-pore-size polycarbonate filters for the Mediterranean sample, 2-μm-pore-size polycarbonate filters for the North Atlantic samples and 1.6-μm-pore-size GF/A glass fiber filters for the Antarctic samples. The prefilters and the Sterivex units were covered with lysis buffer (40 mM EDTA, 50 mM Tris-HCl, 0.75 M sucrose) and frozen at −70°C until nucleic acid was extracted. An aliquot of seawater was fixed with glutaraldehyde to obtain epifluorescence counts for heterotrophic flagellates (37). Subsamples of the whole water and the filtrate after passage through the prefilter were used for chlorophyll (Chl a) and cytometry determinations. Approximately 100 ml of sample was filtered through GF/F filters, and the Chl a concentration was determined by measuring the fluorescence in acetone extracts with a Turner Designs fluorometer (32). Subsamples used for flow cytometry counting were collected by fixing 1.2 ml of seawater with glutaraldehyde-paraformaldehyde (final concentration, 0.05 and 1%, respectively). Populations of Synechococcus, Prochlorococcus, and photosynthetic picoeukaryotes were distinguished by their distinct size and pigment properties by using a FACScalibur flow cytometer (Becton Dickinson) as explained by Olson et al. (31). Strictly speaking, the picoeukaryotes comprise organisms that are between 0.2 and 2 μm in diameter, but here we use the term loosely to include the larger organisms analyzed in the Mediterranean sample (diameter, 0.2 to 5 μm). Moreover, flow cytometry detects the most abundant photosynthetic eukaryotes, often including organisms that are more than 2 μm in diameter.
TABLE 1.
Characteristics of the samples used to generate the five libraries of eukaryotic 18S rRNA genes
| Library | System | Characteristics | Coordinates | Date (day/mo/yr) | Temp (°C) | Fraction analyzed (μm) | Chl a concn (μg liter−1) | |
|---|---|---|---|---|---|---|---|---|
| Total | In fraction | |||||||
| ME-1 | Mediterranean (Alborán Sea) | Upwelling | 36°14′N, 4°15′W | 9/11/97 | 18.0 | 0.2–5 | 0.87 | 0.72 |
| ANT37 | Antarctica (Weddell Sea) | Ice edge | 60°32′S, 44°12′W | 26/1/98 | −1.8 | 0.2–1.6 | 0.35 | 0.01 |
| ANT12 | Antarctica (Scotia Sea) | Open ocean | 58°16′S, 44°27′W | 23/1/98 | 1.9 | 0.2–1.6 | 0.29 | 0.02 |
| NA11 | North Atlantic | Within eddy | 59°30′N, 21°8′W | 14/6/98 | 11.2 | 0.2–2 | 0.87 | 0.22 |
| NA37 | North Atlantic | Outside eddy | 59°34′N, 21°3′W | 21/6/98 | 10.5 | 0.2–2 | 0.59 | 0.19 |
Nucleic acid extraction.
Nucleic acid extraction started with addition of lysozyme (final concentration, 1 mg ml−1) and incubation of the Sterivex units at 37°C for 45 min. Then, sodium dodecyl sulfate (final concentration, 1%) and proteinase K (final concentration, 0.2 mg ml−1) were added, and the Sterivex units were incubated at 55°C for 60 min. Lysates were recovered from the Sterivex units with a syringe. Nucleic acids were extracted with phenol-chloroform-isoamyl alcohol (25:24:1), and the residual phenol was removed with chloroform-isoamyl alcohol (24:1). Nucleic acid extracts were purified further, desalted, and concentrated with a Centricon-100 concentrator (Millipore). DNA integrity was checked by agarose gel electrophoresis, and DNA yield was quantified by a Hoechst dye fluorescence assay (35). Nucleic acid extracts were stored at −70°C until analysis.
Eukaryotic rDNA genetic libraries.
Eukaryotic 18S rRNA genes were amplified by PCR with eukaryote-specific primers EukA, EukB (26), and 326f (22). Most libraries were constructed with the 326f-EukB primer combination (1,420-bp insert); the only exception was the ME1 library, which was constructed with primers EukA and EukB (1,780-bp insert). The PCR mixtures (100 μl) each contained 10 to 100 ng of environmental DNA as the template, each deoxynucleoside triphosphate at a concentration of 200 μM, 1.5 mM MgCl2, each primer at a concentration of 0.3 μM, 2.5 U of Taq DNA polymerase (Gibco BRL), and the PCR buffer supplied with the enzyme. Reactions were carried out in an automated thermocycler (Genius; Techne) with the following cycle: an initial denaturation at 94°C for 3 min, 30 cycles of denaturation at 94°C for 45 s, annealing at 55°C for 1 min, and extension at 72°C for 3 min, and a final extension at 72°C for 5 min. Amplified rRNA gene products from several individual PCRs were pooled (four 50-μl samples or two 100-μl samples), ethanol precipitated, and resuspended in 20 μl of sterile water. An aliquot of each concentrated PCR product preparation was ligated into the prepared vector (pCR 2.1) supplied with a TA cloning kit (Invitrogen) by following the manufacturer's recommendations. Putative positive colonies were picked, transferred to a multiwell plate containing Luria-Bertani medium and 7% glycerol, and stored at −70°C.
RFLP analysis.
The presence of the 18S rDNA insert in colonies was checked by PCR reamplification with primers 326f and EukB by using a small aliquot of a culture as the template. PCR amplification products containing the right size of insert were digested with 1 U of restriction enzyme _Hae_III (Gibco BRL) μl−1 for 6 to 12 h at 37°C. The digested products were separated by electrophoresis at 80 V for 2 to 3 h in a 2.5% low-melting-point agarose gel. A 50-bp DNA ladder (Gibco BRL) was included in each gel to aid in visual comparisons of the RFLP patterns of clones appearing in different gels. When ambiguities appeared, clones were electrophoresed simultaneously in the same agarose gel. Clones that produced the same RFLP pattern (DNA fragments of the same size) were grouped together and were considered members of the same operational taxonomic unit (OTU). Coverage values were calculated for each library by using the relative distribution of OTUs and the equation described by Good (14).
rDNA sequencing.
