evolution – NIH Director's Blog (original) (raw)
New Technology Opens Evolutionary Window into Brain Development
Posted on October 28th, 2021 by Dr. Francis Collins

One of the great mysteries in biology is how we humans ended up with such large, complex brains. In search of clues, researchers have spent years studying the protein-coding genes activated during neurodevelopment. But some answers may also be hiding in non-coding regions of the human genome, where sequences called regulatory elements increase or decrease the activity of genes.
A fascinating example involves a type of regulatory element called a human accelerated region (HAR). Although “human” is part of this element’s name, it turns out that the genomes of all vertebrates—not just humans—contain the DNA segments now designated as HARs.
In most organisms, HARs show a relatively low rate of mutation, which means these regulatory elements have been highly conserved across species throughout evolutionary time [1]. The big exception is Homo sapiens, in which HARs have exhibited a much higher rate of mutations.
The accelerated rate of HARs mutations observed in humans suggest that, over the course of very long periods of time, these genomic changes might have provided our species with some sort of evolutionary advantage. What might that be? Many have speculated the advantage might involve the brain because HARs are often associated with genes involved in neurodevelopment. Now, in a paper published in the journal Neuron, an NIH-supported team confirms that’s indeed the case [2].
In the new work, researchers found that about half of the HARs in the human genome influence the activity, or expression, of protein-coding genes in neural cells and tissues during the brain’s development [3]. The researchers say their study—the most comprehensive to date of the 3,171 HARs in the human genome—firmly establishes that this type of regulatory element helps to drive patterns of neurodevelopmental gene activity specific to humans.
Yet to be determined is precisely how HARs affect the development of the human brain. The quest to uncover these details will no doubt shed new light on fundamental questions about the brain, its billions of neurons, and their trillions of interconnections. For example, why does human neural development span decades, longer than the life spans of most primates and other mammals? Answering such questions could also reveal new clues into a range of cognitive and behavioral disorders. In fact, early research has already made tentative links between HARs and neurodevelopmental conditions such as autism spectrum disorder and schizophrenia [3].
The latest work was led by Kelly Girskis, Andrew Stergachis, and Ellen DeGennaro, all of whom were in the lab of Christopher Walsh while working on the project. An NIH grantee, Walsh is director of the Allen Discovery Center for Brain Evolution at Boston Children’s Hospital and Harvard Medical School, which is supported by the Paul G. Allen Foundation Frontiers Group, and is an Investigator of the Howard Hughes Medical Institute.
Though HARs have been studied since 2006, one of the big challenges in systematically assessing them has been technological. The average length of a HAR is about 269 bases of DNA, but current technologies for assessing function can only easily analyze DNA molecules that span 150 bases or less.
Ryan Doan, who was then in the Walsh Lab, and his colleagues solved the problem by creating a new machine called CaptureMPRA. (MPRA is short for “massively parallel reporter assays.”) This technological advance cleverly barcodes HARs and, more importantly, makes it possible to analyze HARs up to about 500 bases in length.
Using CaptureMPRA technology in tandem with cell culture studies, researchers rolled up their sleeves and conducted comprehensive, full-sequence analyses of more than 3,000 HARs. In their initial studies, primarily in neural cells, they found nearly half of human HARs are active to drive gene expression in cell culture. Of those, 42 percent proved to have increased ability to enhance gene expression compared to their orthologues, or counterparts, in chimpanzees.
Next, the team integrated these data with an existing epigenetic dataset derived from developing human brain cells, as well as additional datasets generated from sorted brain cell types. They found that many HARs appeared to have the ability to increase the activity of protein-coding genes, while a smaller—but very significant—subset of the HARs appeared to be enhancing gene expression specifically in neural progenitor cells, which are responsible for making various neural cell types.
The data suggest that as the human HAR sequences mutated and diverged from other mammals, they increased their ability to enhance or sometimes suppress the activity of certain genes in neural cells. To illustrate this point, the researchers focused on two HARs that appear to interact specifically with a gene referred to as R17. This gene can have highly variable gene expression patterns not only in different human cell types, but also in cells from other vertebrates and non-vertebrates.
In the human cerebral cortex, the outermost part of the brain that’s responsible for complex behaviors, R17 is expressed only in neural progenitor cells and only at specific time points. The researchers found that R17 slows the progression of neural progenitor cells through the cell cycle. That might seem strange, given the billions of neurons that need to be made in the cortex. But it’s consistent with the biology. In the human, it takes more than 130 days for the cortex to complete development, compared to about seven days in the mouse.
