Health – Page 2 – NIH Director's Blog (original) (raw)
Study of Protective Gene Variant Provides Insight into Delaying Onset of Alzheimer’s Dementia
Posted on July 18th, 2024 by Dr. Monica M. Bertagnolli

Credit: Donny Bliss/NIH
Alzheimer’s disease is currently the seventh leading cause of death in the U.S. While your likelihood of developing Alzheimer’s-related cognitive impairment increases with age, risk for this disease and age of its onset depend on many factors, including the genes you carry. An intriguing new study suggests that having just one copy of a protective gene variant may be enough to delay cognitive impairment from this devastating disease in individuals who are otherwise genetically predisposed to developing early-onset Alzheimer’s dementia.
The findings, from a study supported in part by NIH and reported in The New England Journal of Medicine, offer important insights into the genetic factors and underlying pathways involved in Alzheimer’s dementia.1 While much more study is needed, the findings have potential implications for treatments that could one day work like this gene variant does to delay or perhaps even prevent Alzheimer’s dementia.
This research comes from an international team including Yakeel Quiroz, Massachusetts General Hospital, Boston; Joseph Arboleda-Velasquez, Mass Eye and Ear, Boston; and Francisco Lopera, University of Antioquia, Colombia. For the last 40 years, Lopera has been studying a Colombian family of about 6,000 blood relatives, 1,200 of whom carry a mutation known as Paisa (or Presenilin-1 E280A) that predisposes them to developing early-onset Alzheimer’s dementia. Those who carry a single copy of this gene variant typically show signs of cognitive decline in their early 40s, progressing to dementia by age 50. They frequently die from dementia-related complications in their 60s.
In 2019, the researchers reported on an extraordinary individual who was an exception to this prognosis.2 Even though she carried the Paisa mutation, she didn’t develop any notable cognitive decline until her late 70s—30 years later than expected. The researchers traced her protection against dementia to two copies of a rare variant of the APOE gene dubbed Christchurch. Further study of her brain after death also found lower levels of inflammation and tau protein, which forms damaging tangles inside neurons in the Alzheimer’s brain.
Christchurch is a rare variant, and it’s far more common for people to carry one copy of the protective variant versus two. Would a single copy of the Christchurch variant offer some protection against Alzheimer’s dementia, too? To find out in the new study, the researchers analyzed data from 27 members of this family carrying a single copy of the Christchurch variant among 1,077 carriers of the Paisa mutation.
The researchers compared Christchurch carriers to those without the protective variant and found the variant did delay the age of onset of Alzheimer’s-related cognitive decline and dementia. The median age at the onset of mild cognitive impairment was 52 in family members with the Christchurch variant, compared to approximately age 47 in a matched group without the variant. Similarly, the median age at the onset of dementia was 54, compared to the median age of 50 in noncarriers.
To learn more, the researchers imaged the brains of two of the individuals who had one copy of Christchurch. The brain scans showed lower levels of tau and more normal metabolic activity in brain areas that are known to play a role in Alzheimer’s. Interestingly, their brains still showed accumulations of amyloid proteins, which form plaques that are another hallmark of Alzheimer’s. The team also analyzed autopsy samples from four deceased individuals with one copy of the Christchurch variant and found that blood vessels in their brains appeared healthier, which may help to explain the protective effects of Christchurch. The findings suggest a significant role for blood vessel health in protecting the brain from cognitive decline, as well as a role for disease of the brain blood vessels in contributing to cognitive decline and dementia.
The researchers note this study is limited to a relatively small number of people with both the Paisa and Christchurch variants in one group of related individuals. Further studies involving larger and more diverse samples are needed to learn more about this protective gene variant and its effects on the brain in the general population. The hope is these findings may one day yield new approaches to delaying the onset of Alzheimer’s or slowing its progression in millions more people around the world at risk of developing this devastating disease.
References:
[1] Quiroz YT, et al. APOE3 Christchurch Heterozygosity and Autosomal Dominant Alzheimer’s Disease. The New England Journal of Medicine. DOI: 10.1056/NEJMoa2308583 (2024).
[2] Arboleda-Velasquez JF, et al. Resistance to autosomal dominant Alzheimer’s disease in an APOE3 Christchurch homozygote: a case report. Nature Medicine. DOI: 10.1038/s41591-019-0611-3 (2019).
NIH Support: National Institute on Aging, National Institute of Neurological Disorders and Stroke
Posted In: Health, News, Science
Tags: aging, Alzheimer’s disease, brain, cognitive decline, dementia, DNA, gene variants, genetics, neurological disease
Sequencing Technique Detects Earliest Signs of Genetic Mutations Underlying Cancer, Aging, and More
Posted on July 11th, 2024 by Dr. Monica M. Bertagnolli

Every day, billions of cells in your body divide, each producing two daughter cells. It’s an essential process for your tissues and organs to renew themselves and remain healthy. To do it, cells must first duplicate their DNA to ensure that each daughter cell gets an accurate copy. In this process, mistakes are inevitably made. Most DNA errors are accurately fixed and do not lead to mutations. But when small errors akin to single-letter typos aren’t corrected, they can become permanent in a cell and multiplied with each subsequent cell division. Even cells that don’t divide, such as neurons in your brain, acquire damage and mutations in their DNA with age. As a result, your tissues contain collections of cells with distinct mutations that accumulate over time.