Double-stranded plasmid DNAs from selected clones were extracted with a QIAprep miniprep kit (QIAGEN). Sequencing reactions were performed with a Thermo SEQUENASE v.2 kit (Amersham, U.S. Biochemicals) and an ABI PRISM model 377 (v. 3.3) automated sequencer. A single reaction with primer 326f was performed for each clone, which resulted in a 550 to 750-bp sequence. Sequences were subjected to a BLAST search (1) to determine the first phylogenetic affiliation and to the CHECK-CHIMERA command (25) to determine potential chimeric artifacts. Sequences were aligned with about 3,200 homologous eukaryotic 18S rRNA primary structures by using the automatic alignment tool of the ARB program package (http://www.mikro.biologie.tu-muenchen.de). Then partial sequences were inserted into the optimized tree derived from complete sequence data by using the Quick add using parsimony tool, which did not affect the initial tree topology. The resulting tree was pruned to save space; only the closest relatives of our clones were retained. Since the similarity value obtained in a BLAST analysis often is for only a fraction of the sequence submitted, similarity values for new and database sequences were calculated by using the aligned ARB file.
DGGE.
Eukaryotic 18S rRNA genes were amplified with eukaryote-specific primers Euk1A and Euk516r-GC, which amplify an approximately 560-bp fragment (9). The PCR program included an initial denaturation at 94°C for 130 s and 35 cycles of denaturation at 94°C for 30 s, annealing at 56°C for 45 s, and extension at 72°C for 130 s. The PCR products were quantified with a Low DNA Mass Ladder (Gibco BRL) by performing agarose gel electrophoresis with a DGGE-2000 system (CBS Scientific Company). A 0.75-mm-thick 6% polyacrylamide gel was cast by mixing two stock solutions containing 45 and 65% DNA denaturant agent (100% was defined as 7 M urea and 40% deionized formamide). Approximately 800 ng of PCR product was applied to each lane in the gel. Electrophoresis was performed at 100 V and 60°C for 16 h in 1× TAE buffer (40 mM Tris base, 20 mM sodium acetate, 1 mM EDTA; pH 7.4). The DGGE gel was stained with GelStar (FMC BioProducts) for 30 min, rinsed with 1× TAE buffer, and visualized with UV radiation by using a Fluor-S MultiImager and the MultiAnalyst imaging software (Bio-Rad). The presence and intensity of DGGE bands were estimated by image analysis as previously described (41).
Nucleotide sequence accession numbers.
Nucleotide sequences determined in this study have been deposited in the GenBank database under accession numbers AF363153 to AF363228.
RESULTS AND DISCUSSION
The aim of this study was to analyze the phylogenetic composition of the marine planktonic picoeukaryotes, a ubiquitous, heterogeneous, poorly identified assemblage. We present the results obtained with a molecular approach, gene cloning and sequencing of SSU rRNA genes, that has been used very successfully to identify marine bacteria and archaea (8, 13) and has only recently been applied to marine eukaryotes (24, 29). We analyzed clone libraries from five surface samples taken in three distant marine regions, the Mediterranean Sea (library ME1), the Southern Ocean (libraries ANT37 and ANT12), and the North Atlantic Ocean (libraries NA11 and NA37). These samples exhibited a wide range of in situ temperatures, from 18°C in the Mediterranean Sea to −1.8°C in the Weddell Sea (Table 1). They also differed in terms of the composition of the phototrophic picoplankton; picoeukaryotes were present in all three systems, together with Synechococcus in the Mediterranean and Atlantic samples and Prochlorococcus in the Mediterranean sample (Table 2). Therefore, the physical and biological parameters of the five samples analyzed were very different.
TABLE 2.
Concentrations of heterotrophic and phototrophic picoeukaryotes, Prochlorococcus and Synechococcus in whole water and in the fractions passing through the prefilters for the samples used to generate eukaryotic genetic libraries
| Library | Sample | Concn (103 cells ml−1) ofa: | ||||||
|---|---|---|---|---|---|---|---|---|
| Picoeukaryotes | Prochlorococcus | Synechococcus | ||||||
| Heterotrophic | Phototrophic | P1 | P2 | P3 | ||||
| ME1 | Whole water | 1.5 | 13.9 | 12.7 | 12.9 | |||
| <5-μm fraction | NDb | 14.1 | 14.8 | 12.7 | ||||
| ANT37 | Whole water | 0.4 | 9.9 | 2.5 | 5.2 | 2.2 | 0 | 0 |
| <1.6-μm fraction | ND | 0.5 | 0.4 | 0.1 | 0 | 0 | 0 | |
| ANT12 | Whole water | 0.7 | 3.2 | 1.3 | 1.5 | 0.4 | 0 | 0 |
| <1.6-μm fraction | ND | 1.0 | 0.8 | 0.2 | 0.0 | 0 | 0 | |
| NA11 | Whole water | ND | 6.6 | 4.8 | 1.8 | 0 | 4.6 | |
| <2-μm fraction | ND | 6.6 | 4.6 | 2.0 | 0 | 2.6 | ||
| NA37 | Whole water | ND | 3.2 | 0 | 7.3 | |||
| <2-μm fraction | ND | 4.6 | 0 | 6.8 |
In contrast to marine bacteria and archaea, the planktonic eukaryotes cover a broad size spectrum; they vary from microns to millimeters in diameter. Therefore, the approach used to collect picoeukaryotes is very important. Picoplanktonic biomass was obtained by prefiltering a sample and collecting the organisms that passed through the prefilter. The performance of this size fractionation technique for the five samples used to construct clone libraries was assessed by carrying out Chl a (Table 1), flow cytometry (Table 2), and molecular fingerprinting (Fig. 1) analyses. For the Mediterranean sample, filtration through a 5-μm-pore-size filter resulted in a slight reduction in the level of Chl a (Table 1) but no reduction in the level of phototrophic picoeukaryotes (Table 2). For the North Atlantic samples filtration through a 2-μm-pore-size filter resulted in a significant reduction in the level of Chl a, but it had no effect on phototrophic picoeukaryote abundance. For the Antarctic samples, filtration through a 1.6-μm-pore-size filter resulted in a dramatic decrease in the level of Chl a (only 3 to 7% passed through the filter) and in the level of phototrophic picoeukaryotes (5 to 31% passed through the filter). When possible, distinct picoeukaryotic populations were distinguished on the cytometry graph and analyzed separately (Table 2). The two populations detected in the NA11 sample were not affected by prefiltration, whereas the abundance of the three populations detected in the Antarctic samples decreased after filtration and there was a more pronounced effect on the largest of the three populations (P3). Therefore, the fractions analyzed appeared to contain all of the phototrophic picoeukaryotes for the Mediterranean and Atlantic samples and only a fraction of the phototrophic picoeukaryotes for the Antarctic samples.