Clearly, to learn more about how the human brain evolved, researchers will need to look for clues in many parts of the genome at once, including its non-coding regions. To help researchers navigate this challenging terrain, the Walsh team has created an online resource displaying their comprehensive HAR data. It will appear soon, under the name HAR Hub, on the University of California Santa Cruz Genome Browser.
References:
[1] An RNA gene expressed during cortical development evolved rapidly in humans. Pollard KS, Salama SR, Lambert N, Lambot MA, Coppens S, Pedersen JS, Katzman S, King B, Onodera C, Siepel A, Kern AD, Dehay C, Igel H, Ares M Jr, Vanderhaeghen P, Haussler D. Nature. 2006 Sep 14;443(7108):167-72.
[2] Rewiring of human neurodevelopmental gene regulatory programs by human accelerated regions. Girskis KM, Stergachis AB, DeGennaro EM, Doan RN, Qian X, Johnson MB, Wang PP, Sejourne GM, Nagy MA, Pollina EA, Sousa AMM, Shin T, Kenny CJ, Scotellaro JL, Debo BM, Gonzalez DM, Rento LM, Yeh RC, Song JHT, Beaudin M, Fan J, Kharchenko PV, Sestan N, Greenberg ME, Walsh CA. Neuron. 2021 Aug 25:S0896-6273(21)00580-8.
[3] Mutations in human accelerated regions disrupt cognition and social behavior. Doan RN, Bae BI, Cubelos B, Chang C, Hossain AA, Al-Saad S, Mukaddes NM, Oner O, Al-Saffar M, Balkhy S, Gascon GG; Homozygosity Mapping Consortium for Autism, Nieto M, Walsh CA. Cell. 2016 Oct 6;167(2):341-354.
Links:
Christopher Walsh Laboratory (Boston Children’s Hospital and Harvard Medical School)
The Paul G. Allen Foundation Frontiers Group (Seattle)
NIH Support: National Institute of Neurological Disorders and Stroke; National Institute of Mental Health; National Institute of General Medical Sciences; National Cancer Institute
Posted In: News
Tags: Autism Spectrum Disorder, brain, brain development, CaptureMPRA, cerebral cortex, epigenetics, evolution, evolutionary biology, genomics, HAR, human accelerated region, human evolution, mutation rate, mutations, neural progenitor cells, neurodevelopment, neurons, non-coding DNA, R17, regulatory elements, schizophrenia, UC Santa Cruz Genome Browser
Understanding Neuronal Diversity in the Spinal Cord
Posted on May 20th, 2021 by Dr. Francis Collins
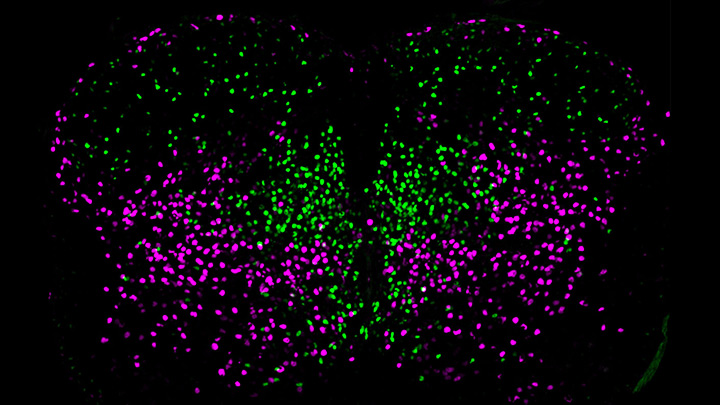
Credit: Salk Institute, La Jolla, CA
The spinal cord, as a key part of our body’s central nervous system, contains millions of neurons that actively convey sensory and motor (movement) information to and from the brain. Scientists have long sorted these spinal neurons into what they call “cardinal” classes, a classification system based primarily on the developmental origin of each nerve cell. Now, by taking advantage of the power of single-cell genetic analysis, they’re finding that spinal neurons are more diverse than once thought.