While many of these small errors will show no obvious consequences, others can lead to cancer and other health conditions. Now, a new DNA sequencing technique, described in Natureand developed through research supported by NIH, promises to detect early DNA changes before they become permanent mutations in a cell’s genome. The method, called Hairpin Duplex Enhanced Fidelity Sequencing (HiDEF-seq), could advance our understanding of how and why mutations arise, with potentially important implications for our health. For example, the ability to identify signs that precede mutations may help predict a person’s health risks based on genetic predispositions, environmental exposures, or other factors.
The HiDEF-seq technique comes from an international team led by Gilad Evrony at NYU Grossman School of Medicine’s Center for Human Genetics and Genomics in New York City. To understand how the method works, it helps to remember that each DNA molecule stores genetic information in the form of two complementary strands made up of four molecular “letters,” or chemical bases. Those bases are adenine (A), thymine (T), guanine (G), and cytosine (C). The sequence of about three billion As, Ts, Gs, and Cs in human DNA’s two strands generally should match up, such that As pair with Ts and Gs with Cs.
The first step in which DNA mutations arise usually involves a change in only one of the two DNA strands. Those single-strand errors only become permanent mutations in both strands when a cell’s copying machinery fails to detect the mistake before the cell divides again, or when the cell’s DNA repair machinery makes a mistake in the correction process. However, because other methods to sequence DNA can’t accurately detect changes that are in only one of the DNA strands, it hasn’t been possible for researchers to study this process in detail. This is where HiDEF-seq comes in.
The researchers wanted to develop an approach for directly sequencing single DNA molecules to detect these early-stage DNA errors. Detecting changes that are in only one of the two DNA strands requires an extremely high degree of sequencing accuracy, with less than one error per one billion bases, so the team devised a method to read DNA with higher precision than was previously possible. To put HiDEF-seq to the test, they profiled 134 samples from various human tissues, including those from people with syndromes that predisposed them to cancer due to an unusually high number of new mutations.
The research team found they could use HiDEF-seq to identify changes present in only one of the two DNA strands that were the precursors to mutational events. For example, they identified places where a C was mistakenly paired with a T instead of a G. As expected, those early changes in DNA turned up more often in people with syndromes that increase their risk of cancer than in those without. The patterns of those single-strand DNA changes also looked a lot like the patterns of double-strand DNA mutations seen in people with these syndromes, suggesting that the HiDEF-seq method was indeed seeing the precursors to mutations.
The method can also detect a common form of DNA damage called cytosine deamination, in which cytosine is converted to a different base called uracil (U), which is another source of mutations. Experiments in human sperm, which rarely pick up mutations compared to other cell types, showed a pattern of cytosine deamination that closely matched damage caused by heating healthy DNA in the lab. This led the researchers to suggest that the damage to DNA happens similarly in both situations.
The researchers have already begun to produce a catalog of the various single-strand DNA errors they’ve uncovered. They suggest that HiDEF-seq may allow new ways to monitor the everyday effects of environmental exposures or other insults on our DNA and shed light on the balance in cells between DNA damage, repair, and replication. Along the way, this new technique will enable the continued study of DNA damage and the origins of mutations in a way that hasn’t been possible before.
Reference:
Liu MH, et al. Single-strand mismatch and damage patterns revealed by single-molecule DNA sequencing. Nature. DOI: 10.1038/s41586-024-07532-8 (2024).
NIH Support: Common Fund, Eunice Kennedy Shriver National Institute of Child Health and Human Development, National Institute on Aging, National Institute of Neurological Disorders and Stroke, National Cancer Institute
Posted In: Health, News, Science
Tags: aging, cancer, cell division, DNA, DNA damage, DNA sequencing, genetics, genomics, mutations
Molecular Portrait of Key Driver of Pancreatic Cancer Offers Hope for Continued Treatment Advances
Posted on June 27th, 2024 by Dr. Monica M. Bertagnolli

Credit: magicmine/Adobe Stock
Cancer arises when changes in genes that normally control cell division lead to unchecked growth at the expense of healthy tissues. One of the most common genetic alterations across human cancers—occurring in 95% of pancreatic cancers but also many non-small cell lung cancers, colorectal cancers, and others—is in a gene known as KRAS. While promising new treatments targeting KRAS to shrink cancerous tumors have recently gained approval, less than 40% of pancreatic cancers respond to treatment with KRAS inhibitors for reasons that aren’t well understood.
There’s much more to learn about how KRAS spurs cancer growth—and how KRAS-mutant cancers resist treatment with existing KRAS inhibitors. To address this need, researchers behind two studies in Science have established the most comprehensive molecular portrait yet of the workings of KRAS and how its many downstream impacts may influence outcomes for people with pancreatic cancer.1,2 The findings could lead to new treatment approaches, including ways to potentially guide treatment for individuals with pancreatic cancer, the third leading cause of cancer-related death in the U.S.