FIG. 1.

DGGE gel separating eukaryotic 18S rDNA fragments from the populations retained on prefilters and from the populations appearing in filtrates from the five samples used to generate genetic libraries. The filtrate samples analyzed by using genetic libraries are circled.
We then checked whether the eukaryotes that passed through the prefilter (and thus were analyzed in the clone library) were phylogenetically different from the eukaryotes that were retained in the prefilter. It is well known that filters allow passage of cells larger than their nominal pore sizes and that filters can clog, which results in retention of smaller cells. The DGGE fingerprints obtained with eukaryote-specific primers for both size fractions were very different for the five samples analyzed (Fig. 1). On average, 66% of the bands that appeared in the larger-fraction fingerprint (accounting for 45% of the total band intensity) were not found in the smaller-fraction fingerprint, indicating that many populations were totally retained in the prefilter (Fig. 2). Conversely, on average, one-half of the bands that appeared in the smaller-fraction fingerprint (accounting for 32% of the band intensity) were unique to this fraction, indicating that many populations completely passed through the prefilters used. Although the prefiltration method was not perfect and some other populations appeared in both size fractions, this method appeared to enrich the smallest cells and exclude most large eukaryotes. Therefore, we were confident that we were analyzing mostly picoeukaryotes.
FIG. 2.
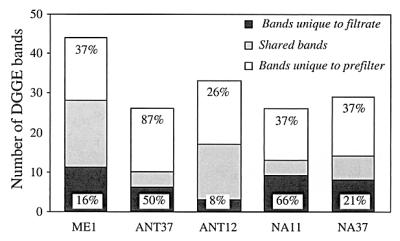
Numbers of bands that were unique to the filtrates, that were shared, and that were unique to the prefilters for the five samples used to generate genetic libraries after quantitative analysis of the DGGE gel shown in Fig. 1. The values above the bars for unique bands are the percentages of band intensity accounted for by these bands in the DGGE profile.
The five libraries of picoeukaryotic rRNA genes were first screened by RFLP analysis, which grouped clones into discrete OTUs (Table 3). An OTU comprising 33 clones in library ME1 was identical to the appendicularian Oikopleura, and an OTU comprising 43 clones in library NA11 was similar to a copepod. These metazoan OTUs were clearly artifacts of the prefiltration step and were thus excluded from further analyses. Apart from these two OTUs, we analyzed a total of 225 clones, which yielded 76 different OTUs. The coverage values, calculated from the relative distribution of OTUs in each library, were relatively high, ranging from 47% in library NA11, in which few clones were obtained, to 82% in library ANT12 (Table 3). These high values indicated that most of the diversity at the level examined had been sampled; only a few more OTUs would be recovered by analyzing more clones. About one-half of the OTUs that appeared in each library were unique to that library (Table 3), whereas the remaining OTUs appeared in two or more libraries, indicating that there was potential overlap of picoeukaryotic phylotypes among samples.
TABLE 3.
Results of RFLP analysis of the five genetic libraries
| Library | No. of clones | No. of OTUs | Coverage (%) | No. of OTUs unique to library | No. of OTUs found in libraries from the same system | No. of OTUs found in libraries from different systems |
|---|---|---|---|---|---|---|
| ME1 | 63 | 28 | 81 | 17 | NAa | 11 |
| ANT37 | 58 | 29 | 66 | 20 | 4 | 5 |
| ANT12 | 67 | 23 | 82 | 8 | 4 | 11 |
| NA11 | 17 | 12 | 47 | 7 | 1 | 4 |
| NA37 | 20 | 10 | 75 | 5 | 1 | 4 |
In the ME1 and ANT37 libraries one clone of each OTU was partially sequenced. When clones from different libraries belonging to the same OTU were compared, they were found to be very similar (average similarity for 10 cases examined, 98.6% [data not shown]). Thus, clones from the remaining three libraries were affiliated with an OTU found in ME1 or ANT37, and only clones representing new OTUs were sequenced. The affiliations of clones from each library with the 76 defined OTUs and the closest match in the database for the clone representing each OTU are shown in Table 4. These clones and database sequences are compared in a phylogenetic tree in Fig. 3. All of the clones were affiliated with eukaryotes, demonstrating the specificity of the eukaryotic primers used to construct the clone libraries. The clones are also widely distributed on the eukaryotic tree, showing the ability of the primers to recover distantly related phylogenetic groups. Some clones were very similar to previously isolated organisms, mainly organisms affiliated with groups having known picoeukaryotic representatives, such as prasinophytes, prymnesiophytes, and pelagophytes. Other clones seemed to represent new phylotypes in well-defined phylogenetic groups or even novel phylogenetic lineages.
TABLE 4.
Number of clones belonging to each OTU in genetic libraries and phylogenetic affiliations of the representative clones sequenced
| Taxon | OTU | No. of clones in libraries | Clone | Closest relative (% similarity) | ||
|---|---|---|---|---|---|---|
| ME1 | ANT37 | ANT12 | NA11 | NA37 | ||
| Prasinophytes | 1 | 6 | ME1-1 | Ostreococcus tauri (98.1) | ||
| 2 | 8 | 16 | 5 | ME1-2 | Mantoniella squamata (96.8) | |
| 3 | 2 | 1 | 3 | ME1-3 | Ostreococcus tauri (94.1) | |
| 4 | 1 | ANT37-3 | Mantoniella squamata (99.9) | |||
| 5 | 1 | ANT37-4 | Mantoniella squamata (96.1) | |||
| 6 | 1 | NA11-1 | Micromonas pusilla (99.0) | |||
| 7 | 2 | NA37-1 | Micromonas pusilla (99.1) | |||
| Prymnesiophytes | 8 | 1 | 2 | ME1-4 | Prymnesium patelliferum (94.3) | |
| 9 | 5 | 16 | 1 | ANT37-5 | Phaeocystis antarctica (99.8) | |
| 10 | 1 | ANT12-2 | Phaeocystis antarctica (97.6) | |||
| 11 | 1 | NA11-2 | Emiliania huxleyi (96.0) | |||
| 12 | 1 | NA11-7 | Emiliania huxleyi (95.6) | |||
| Diatoms | 13 | 3 | 1 | ME1-14 | Papiliocellulus elegans (95.5) | |
| 14 | 1 | ME1-15 | Chaetoceros rostratus (96.7) | |||
| 15 | 2 | 1 | ME1-16 | Skeletonema costatum (96.8) | ||
| 16 | 1 | ANT37-10 | Corethron criophilum (95.9) | |||
| 17 | 5 | ANT37-11 | Corethron criophilum (89.0) | |||
| 18 | 1 | ANT37-12 | Corethron criophilum (95.3) | |||
| 19 | 2 | ANT37-9 | Chaetoceros sp. (86.7) | |||
| 20 | 1 | ANT12-3 | Pseudonitzschia multiseries (98.6) | |||
| Dinophytes | 21 | 1 | ME1-8 | Lepidodinium viride (76.0) | ||
| 22 | 2 | 1 | 3 | 6 | ANT37-6 | Gymnodinium mikimotoi (96.7) |
| 23 | 1 | ANT37-8 | Lepidodinium viride (98.5) | |||
| Pelagophytes | 24 | 1 | 6 | ME1-27 | Pelagomonas calceolata (100) | |
| Glaucocystophytes | 25 | 1 | ANT12-4 | Cyanophora paradoxa (84.9) | ||
| 26 | 2 | ANT37-26 | Cyanophora paradoxa (80.0) | |||
| 27 | 1 | ANT37-27 | Cyanophora paradoxa (85.8) | |||
| Dictyochales | 28 | 1 | ANT37-15 | Dictyocha speculum (90.3) | ||
| 29 | 1 | NA11-6 | Dictyocha speculum (91.0) | |||
| 30 | 1 | NA37-4 | Dictyocha speculum (91.5) | |||
| Cryptophytes | 31 | 3 | ME1-5 | Geminigera cryophila (98.5) | ||
| Eustigmatophytes | 32 | 2 | ME1-25 | Nannochloropsis sp. (89.0) | ||
| Bolidophytes | 33 | 1 | ANT37-30 | Bolidomonas pacifica (94.6) | ||
| Novel stramenopiles | 34 | 1 | ME1-17 | Hyphochytrium catenoides (90.8) | ||
| 35 | 3 | ME1-18 | Hyphochytrium catenoides (87.6) | |||
| 36 | 2 | ME1-19 | Hyphochytrium catenoides (87.8) | |||
| 37 | 2 | ME1-20 | Hyphochytrium catenoides (88.3) | |||
| 38 | 1 | ME1-21 | Hyphochytrium catenoides (90.9) | |||
| 39 | 1 | ME1-22 | Hyphochytrium catenoides (90.3) | |||
| 40 | 1 | 5 | ANT12-6 | Hyphochytrium catenoides (89.2) | ||
| 41 | 6 | ANT12-7 | Hyphochytrium catenoides (88.8) | |||
| 42 | 1 | 5 | ANT12-24 | Hyphochytrium catenoides (89.4) | ||
| 43 | 1 | ANT12-9 | Hyphochytrium catenoides (88.5) | |||
| 44 | 2 | 2 | ANT12-10 | Hyphochytrium catenoides (88.0) | ||
| 45 | 1 | 1 | ANT12-11 | Hyphochytrium catenoides (93.7) | ||
| 46 | 1 | ANT37-19 | Hyphochytrium catenoides (90.0) | |||
| 47 | 1 | ANT37-20 | Hyphochytrium catenoides (85.5) | |||
| 48 | 1 | ANT37-21 | Hyphochytrium catenoides (92.9) | |||
| 48 | 1 | ANT37-22 | Hyphochytrium catenoides (85.4) | |||
| 50 | 1 | ANT37-23 | Hyphochytrium catenoides (90.3) | |||
| 51 | 1 | NA11-4 | Hyphochytrium catenoides (86.4) | |||
| 52 | 3 | 1 | NA11-5 | Hyphochytrium catenoides (87.7) | ||
| 53 | 2 | ANT37-13 | Hyphochytrium catenoides (87.7) | |||
| 54 | 1 | ANT37-16 | Hyphochytrium catenoides (87.5) | |||
| 55 | 2 | 1 | ME1-24 | Hyphochytrium catenoides (90.2) | ||
| 56 | 1 | ANT12-8 | Hyphochytrium catenoides (88.9) | |||
| 57 | 1 | ANT12-5 | Hyphochytrium catenoides (87.4) | |||
| Novel alveolates | 58 | 6 | 1 | 3 | ME1-6 | Pentapharsodinium tyrrhenicum (87.2) |
| 59 | 2 | ME1-7 | Heterocapsa triquetra (87.8) | |||
| 60 | 1 | ME1-9 | Heterocapsa triquetra (89.3) | |||
| 61 | 3 | ME1-10 | Heterocapsa triquetra (85.8) | |||
| 62 | 2 | NA37-2 | Gymnodinium catenatum (89.2) | |||
| 63 | 1 | NA37-3 | Lepidodinium viride (89.3) | |||
| Ciliates | 64 | 2 | 1 | 1 | ME1-11 | Strombidium purpureum (93.3) |
| 65 | 1 | ME1-12 | Oxytricha granulifera (92.2) | |||
| 66 | 1 | 2 | ME1-13 | Oxytricha granulifera (87.5) | ||
| 67 | 1 | ME1-31 | Oxytricha granulifera (91.3) | |||
| 68 | 1 | NA11-3 | Oxytricha granulifera (88.1) | |||
| 69 | 1 | ANT37-24 | Oxytricha granulifera (93.5) | |||
| Chrysophytes | 70 | 3 | 2 | ME1-23 | Paraphysomonas foraminifera (98.7) | |
| 71 | 1 | NA11-11 | Paraphysomonas foraminifera (95.3) | |||
| Cercomonads | 72 | 1 | ME1-26 | Heteromita globosa (88.1) | ||
| 73 | 1 | ANT12-14 | Thaumatomonas sp. (82.9) | |||
| 74 | 1 | ANT37-28 | Thaumatomonas sp. (89.6) | |||
| 75 | 2 | NA37-5 | Cercomonas strain ATCC 50318 (83.0) | |||
| Fungi | 76 | 2 | 3 | ANT12-13 | Stenocybe pullatula (80.0) | |
| Metazoans | 77 | 36 | Oikopleura sp. (99.2) | |||
| 78 | 43 | Cancrincola plumipes (94.0) |
FIG. 3.

Phylogenetic tree for partial sequences of environmental clones and the most closely related cultured organisms. Environmental clones are indicated by boldface type and each clone is designated by the library designation followed by a number. One clone representing each different OTU detected by the RFLP analysis of the five genetic libraries is included. The bar indicates 10% estimated sequence divergence. The number of clones in each genetic library belonging to each phylogenetic group is indicated on the right.