These studies, supported in part by NIH, come from a team led by Channing Der and Adrienne Cox, together with Jeffrey Klomp, Clint Stalnecker, and Jennifer Klomp, at the Lineberger Comprehensive Cancer Center, University of North Carolina at Chapel Hill. The researchers were inspired in part by the Food and Drug Administration’s recent approval of treatments that block a mutated version of KRAS that drives many pancreatic cancers. The team was also motivated by the realization that many patients whose cancers initially respond to the new treatments relapse rather quickly as the cancers find ways to reactivate underlying growth pathways.
The researchers wanted to know more about KRAS and its influence on another essential pathway, including a protein called ERK, by defining all the genes that are actively transcribed into proteins in KRAS-mutant cancer cell lines and tumors. Their findings show that changes in KRAS signaling drive cancer growth mainly though the ERK network. In turn, ERK regulates many other genes to determine which ones are switched on or off while also influencing the activity of many other proteins. This shows that the effect of mutant KRAS on the ERK protein alone leads to widespread effects on the activity of thousands of other genes and proteins. The study uncovered underlying mechanisms that affected multiple stages of the cell cycle that leads to cell division.
Kinases, including ERK, alter the activity of other proteins by the addition of a chemical phosphate group. One of the things that makes ERK unique is that it activates a wide range of functionally distinct proteins, both directly and indirectly. To learn more about its influence, the team explored the many proteins ERK chemically modifies in pancreatic cancers that rely on mutant KRAS. Altogether, they found more than 2,100 proteins that were modified by activated ERK, more than half of which had not been associated with this protein before. Because activation of this pathway is so important for the response to KRAS inhibitors, the findings promise to help elucidate the underlying mechanisms involved in treatment responses as well as drug resistance.
Importantly, the researchers found that the molecular signatures they’ve uncovered may predict tumor responses in patients treated with KRAS inhibitors or ERK inhibitors. Based on their findings, they suspect that the reason so many pancreatic cancers don’t respond to KRAS inhibitors may be because the drugs simply don’t block KRAS well enough—and not because the cancers no longer depend on KRAS signals for their growth. The researchers suggest it may be beneficial to monitor these underlying molecular pathways in patients to better understand treatment outcomes and guide treatment decisions.
The team plans to continue exploring the role of these and other important drivers of cancer growth and treatment resistance. Ultimately, their goal is to help advance the development of the next generation of KRAS inhibitors that will work even better for many more people with pancreatic or other KRAS-driven cancers.
References:
[1] Klomp JA, et al. Defining the KRAS- and ERK-dependent transcriptome in KRAS-mutant cancers. Science. DOI: 10.1126/science.adk0775 (2024).
[2] Klomp JE, et al. Determining the ERK-regulated phosphoproteome driving KRAS-mutant cancer. Science. DOI: 10.1126/science.adk0850 (2024).
NIH Support: National Cancer Institute, National Institute of General Medical Sciences
Antibiotic Compound Kills Hard-to-Treat, Infectious Bacteria While Sparing Healthy Bacteria in the Gut
Posted on June 20th, 2024 by Dr. Monica M. Bertagnolli
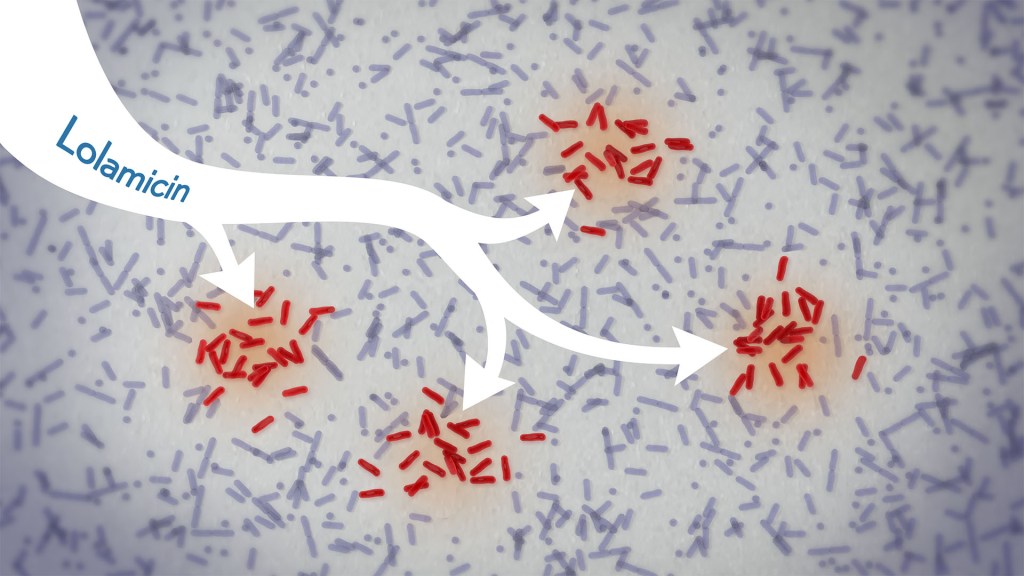
In a new study, an antibiotic compound, lolamicin, targeted infectious, gram-negative bacteria without harming the gut microbiome. Credit: Donny Bliss/NIH
Drug-resistant bacteria are responsible for a rise in serious, hospital-acquired infections, including pneumonia and sepsis. Many of these bacteria are classified as “gram-negative,” and are harder to kill than “gram-positive” bacteria. Unfortunately, the limited number of antibiotics that can help combat these dangerous infections can also damage healthy microbes in the gut, leaving people at risk for other, potentially life-threatening infections. Such antibiotic-induced disruption has also been linked in studies to irritable bowel syndrome, colon cancer, and many other health conditions.