The fact that clones belonging to the same OTU were seldom identical indicated that we were underestimating the true diversity by sequencing only one clone of each OTU. However, this was the approach chosen since we were more interested in broad identification of the picoeukaryotic phylotypes present in different marine environments than in a detailed list of species. Moreover, only partial sequences were obtained (at least one-third of the 18S rRNA gene), and the phylogenetic affiliations of the clones and the percentages of similarity calculated were not as precise as they would have been if we had sequenced the whole gene. It is clear, however, that partial sequences are sufficient to infer the positions of clones in a given line of descent (46). Finally, the clonal representation of a group does not necessarily reflect its precise abundance in nature, given the potential biases inherent with PCR-based methods (50). For this reason we refer here to the percentages of clones in the libraries, which are useful values for comparing the distributions of groups among libraries.
Phototrophic picoeukaryotic isolates generally belong to the classes Prasinophyceae, Chlorophyceae, Prymnesiophyceae, and Pelagophyceae (17, 38, 44, 47). The relevance of these isolates in natural assemblages is uncertain, given the biases that occur when microorganisms are cultured (2, 16, 23). A significant number of clones in our libraries were affiliated with these classes; however, there was a conspicuous absence of chlorophytes. Our clones exhibited rather high similarities with picoeukaryotic isolates (between 94.1 and 100%; average, 97.4%), suggesting that the cultures available are fair representatives of natural phototrophic picoeukaryotes. The prasinophyte group was the most abundant and widespread algal group in our libraries and was represented by 46 clones and seven OTUs. These clones were present in all libraries and were dominant in ME1 and ANT37. The two Atlantic libraries contained clones very similar to Micromonas pusilla. In the other three libraries all clones were most similar to Mantoniella squamata or Ostreococcus tauri. In particular, one OTU that exhibited 96.8% similarity to M. squamata was represented 8 times in the Mediterranean library and 16 and 5 times in the two Antarctic libraries. Clones belonging to this OTU obtained from systems separated by thousands of kilometers were very similar (98.8%), indicating that very similar phylotypes are widely distributed. The second most abundant OTU had a phylogenetic position between Ostreococcus and Mantoniella and was also widely distributed. Finally, an OTU very similar to O. tauri appeared six times, but only in the ME1 library. It is perhaps not coincidental that O. tauri was described from a Mediterranean lagoon (7). Our culture-independent data confirm the importance of prasinophytes in marine picoplankton, in which their marker pigment prasinoxanthin is found widely (18, 36), and indicate that their diversity is relatively high since several phylotypes coexisted in the same sample.
The prymnesiophytes were represented by 28 clones belonging to five OTUs. They were abundant in the two Antarctic libraries but rare in the other three libraries. The most abundant OTU, with 5 clones in ANT37 and 16 clones in ANT12, was almost identical to Phaeocystis antarctica. The presence of this organism is expected in Antarctic waters, where it frequently forms large blooms, many times consisting of the colonial form. Unicellular flagellated forms of P. antarctica were probably responsible for the sequences detected. It must be noted that prefiltration of the Antarctic samples removed many phototrophic picoeukaryotes, and thus, a large fraction of the natural assemblage remained undescribed. In the Atlantic libraries two clones were moderately related to Emiliania huxleyi. This prymnesiophyte accounted for up to 60% of the phytoplankton biomass in the Atlantic samples (unpublished results) but was obviously effectively removed by the prefiltration step. Finally, a few clones in the ME1 and ANT12 libraries were moderately related to Prymnesium patelliferum.
Members of the Bacillariophyceae (diatoms) were represented by 18 clones belonging to eight OTUs. Most OTUs were found only in one library, indicating that a different diatom assemblage occurred in each marine region. In ANT37 most diatom clones were affiliated with Corethron criophilum, and one clone showed a very low level of similarity to Chaetoceros. The three OTUs in ME1 exhibited relatively high levels of similarity to Papiliocellulus elegans, Chaetoceros rostratus, and Skeletonema costatum, and one clone in ANT12 was very similar to Pseudonitzschia multiseries. Although most known diatoms tend to be larger than the size analyzed here, very small diatoms have been described (47). In an HPLC study of the distribution of size-fractionated pigments in the Arabian Sea, Latasa and Bidigare (18) found that between 70 and 92% of the marker pigment fucoxanthin occurred in the <2-μm fraction during the Spring Intermonsoon and between 26 and 85% of this pigment occurred in this fraction during the Southwest Monsoon in the two most open sea stations. The retrieval of diatom genes in our study is consistent with the presence of the marker pigment in the small-size fraction. Another possible explanation is cell breakage during prefiltration or squeezing of cells through the filter.
Dinoflagellates were represented by 14 clones in three different OTUs and accounted for a significant fraction of the eukaryotic clones in the North Atlantic libraries. One OTU that was most similar to Gymnodinium mikimotoi appeared in all Antarctic and North Atlantic libraries. A single clone recovered from ANT37 was very similar to Lepidodinium viride, whereas another clone in ME1 was very different from any known dinoflagellate. Dinoflagellates tend to be large and conspicuous organisms, and there is not any known form of picoplanktonic size. Dinoflagellates might be overrepresented in genetic libraries because they have larger genomes than members of other phytoplankton groups (40) and therefore potentially higher rRNA gene copy numbers (6). Like diatoms, their presence in genetic libraries might be due to inefficient prefiltration or the existence of unknown picodinoflagellates. In the study mentioned above, Latasa and Bidigare (18) found that often more than 50% (and up to 75%) of peridinin, the marker pigment of dinoflagellates, appeared in the <2-μm fraction.
The remaining phytoplankton groups were minor components of our libraries (Table 4). One OTU represented by one clone in ME1 and six clones in ANT12 was affiliated with the Pelagophyceae, and the sequenced clone was 100% similar to Pelagomonas calceolata. Three ME1 clones were very similar to the cryptophyte Geminigera criophila, and a single clone in ANT37 was moderately affiliated with the recently described picoeukaryote Bolidomonas pacifica. The closest relative of four clones in the Antarctic libraries was the glaucocystophyte Cyanophora paradoxa, but the similarity was so low (80.0 to 85.8%) that even an affiliation with this algal class is uncertain. The same is true for three clones in the ANT37 and Atlantic libraries affiliated with Dictyocha speculum (similarities, around 90%) and two ME1 clones distantly related to the eustigmatophyte Nannochloropsis (89.0%).
Among the clearly heterotrophic groups we found clones belonging to the Ciliophora, the cercomonads, and the fungi (Fig. 3). Clones clustering with the ciliates were present in all libraries (11 clones and six OTUs); one-half of them were in the ME1 library, which was constructed by using the prefilter with larger pores. These sequences were rather distantly related to database sequences (the levels of similarity were between 87.5 and 93.5%) and thus belong to new organisms. Five clones representing four different OTUs were distantly related to the cercomonads (the levels of similarity were between 83.0 and 89.6%), and low amounts of these clones were detected in the three systems. Five Antarctic clones were affiliated with the fungi and exhibited rather low levels of similarity to any known organism (80.0%). Six clones found in the three systems were affiliated with the class Chrysophyceae, which is known to contain mostly phototrophic organisms but also some heterotrophs. These clones were closely related to Paraphysomonas foraminifera and thus likely are heterotrophic flagellates and not phototrophic organisms.
A significant number of clones in the libraries did not show a close affiliation with any known class of organisms and formed two novel phylogenetic lineages. Novel stramenopiles were the more abundant lineages of the two (Fig. 3). These sequences, representing 53 clones and 24 OTUs, accounted for a significant fraction of the clones in each library: 19% in ME1, 19% in ANT37, 34% in ANT12, 36% in NA11, and 5% in NA37. They clustered in the basal branches of the stramenopile line of descent; the sequence of the fungus-like organism Hyphochytrium catenoides was the most similar sequence in the database, but the levels of similarity were always very low. The novel stramenopile clones were more similar to each other than to any other sequence and showed a relatively high degree of genetic diversity; separate clusters were apparent (Fig. 3). These clusters did not necessarily correspond to different phylotypes found in different samples. Instead, some phylotypes had a wide geographic distribution; for instance, a clone from the Mediterranean Sea (ME1-19) was almost identical (99.5% similarity) to a clone from the North Atlantic (NA11-4).
The stramenopiles (34) form a monophyletic group that is extremely diverse in terms of metabolism and cell type and includes algal cells, fungus-like cells, and heterotrophic flagellates. Phylogenetic relationships determined by using 18S rDNA sequences suggest that stramenopiles were initially heterotrophic and acquired a chloroplast at a certain point in evolution (19). Although the exact position of the novel stramenopile sequences within the different heterotrophic branches could not be unambiguously resolved, our phylogenetic analyses indicated that these organisms appeared before the chloroplast was acquired. Thus, the new sequences probably belong to heterotrophic organisms, and we hypothesize that they account for the bulk of the heterotrophic flagellates in the oceans (10). In open ocean waters, heterotrophic flagellates are mainly cells less than 2 or 3 μm in diameter (5, 42) and might be as abundant as phototrophic picoeukaryotes. Therefore, we expected these organisms to be included in our clone libraries, but we detected very few clones related to known heterotrophic flagellates (10, 48); only five clones were distantly related to cercomonads and six clones were affiliated with the chrysomonad Paraphysomonas. This is not surprising, since in a previous study it was shown that Paraphysomonas imperforata systematically dominated enrichment cultures from coastal samples but accounted for less than 1% of the heterotrophic flagellates in the natural system (23). The novel stramenopile sequences are also distantly related to known heterotrophic flagellates, such as Developayella elegans or the bicosoecids. In addition, there is reasonable agreement between the percentage of heterotrophic flagellate cells (based on the total number of picoeukaryotic cells) and the percentage of novel stramenopile clones (based on the total number of clones); these values are 10 and 19% in ME1, 11 and 34% in ANT12, and 4 and 19% in ANT37, respectively.
Novel alveolates formed the second novel lineage that was abundant in our libraries. They were represented by 19 clones and six OTUs and were recovered only from the Mediterranean and North Atlantic samples. Perhaps they were excluded from the Antarctic samples by the more drastic prefiltration technique used. The Mediterranean library contained the greatest diversity of marine alveolates, with 12 clones and four OTUs, whereas the Atlantic libraries (especially NA37) contained large percentages of marine alveolates, given the low number of clones analyzed. The novel alveolate sequences clustered in the basal part of the dinoflagellate clade and exhibited very low levels of similarity (76.0 to 89.3%) to dinoflagellate sequences. Their intermediate position between dinoflagellates and the newly established phylum Perkinsozoa, which contains marine parasites, did not allow us to hypothesize about their role in planktonic systems. Similar sequences have also been found in other picoeukaryotic genetic libraries from a surface sample (29) and deep samples (24).
Our results uncovered several patterns related to the diversity of the smallest eukaryotic plankton in the ocean. First, the diversity of picoeukaryotes in a single sample was great, and the organisms belonged to very different phylogenetic groups. Second, prasinophytes were very important in all the libraries, and this group may be the most widespread and abundant group of small phytoplankton in the ocean. Although quantitative data from PCR-based methods should be regarded with caution (50), two additional PCR-based methods (involving the use of different primers) applied to the Mediterranean sample also showed the dominance of the prasinophytes (9). Moreover, HPLC data also showed that there was a high proportion of Chl _b_-containing algae (including prasinophytes) in the same sample (9). Thus, the conclusion that prasinophytes are abundant in the oceans seems to be robust. Third, a large number of novel alveolate sequences (unrelated to known sequences) were relatively abundant in all libraries. And fourth, clones belonging to novel lineages of stramenopiles were present at very high frequencies in all libraries; these lineages appeared to branch among the basal heterotrophic groups of stramenopiles and may have important roles in the dynamics of the plankton.
ACKNOWLEDGMENTS
This work was funded by EU contracts MIDAS (MAS3-CT97-00154) and PICODIV (EVK3-CT1999-00021). The North Atlantic samples were collected during the ACSOE NAE cruise of the RRS Discovery funded by the British NERC and Spanish CICYT through grant MAR97-1885-E; the Mediterranean sample was collected during the MATER cruise of the B/O García del Cid funded by EU grant MATER (MAS3-CT96-0051); and the Antarctic samples were gathered during the E-DOVETAIL cruise of the B.I.O. Hespérides funded by Spanish CICYT grant ANT96-0866.
We thank Marta Estrada and Mikel Latasa for helpful comments and Josep M. Gasol for help with flow cytometry.