There’s a great need for more targeted antibiotics capable of fending off infectious gram-negative bacteria while sparing the community of microbes in the gut, collectively known as the gut microbiome. Now, in findings reported in the journal Nature, a research team has demonstrated a promising candidate for the job. While the antibiotic hasn’t yet been tested in people, the findings in cell cultures suggest it could work against more than 130 drug-resistant bacterial strains. What’s more, the study, supported in part by NIH, shows that this compound, when given to infected mice, thwarts potentially life-threatening bacteria while leaving the animals’ gut microbiomes intact.
One reason it’s been difficult to find antibiotics that work against gram-negative bacteria without killing too many benign bacteria is that most promising targets for gram-negative bacteria are shared by gram-positive bacteria. But the team, led by Paul Hergenrother and Kristen Muñoz at the University of Illinois Urbana-Champaign, recognized an intriguing target for more specific microbiome-sparing antibiotics in a collection of proteins that gram-negative bacteria depend on to transport lipoproteins between their inner and outer cell membranes. Gram-positive bacteria, with only one cell membrane, don’t require lipoprotein transport and therefore lack these proteins.
The researchers knew that this lipoprotein-transport system, known as the Lol system, is required for infectious E. coli bacteria to live and grow. It’s also found in many other infectious gram-negative bacteria. They thought that compounds aimed at this system might be doubly selective—specifically targeting hard-to-treat gram-negative bacteria and leaving the gut microbiome relatively unscathed. They also knew other drugs targeting the Lol system had showed some promise in selectively targeting gram-negative bacteria. However, these antibiotics didn’t work well enough on their own to fight infections.
In the new study, the researchers tinkered with the design of those compounds in search of one that might work better. It led them to a compound, which they call lolamicin, that they found could selectively target three different types of infectious gram-negative bacteria (E. coli, Klebsiella pneumoniae, and Enterobacter cloacae) in the lab. They also found that the antibiotic at high doses killed up to 90% of multidrug-resistant strains of those infectious bacteria in cell cultures.
In additional experiments, the researchers wanted to see how well lolamicin would treat infected mice. They found that treatment with the antibiotic was well tolerated by the animals. They went on to show in mouse models of acute pneumonia and sepsis that oral treatment with lolamicin reduced the number of infectious bacteria. When mice with sepsis were treated with lolamicin, all of them survived. Lolamicin treatment also rescued 70% of mice with pneumonia infection.
While treatment with amoxicillin, a broad-spectrum antibiotic, and clindamycin, an antibiotic that only targets gram-positive bacteria, disrupted the assemblage of healthy microbes living in the mouse gut, the researchers found that treatment with lolamicin did not. They saw no big changes in the microbial community present in the mouse gut after three days of lolamicin treatment. As a result, unlike mice treated with the other two antibiotics, mice treated with lolamicin were protected from secondary infection by Clostridioides difficile, a bacterium that can infect the colon to cause diarrhea and life-threatening tissue damage.
These new findings, while promising, are at an early stage of drug discovery and development, and much more study is needed before this compound could be tested in people. It will also be important to learn how rapidly infectious gram-negative bacteria may develop resistance to lolamicin. Nevertheless, these findings suggest it may be possible to further develop lolamicin or related antibiotic compounds targeting the Lol system to treat dangerous gram-negative infections without harming the microbiome.
Reference:
Muñoz KA, et al. A Gram-negative-selective antibiotic that spares the gut microbiome. Nature. DOI: 10.1038/s41586-024-07502-0 (2024).
NIH Support: National Institute of Allergy and Infectious Diseases
Insights into Molecular Basis of PTSD and Major Depression Could One Day Aid in Diagnosis and Treatment
Posted on June 13th, 2024 by Dr. Monica M. Bertagnolli

Credit: S. Thomas Carmichael/UCLA, Yuliia/Adobe Stock
We know stress can take a toll on our mental health. Yet, it’s unclear why some people develop stress-related mental health disorders and others don’t. The risk for developing a stress-related mental health disorder such as post-traumatic stress disorder (PTSD) or major depressive disorder (MDD) depends on a complex interplay between the genetic vulnerabilities we are born with and the impact of traumatic stress we experience over our lifetimes.
Given this complexity, it’s been difficult for researchers to pinpoint the underlying biological pathways in the body that ultimately produce changes associated with PTSD, major depression, or other mental health conditions. Now, a study reported in a special issue of Science on decoding the brain uses a comprehensive approach to examine multiple biological processes across brain regions, cell types, and blood to elucidate this complexity. It’s an unprecedented effort to understand in a more holistic way the essential biological networks involved in PTSD and MDD.
While earlier studies looked at stress hormones, the immune system, and other molecular signatures of stress in blood samples, what had been largely missing from the picture of PTSD and MDD were links between those changes in the body and changes in the brain. To get a more complete picture, a multisite research team led by Nikolaos P. Daskalakis and Kerry Ressler of McLean Hospital, Belmont, MA, developed a vast molecular dataset including DNA variants, RNA, proteins, and chemical modifications to DNA. This “multi-omic” dataset was generated by the NIH-supported PTSD Brainomics Project of the PsychENCODE Consortium, and included postmortem data from 231 individuals with PTSD and/or MDD, as well as from individuals who didn’t have known mental health conditions.
In the study, the researchers looked at three essential brain regions: the medial prefrontal cortex (mPFC), the hippocampal dentate gyrus, and the central nucleus of the amygdala. They conducted single-cell RNA sequencing analysis of 118 dorsolateral prefrontal cortex (dlPFC) samples to look at cell-type-specific patterns and evaluated protein changes in the blood of more than 50,000 UK Biobank samples to look for biomarkers of stress-related disorders. After identifying key brain-based genes whose expression was altered in PTSD and/or MDD, the researchers compared them to genes linked to increased risk for these conditions.
Among many findings, the study results show an important role for the mPFC in both stress-related conditions, which is interesting, as the mPFC is essential for integrating signals from other brain areas and is known to play a role in cognitive processes, emotional regulation, motivation, and sociability. The findings also highlight important roles for molecular pathways known to play a role in immune function, the regulation of neurons and neural connections, and stress hormones. The single-cell RNA sequencing in the dlPFC also uncovered dysregulated stress-related signals in neurons and other brain cell types.
Furthermore, the findings reveal shared changes in gene activity between PTSD and MDD, as well as notable differences in the patterns of methyl marks on the DNA, suggesting changes in the way genes are switched on or off, and at the level of cell-type-specific gene activity. The researchers also found that history of childhood trauma and suicide were drivers of molecular changes in both disorders.
The data point to a short list of proteins that may be important in regulating key genetic pathways underlying these disorders. They also reveal links to gene networks related to aging, inflammation, stress, and more. Similarities in disease signals in the brain and blood suggest that blood-based tests might one day offer an additional avenue for assessing these disorders. Interestingly, there was little overlap between PTSD and MDD risk genes and those involved in the underlying molecular-level changes in the brains of people with one or both conditions. This shows that there’s a need for more research into how genetic risk factors are related to molecular-level disease processes.
There’s clearly much more to discover in the years ahead. But these insights already point to important roles for known stress-related pathways in fundamental brain changes underlying PTSD and MDD, while also revealing more novel pathways as potentially promising new treatment targets. With further study, the researchers hope these findings can also begin to answer vexing questions, such as why some people develop PTSD or major depression after stressful events and others don’t.
Reference:
Daskalakis NP, et al. Systems biology dissection of PTSD and MDD across brain regions, cell types, and blood. Science. DOI: 10.1126/science.adh3707 (2024).
This paper is part of a larger collection of studies from the PsychENCODE Consortium looking at the underlying mechanisms of neuropsychiatric diseases.
NIH Support: National Institute of Mental Health
Posted In: Health, News, Science
Tags: biomarkers, brain, depression, mental health, mental health disorders, molecular signature, multiomics, neuroscience, post-traumatic stress disorder, PTSD, stress
Study Suggests Computerized Brain Implant Could One Day Decode Internal Speech for Those Who Can No Longer Speak
Posted on June 6th, 2024 by Dr. Monica M. Bertagnolli

Credit: Tom Merton/KOTO/Adobe Stock
The ability to communicate using only your thoughts might sound like the stuff of science fiction. But for people who don’t have the ability to speak or move due to injury or disease, there’s great hope that this may one day be possible using brain-computer interfaces (BCIs) that can “read” relevant brain signals and translate them into written or spoken words. A research team has made a preliminary advance in this direction by showing for the first time that a computerized brain implant can decode internal speech with minimal training.
In the new NIH-supported study, researchers implanted such a device in a brain area known to be important for representing spoken words called the supramarginal gyrus in two people with tetraplegia, a condition marked by full body paralysis from the neck down due to cervical spinal cord injury. The researchers found that the device could decode several words the participants “spoke” only in their minds. While we are far from using such a device to decode whole sentences or even phrases, and the exact mechanisms of internal speech are still under study, the findings, reported in Nature Human Behavior, are notable because it had been unclear whether the brain signals involved in thinking words could be reproducibly translated.
The findings come from a team led by Richard Andersen at the California Institute of Technology, Pasadena, CA, and Sarah Wandelt, now at the Feinstein Institutes for Medical Research in Manhasset, NY, and the study was supported by the NIH _Brain Research Through Advancing Innovative Neurotechnologies_® (BRAIN) Initiative Research Opportunities in Humans program. Though earlier research had shown that brain implants could decode vocalized, attempted, and mimed speech, it had yet to be seen whether internal speech could be similarly decoded.
An earlier advance in decoding speech signals from the brain came in 2022, when the researchers reported they could accurately predict the words that a person with tetraplegia was thinking using a BCI. In the new study, they’ve shown that the device works in a second person with tetraplegia. The finding is an indication that the approach can work in different individuals and doesn’t depend on the brain characteristics of a particular person or the precise orientation of the implant in their brain.