REFERENCES
- 1.Altschul S F, Madden T L, Schäffer A A, Zhang J, Zhang Z, Miller W, Lipman D J. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 1997;25:3389–3402. doi: 10.1093/nar/25.17.3389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Amann R I, Ludwig W, Schleifer K H. Phylogenetic identification and in situ detection of individual microbial cells without cultivation. Microbiol Rev. 1995;59:143–169. doi: 10.1128/mr.59.1.143-169.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Andersen R A, Saunders G W, Paskind M P, Sexton J P. Ultrastructure and 18S rRNA gene sequence for Pelagomonas calceolata gen. et sp. nov. and the description of a new algal class, the Pelagophyceae classis nov. J Phycol. 1993;29:701–715. [Google Scholar]
- 4.Andersen R A, Bidigare R R, Keller M D, Latasa M. A comparison of HPLC pigment signatures and electron microscopic observations for oligotrophic waters of the North Atlantic and Pacific oceans. Deep-Sea Res II. 1996;43:517–537. [Google Scholar]
- 5.Caron D A, Peele E R, Lim E L, Dennett M R. Picoplankton and nanoplankton and their trophic coupling in the surface waters of the Sargasso Sea south of Bermuda. Limnol Oceanogr. 1999;44:259–272. [Google Scholar]
- 6.Cavalier-Smith T. Eukaryote gene numbers, non-coding DNA and genome size. In: Cavalier-Smith T, editor. The evolution of genome size. Chichester, United Kingdom: Wiley; 1985. pp. 69–103. [Google Scholar]
- 7.Courties C, Vaquer A, Trousselier M, Lautier J, Chrétiennot-Dinet M-J, Neveux J, Machado C, Claustre H. Smallest eukaryotic organism. Nature. 1994;370:255. [Google Scholar]
- 8.DeLong E F. Archaea in coastal marine environments. Proc Natl Acad Sci USA. 1992;89:5685–5689. doi: 10.1073/pnas.89.12.5685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Díez B, Pedrós-Alió C, Marsh T L, Massana R. Application of denaturing gradient gel electrophoresis (DGGE) to study the diversity of marine picoeukaryotic assemblages and comparison of DGGE with other molecular techniques. Appl Environ Microbiol. 2001;67:2942–2951. doi: 10.1128/AEM.67.7.2942-2951.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Fenchel T. The ecology of heterotrophic microflagellates. Adv Microb Ecol. 1986;9:57–97. [Google Scholar]
- 11.Fogg G E. Some comments on picoplankton and its importance in the pelagic ecosystem. Aquat Microb Ecol. 1995;9:33–39. [Google Scholar]
- 12.Fuhrman J A, McCallum K, Davis A A. Novel major archaebacterial group from marine plankton. Nature. 1992;356:148–149. doi: 10.1038/356148a0. [DOI] [PubMed] [Google Scholar]
- 13.Giovannoni S J, Britschgi T B, Moyer C L, Field K G. Genetic diversity in Sargasso Sea bacterioplankton. Nature. 1990;345:60–63. doi: 10.1038/345060a0. [DOI] [PubMed] [Google Scholar]
- 14.Good I J. The population frequencies of species and the estimation of the population parameters. Biometrika. 1953;40:237–264. [Google Scholar]
- 15.Guillou L, Chrétiennot-Dinet M-J, Medlin L K, Claustre H, Loiseaux-de Goër S, Vaulot D. Bolidomonas: a new genus with two species belonging to a new algal class, the Bolidophyceae (Heterokonta) J Phycol. 1999;35:368–381. [Google Scholar]
- 16.Guillou L, Moon-van der Staay S Y, Claustre H, Partensky F, Vaulot D. Diversity and abundance of Bolidophyceae (Heterokonta) in two oceanic regions. Appl Environ Microbiol. 1999;65:4528–4536. doi: 10.1128/aem.65.10.4528-4536.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hooks C E, Bidigare R R, Keller M D, Guillard R R L. Coccoid eukaryotic marine ultraplankters with four different HPLC pigment signatures. J Phycol. 1988;24:571–580. [Google Scholar]
- 18.Latasa M, Bidigare R R. A comparison of phytoplankton populations of the Arabian Sea during the Spring Intermonsoon and Southwest Monsoon of 1995 as described by HPLC-analyzed pigments. Deep-Sea Res II. 1998;45:2133–2170. [Google Scholar]
- 19.Leipe D D, Tong S M, Goggin C L, Slemenda S B, Pieniazek N J, Sogin M L. 16S-like rDNA sequences from Developayella elegans, Labyrinthuloides haliotidis, and Proteromonas lacertae confirm that the stramenopiles are a primarily heterotrophic group. Eur J Protistol. 1996;32:449–458. [Google Scholar]
- 20.Letelier R M, Bidigare R R, Hebel D V, Ondrusek M E, Winn C D, Karl D M. Temporal variability of phytoplankton community structure at the U.S.-JGOFS time-series Station ALOHA (22°45′N, 158°00′W) based on HPLC pigment analysis. Limnol Oceanogr. 1993;38:1420–1437. [Google Scholar]
- 21.Li W K W. Primary production of prochlorophytes, cyanobacteria, and eucaryotic ultraphytoplankton: measurements from flow cytometric sorting. Limnol Oceanogr. 1994;39:169–175. [Google Scholar]
- 22.Lim E L, Amaral L A, Caron D A, DeLong E F. Application of rRNA-based probes for observing marine nanoplanktonic protists. Appl Environ Microbiol. 1993;59:1647–1655. doi: 10.1128/aem.59.5.1647-1655.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lim E L, Dennet M R, Caron D A. The ecology of Paraphysomonas imperforata based on studies employing oligonucleotide probe identification in coastal water samples and enrichment cultures. Limnol Oceanogr. 1999;44:37–51. [Google Scholar]
- 24.López-García P, Rodríguez-Valera F, Pedrós-Alió C, Moreira D. Unexpected diversity of small eukaryotes in deep-sea Antarctic plankton. Nature. 2001;409:603–607. doi: 10.1038/35054537. [DOI] [PubMed] [Google Scholar]
- 25.Maidak B L, Cole J R, Lilburn T G, Parker C T, Jr, Saxman P R, Stredwick J M, Garrity G M, Li B, Olsen G J, Pramanik S, Schmidt T M, Tiedje J M. The RDP (Ribosomal Database Project) continues. Nucleic Acids Res. 2000;28:173–174. doi: 10.1093/nar/28.1.173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Medlin L, Elwood H J, Stickel S, Sogin M L. The characterization of enzymatically amplified eukaryotic 16S-like rRNA-coding regions. Gene. 1988;71:491–499. doi: 10.1016/0378-1119(88)90066-2. [DOI] [PubMed] [Google Scholar]