In the study, the researchers trained their device to recognize brain patterns associated with certain internal “spoken” words including six actual words (battlefield, cowboy, python, spoon, swimming, and telephone) and two nonsense words (nifzig and bindip). During three sessions, the researchers flashed words on a screen and asked each participant to think about “saying” the words without speaking or moving. The BCI then used measurements of brain activity during the sessions and a computer model to predict the words being “spoken” internally.
The researchers found that in this task the device could decode the words with an average accuracy of 79% with the first participant and 23% with the second participant. They noted that the second participant had fewer unique patterns of brain activity associated with the different words, which may explain the lower results. Nevertheless, the findings show that the brain region in question generally contains signals for internal speech, although there is likely also variation among people in how thoughts of particular words are represented in patterns of brain activity. Furthermore, the device’s ability to decode nonsense words suggests that the words are represented in this part of the brain phonetically and not necessarily based on their meanings.
While there is much more to learn about how to decode internal speech more reliably across individuals, the findings offer proof-of-concept for a high-performance internal speech BCI. The new research adds to a growing portfolio of rapidly advancing technologies supported by the BRAIN Initiative that could one day routinely restore the ability to communicate for those who can no longer speak or even move, including people with brain injuries, paralysis, or diseases such as amyotrophic lateral sclerosis (ALS).
Reference:
Wandelt SK, et al. Representation of internal speech by single neurons in human supramarginal gyrus. Nature Human Behaviour. DOI: 10.1038/s41562-024-01867-y (2024).
NIH Support: NIH BRAIN Initiative
Posted In: Health, News, Science
Tags: amyotrophic lateral sclerosis, brain, BRAIN Initiative, brain-computer interfaces, internal speech decoder, neuroscience, paralysis, speech, tetraplegia, thoughts
Addressing Graduates Embarking on Careers in Medicine and Public Health
Posted on May 28th, 2024 by Dr. Monica M. Bertagnolli

NIH Director Monica Bertagnolli giving a commencement address at Columbia University Mailman School of Public Health on May 15, 2024. Credit: Leslye Smith
Earlier this month, I had the honor of giving two commencement addresses, one at Columbia University Mailman School of Public Health, and the other at my alma mater University of Utah School of Medicine. I was also able to spend some time at both schools, meeting with faculty and taking part in some fascinating discussions. Congratulations to the graduates! I have great confidence in them as they embrace the challenging and rewarding paths of public health and medicine. They give me great hope for the future. Here are a few photos from my visits.

Visiting with Columbia Mailman School of Public Health Dean Linda Fried (center) and Department of Sociomedical Sciences Chair Kathleen Sikkema (right). Credit: Leslye Smith

Visiting a lab at the Huntsman Cancer Institute at the University of Utah. Credit: Huntsman Cancer Institute

Discussing the documentary film “dêtetsi vo’i oninjakan Winding Path,” featuring medical student Jenna Murray (center), on a panel discussion moderated by Maija Holsti, Director of the Native American Summer Research Intern program (left) at the University of Utah School of Medicine. Credit: University of Utah Health

Addressing graduates of the University of Utah School of Medicine on May 17, 2024. Credit: University of Utah Health
Posted In: Creative Minds, Health, Science, Snapshots of Life, Training
Tags: Columbia University, commencement addresses, education, Huntsman Cancer Institute, Mailman School of Public Health, medical careers, public health careers, University of Utah
Speeding the Diagnosis of Rare Genetic Disorders with the Help of Artificial Intelligence
Posted on May 16th, 2024 by Dr. Monica M. Bertagnolli

Credit: Donny Bliss/NIH, Qpt/Shutterstock, taka/Adobe Stock
Millions of children around the world are born each year with severe genetic disorders. Many of these are Mendelian disorders, which are rare genetic conditions caused by mutations in a single gene. But pinpointing the specific gene responsible for a disorder to get a clear diagnosis for an individual can be labor-intensive, and reanalysis of undiagnosed cases is also difficult. As a result, only about 30% of people with a rare genetic disorder get a definitive diagnosis, and on average, it takes 6 years from symptom onset to diagnosis.
Progress is needed to get accurate diagnoses to individuals and families more often and faster, and to create more efficient ways to update genetic diagnoses as new discoveries are made. As an important step in this direction, a team funded in part by NIH has developed a new artificial intelligence (AI) system called AI-MARRVEL (AI-Model organism Aggregated Resources for Rare Variant ExpLoration).1
As reported in NEJM AI by a research team led by Pengfei Liu, Hugo Bellen, and Zhandong Liu at the Baylor College of Medicine and the Jan and Dan Duncan Neurological Research Institute at Texas Children’s Hospital in Houston, AI-MARRVEL relies on a machine learning approach. Machine learning involves using vast quantities of data to train computer systems to become increasingly better at recognizing patterns.
The AI-MARRVEL system was trained using a compendium of data called MARRVEL, previously developed by the research team. MARRVEL integrates genetic information from six human databases and seven model organism databases into one site and includes more than 3.5 million known genetic variants from thousands of healthy individuals as well as those with diagnosed cases of genetic disorders. Using what it has learned from that compendium of data, AI-MARRVEL uses a person’s symptoms and protein-coding genome sequences to narrow down the most likely variants responsible for that person’s genetic condition.
To find out how well it works, the researchers compared the results from AI-MARRVEL to other previously published tools for genetic diagnosis based on three different databases containing established molecular diagnoses from a clinical diagnostic laboratory: Baylor Genetics, the NIH-funded Undiagnosed Diseases Network (UDN), and the Deciphering Developmental Disorders (DDD) project. Overall, the researchers found that AI-MARRVEL consistently made accurate diagnoses in twice as many cases as these other tools.
While hundreds of new disease-causing variants are discovered each year, there’s currently no streamlined way to determine which cases should be reanalyzed when previous sequencing and interpretation failed to identify the cause.2 To see how well AI-MARRVEL does at identifying diagnosable cases from pools of unsolved cases, the researchers designed a confidence metric and found the tool achieved a precision rate of 98% and correctly identified 57% of diagnosable cases out of a collection of 871 cases. The researchers also suggest that AI-MARRVEL could help identify short lists of possible gene candidates in even more potentially solvable cases and then send them on to a panel of experts for follow-up review.
There is some early evidence that AI-MARRVEL could also be put to work in making new discoveries that link novel gene variants to diseases for the first time. In fact, the model already correctly identified two recently reported disease genes in a list of top candidates.
These findings suggest a promising path forward where machine learning could one day make diagnostic decisions in a way that’s comparable to experts, only more efficiently. What’s especially exciting is AI-MARRVEL could have the potential for solving rare disease cases, including those that have remained a mystery for years. The hope is that, by combining the power of AI tools together with the latest sequencing data in the years to come, doctors will be able to get faster diagnoses to many more people with rare genetic disorders.
References:
[1] Mao, D, et al. AI-MARRVEL: A Knowledge-Driven Artificial Intelligence for Molecular Diagnostics of Mendelian Disorders. NEJM AI. DOI: 10.1056/AIoa2300009 (2024).
[2] Liu, P, et al. Reanalysis of Clinical Exome Sequencing Data. N Engl J Med. DOI: 10.1056/NEJMc1812033 (2019).
NIH Support: NIH Common Fund, National Human Genome Research Institute, National Institute of Neurological Disorders and Stroke, Eunice Kennedy Shriver National Institute of Child Health and Human Development
AI Tool Using Single-Cell Data Has Promise for Optimally Matching Cancer Drugs to Patients
Posted on May 9th, 2024 by Dr. Monica M. Bertagnolli

Credit: Donny Bliss/NIH, NicoElNino/Adobe Stock
Precision oncology, in which doctors choose cancer treatment options based on the underlying molecular or genetic signature of individual tumors, has come a long way. The Food and Drug Administration has approved a growing number of tests that look for specific genetic changes that drive cancer growth to match patients to targeted treatments. The NCI-MATCH trial, supported by the National Cancer Institute, in which participants with advanced or rare cancer had their tumors sequenced in search of genetic changes that matched them to a treatment, has also suggested benefits for guiding treatment through genetic sequencing. But there remains a need to better predict treatment responses for people with cancer.
A promising approach is to analyze a tumor’s RNA in addition to its DNA. The idea is to not only better understand underlying genetic changes, but also learn how those changes impact gene activity as measured by RNA sequencing data. A recent study introduces an artificial intelligence (AI)-driven tool, dubbed PERCEPTION (PERsonalized single-Cell Expression-based Planning for Treatments In ONcology), developed by an NIH-led team to do just this.1 This proof-of-concept study, published in Nature Cancer, shows that it’s possible to fine-tune predictions of a patient’s treatment responses from bulk RNA data by zeroing in on what’s happening inside single cells.
One of the challenges in relying on bulk data from tumor samples is they typically include mixtures of like cells known as clones. Because different clones may respond differently to specific drugs, averaging what’s happening in cells across a particular patient’s tumor may not provide a clear picture of how that cancer will respond to a drug. Being able to capture gene activity patterns all the way down to the single-cell level might be a better way to target clones with specific alterations and therefore see better drug responses, but so far, single-cell gene expression data haven’t been widely available.
To explore the potential of single-cell RNA data, a team led by Eytan Ruppin and Alejandro Schäffer at NCI’s Cancer Data Science Laboratory at the NIH Clinical Center in Bethesda, MD, and Sanju Sinha, now at Sanford Burnham Prebys in San Diego, used a technique called transfer learning to train an AI model to predict drug responses. They first used existing bulk RNA sequencing data and then fine-tuned those models using single-cell RNA sequencing data from cell lines and large-scale drug screens. All told, they built AI models for 44 drugs approved by the FDA.
They found that PERCEPTION predicts the success of targeted treatments against cell lines with an accuracy reflected by an AUC score of about 0.8. AUC measures how well a model can distinguish between drug-sensitive and drug-resistant cell lines, with 0.5 being no better than a random guess and 1.0 being perfect accuracy. While there’s room for improvement, the findings show that PERCEPTION works better than earlier methods. The results also extended to single drugs and combination treatments in cultured cells and in cells isolated from patient tumors.