- 27.Moestrup Ø. Further studies of presumedly primitive green algae, including the description of Pedinophyceae class. nov. and Resultor gen. nov. J Phycol. 1991;27:119–133. [Google Scholar]
- 28.Moon-van der Staay S Y, van der Staay G W M, Guillou L, Vaulot D, Claustre H, Medlin L K. Abundance and diversity of prymnesiophytes in the picoplankton community from the equatorial Pacific Ocean inferred from 18S rDNA sequences. Limnol Oceanogr. 2001;45:98–109. [Google Scholar]
- 29.Moon-van der Staay, S. Y., R. De Wachter, and D. Vaulot. Oceanic 18S rDNA sequences from picoplankton reveal unsuspected eukaryotic diversity. Nature **409:**607–610. [DOI] [PubMed]
- 30.Murphy L S, Haugen E M. The distribution and abundance of phototrophic ultraplankton in the North Atlantic. Limnol Oceanogr. 1985;30:47–58. [Google Scholar]
- 31.Olson R J, Zettler E R, DuRand M D. Phytoplankton analysis using flow cytometry. In: Kemp P F, Sherr B F, Sherr E B, Cole J J, editors. Handbook of methods in aquatic microbial ecology. Boca Raton, Fla: Lewis Publishers; 1993. pp. 175–186. [Google Scholar]
- 32.Parsons T R, Maita Y, Lalli C M. A manual of chemical and biological methods for seawater analysis. 1st ed. Oxford, United Kingdom: Pergamon Press; 1984. [Google Scholar]
- 33.Partensky F, Guillou L, Simon N, Vaulot D. Recent advances in the use of molecular techniques to assess the genetic diversity of marine photosynthetic microorganisms. Vie Milieu. 1997;47:367–374. [Google Scholar]
- 34.Patterson D J. Stramenopiles: chromophytes from a protistan perspective. In: Green J C, Leadbeater B S C, Diver W L, editors. Chromophyte algae: problems and perspectives. Oxford, United Kingdom: Clarendon Press; 1989. pp. 357–379. [Google Scholar]
- 35.Paul J H, Myers B. Fluorometric determination of DNA in aquatic microorganisms by use of Hoechst 33258. Appl Environ Microbiol. 1982;43:1393–1399. doi: 10.1128/aem.43.6.1393-1399.1982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Peeken I. Photosynthetic pigment fingerprints as indicators of phytoplankton biomass and development in different water masses of the Southern Ocean during austral spring. Deep-Sea Res II. 1997;44:261–282. [Google Scholar]
- 37.Porter K G, Feig Y S. The use of DAPI for identifying and counting aquatic microflora. Limnol Oceanogr. 1980;25:943–948. [Google Scholar]
- 38.Potter D, Lajeunesse T C, Saunders G W, Andersen R A. Convergent evolution masks extensive biodiversity among marine coccoid picoplankton. Biodivers Conserv. 1997;6:99–107. [Google Scholar]
- 39.Rappé M S, Suzuki M T, Vergin K L, Giovannoni S J. Phylogenetic diversity of ultraplankton plastid small-subunit rRNA genes recovered in environmental nucleic acid samples from the Pacific and Atlantic coasts of the United States. Appl Environ Microbiol. 1998;64:294–303. doi: 10.1128/aem.64.1.294-303.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Rizzo P J. Biochemistry of the dinoflagellate nucleus. In: Taylor F J R, editor. The biology of dinoflagellates. Oxford, United Kingdom: Blackwell Scientific; 1985. pp. 143–173. [Google Scholar]
- 41.Schauer M, Massana R, Pedrós-Alió C. Spatial differences in bacterioplankton composition along the Catalan coast (NW Mediterranean) assessed by molecular fingerprinting. FEMS Microbiol Ecol. 2000;33:51–59. doi: 10.1111/j.1574-6941.2000.tb00726.x. [DOI] [PubMed] [Google Scholar]
- 42.Sherr E B, Sherr B F, Fessenden L. Heterotrophic protists in the Central Arctic Ocean. Deep-Sea Res II. 1997;44:1665–1682. [Google Scholar]
- 43.Sieburth J M, Smetacek V, Lenz J. Pelagic ecosystem structure: heterotrophic compartments of the plankton and their relationship to plankton size fractions. Limnol Oceanogr. 1978;23:1256–1263. [Google Scholar]
- 44.Simon N, Barlow R G, Marie D, Partensky F, Vaulot D. Characterization of oceanic photosynthetic picoeukaryotes by flow cytometry. J Phycol. 1994;30:922–935. [Google Scholar]
- 45.Simon N, LeBot N, Marie D, Partensky F, Vaulot D. Fluorescent in situ hybridization with rRNA-targeted oligonucleotide probes to identify small phytoplankton by flow cytometry. Appl Environ Microbiol. 1995;61:2506–2513. doi: 10.1128/aem.61.7.2506-2513.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Stackebrandt E, Rainey F A. Partial and complete 16S rDNA sequences, their use in generation of 16S rDNA phylogenetic trees and their implications in molecular ecological studies. In: Akkermans A D L, van Elsas J D, de Bruijn F J, editors. Molecular microbial ecology manual. 3.1.1. Dordrecht, The Netherlands: Kluwer Academic Publishers; 1995. pp. 1–17. [Google Scholar]
- 47.Thomsen H A. A survey of the smallest eukaryotic organisms of the marine phytoplankton. Can Bull Fish Aquat Sci. 1986;214:121–158. [Google Scholar]
- 48.Tong S M. Heterotrophic flagellates and other protists from Southampton Water, U.K. Ophelia. 1997;47:71–131. [Google Scholar]
- 49.Van Hannen E J, Van Agterveld M P, Gons H J, Laanbroek H J. Revealing genetic diversity of eukaryotic microorganisms in aquatic environments by denaturing gradient gel electrophoresis. J Phycol. 1998;34:206–213. [Google Scholar]
- 50.Von Wintzingerode F, Goebel U B, Stackebrandt E. Determination of microbial diversity in environmental samples: pitfalls of PCR-based rRNA analysis. FEMS Microbiol Rev. 1997;21:213–229. doi: 10.1111/j.1574-6976.1997.tb00351.x. [DOI] [PubMed] [Google Scholar]