But would the tool accurately predict responses to treatments for patients? To find out, the researchers used their models to predict treatment responses based on clinical trial data for 41 patients with multiple myeloma treated with a combination of four drugs and 33 patients with breast cancer treated with a combination of two drugs. Their findings showed that the model could successfully predict treatment responses in patients, again with an AUC score of about 0.8.
Interestingly, their research shows that having just one clone in a tumor that is resistant to a particular drug is enough to thwart a response to that drug. As a result, the clone with the worst response in a tumor will best explain a person’s overall treatment response. Further study revealed that the model could also predict the development of resistance to treatment in published data from 24 people treated with targeted therapies for non-small cell lung cancer.
The researchers note that the accuracy of their technique will only improve as single-cell RNA sequencing data becomes more widely available for more patients with additional cancer types. To aid in this endeavor, they’ve developed a research website and guide to enable other researchers to use PERCEPTION to build AI models that predict treatment responses. Their hope is, as these findings suggest, that single-cell RNA sequencing data could one day help doctors more precisely match patients to their optimal cancer treatments.
Reference:
[1] Sinha S, et al. PERCEPTION predicts patient response and resistance to treatment using single-cell transcriptomics of their tumors. Nature Cancer. DOI 10.1038/s43018-024-00756-7 (2024).
NIH Support: National Cancer Institute
Machine Learning Study Offers Clues to Why Some People Have Rheumatoid Arthritis Pain Without Inflammation
Posted on May 2nd, 2024 by Dr. Monica M. Bertagnolli

Credit: Yakobchuk Olena/Adobe Stock
About 1.5 million adults in the U.S. are living with rheumatoid arthritis (RA), an autoimmune disease in which the immune system attacks joint tissue, causing inflammation, swelling, and pain. Treatments often do a good job fighting inflammation to slow or even stop joint damage and ease pain. But this doesn’t work for everyone. Many people with RA don’t find pain relief, even with the strongest anti-inflammatory, disease-modifying therapies now available.
Why is that? A new study supported in part by NIH and reported in Science Translational Medicine has an intriguing answer.1 The findings suggest that in some people with RA, the joint lining may direct the growth of pain-sensing neurons to cause pain in the absence of inflammation. This discovery, made possible with the help of machine learning, suggests potential new ways to treat this painful disease.
The findings come from a team led by Fei Wang, Weill Cornell Medicine, New York City, and Dana E. Orange, Rockefeller University, New York City. They were inspired by recent studies showing that RA pain and inflammation don’t always go together. In fact, people with RA who have limited inflammation in some cases report just as much pain as those who have extreme inflammation. As a result, they also tend to get less benefit from anti-inflammatory drugs.
To find out why, the researchers studied the soft tissue, or synovium, lining the spaces of the joints from people with this less common form of RA. They were in search of underlying differences in gene activity to explain the pain without inflammation. They knew it wouldn’t be easy, given the variation in the way people experience and report pain and the limited availability of surgically removed tissue samples. To overcome those roadblocks, they developed a machine learning approach that could pinpoint pain-associated patterns of gene activity in the complex data that would otherwise be too difficult to discern.
Their RNA sequencing analysis turned up 815 genes that were expressed at unusually high levels in the joint tissue of 22 people who had RA pain with low inflammation. They also confirmed this same pattern of gene activity in a second group of patients with early untreated RA and little inflammation.
The researchers went on to find that this pattern was clearest in fibroblast cells (a major cell type of the synovium) which provide the structural framework of the joint space, but become a key driver of inflammation and joint damage in RA. Those fibroblasts also expressed a gene that encodes a protein called netrin-4, which is related to a family of proteins that play a role in the growth of neurons. It led them to wonder whether the joint tissue might be producing substances that could alter pain-sensing nerves to cause pain.
To learn more, they turned to studies in mice. They found that fluid collected from joint fibroblast cell cultures and netrin-4 made mouse neurons sprout new branches carrying pain receptors in the lab. The findings suggested that the RA joint lining was indeed producing substances that could lead to the growth of pain-sensing neurons.
To see if this might play a role in people with RA and little inflammation, they looked closely at the joints. Those images revealed an abundance of blood vessels that could nurture tissue growth. Those vessels were also surrounded by pain-sensing nerve fibers extending toward the joint lining in places where there was an abnormal amount of tissue growth.
The researchers think this process explains why painful, arthritic joints sometimes feel squishy and swollen even when they aren’t inflamed. In future studies, they want to learn more about which sensory neurons are specifically affected, noting that there are about a dozen different types. While much more study is needed, their goal is to find promising new ways to treat RA by targeting this underlying process, giving more people with RA much needed pain relief.
Reference:
[1] Bai Z, et al. Synovial fibroblast gene expression is associated with sensory nerve growth and pain in rheumatoid arthritis. Science Translational Medicine. DOI: 10.1126/scitranslmed.adk3506 (2024).
NIH Support: National Institute of Arthritis and Musculoskeletal and Skin Diseases
Posted In: Health, News, Science
Tags: autoimmune disease, basic research, gene expression, genetics, inflammation, machine learning, neurons, pain, rheumatoid arthritis, treatment