This image helps to visualize the story. Each dot represents the nucleus of a spinal neuron in a mouse; humans have a very similar arrangement. Most of these neurons are involved in the regulation of motor control, but they also differ in important ways. Some are involved in local connections (green), such as those that signal outward to a limb and prompt us to pull away reflexively when we touch painful stimuli, such as a hot frying pan. Others are involved in long-range connections (magenta), relaying commands across spinal segments and even upward to the brain. These enable us, for example, to swing our arms while running to help maintain balance.
It turns out that these two types of spinal neurons also have distinctive genetic signatures. That’s why researchers could label them here in different colors and tell them apart. Being able to distinguish more precisely among spinal neurons will prove useful in identifying precisely which ones are affected by a spinal cord injury or neurodegenerative disease, key information in learning to engineer new tissue to heal the damage.
This image comes from a study, published recently in the journal Science, conducted by an NIH-supported team led by Samuel Pfaff, Salk Institute for Biological Studies, La Jolla, CA. Pfaff and his colleagues, including Peter Osseward and Marito Hayashi, realized that the various classes and subtypes of neurons in our spines arose over the course of evolutionary time. They reasoned that the most-primitive original neurons would have gradually evolved subtypes with more specialized and diverse capabilities. They thought they could infer this evolutionary history by looking for conserved and then distinct, specialized gene-expression signatures in the different neural subtypes.
The researchers turned to single-cell RNA sequencing technologies to look for important similarities and differences in the genes expressed in nearly 7,000 mouse spinal neurons. They then used this vast collection of genomic data to group the neurons into closely related clusters, in much the same way that scientists might group related organisms into an evolutionary family tree based on careful study of their DNA.
The first major gene expression pattern they saw divided the spinal neurons into two types: sensory-related and motor-related. This suggested to them that one of the first steps in spinal cord evolution may have been a division of labor of spinal neurons into those two fundamentally important roles.
Further analyses divided the sensory-related neurons into excitatory neurons, which make neurons more likely to fire; and inhibitory neurons, which dampen neural firing. Then, the researchers zoomed in on motor-related neurons and found something unexpected. They discovered the cells fell into two distinct molecular groups based on whether they had long-range or short-range connections in the body. Researches were even more surprised when further study showed that those distinct connectivity signatures were shared across cardinal classes.
All of this means that, while previously scientists had to use many different genetic tags to narrow in on a particular type of neuron, they can now do it with just two: a previously known tag for cardinal class and the newly discovered genetic tag for long-range vs. short-range connections.
Not only is this newfound ability a great boon to basic neuroscientists, it also could prove useful for translational and clinical researchers trying to determine which specific neurons are affected by a spinal injury or disease. Eventually, it may even point the way to strategies for regrowing just the right set of neurons to repair serious neurologic problems. It’s a vivid reminder that fundamental discoveries, such as this one, often can lead to unexpected and important breakthroughs with potential to make a real difference in people’s lives.
Reference:
[1] Conserved genetic signatures parcellate cardinal spinal neuron classes into local and projection subsets. Osseward PJ 2nd, Amin ND, Moore JD, Temple BA, Barriga BK, Bachmann LC, Beltran F Jr, Gullo M, Clark RC, Driscoll SP, Pfaff SL, Hayashi M. Science. 2021 Apr 23;372(6540):385-393.
Links:
What Are the Parts of the Nervous System? (Eunice Kennedy Shriver National Institute of Child Health and Human Development/NIH)
Spinal Cord Injury (National Institute of Neurological Disorders and Stroke/NIH)
Samuel Pfaff (Salk Institute, La Jolla, CA)
NIH Support: National Institute of Mental Health; National Institute of Neurological Disorders and Stroke; Eunice Kennedy Shriver National Institute of Child Health and Human Development
Posted In: Snapshots of Life
Tags: basic research, brain, cardinal spinal classes, central nervous system, evolution, gene expression, genetic signatures, genomics, motor neurons, neurodegenerative disorders, neurons, neuroscience, peripheral nervous system, regenerative medicine, RNA sequencing, sensory neurons, single cell analysis, spinal cord, spinal cord injuries, spinal neurons, tissue engineering
An Evolutionary Guide to New Immunotherapies
Posted on April 29th, 2021 by Dr. Francis Collins

Credit: Dave Titensor, University of Utah, Salt Lake City
One of the best ways to learn how something works is to understand how it’s built. How it came to be. That’s true not only if you play a guitar or repair motorcycle engines, but also if you study the biological systems that make life possible. Evolutionary studies, comparing the development of these systems across animals and organisms, are now leading to many unexpected biological discoveries and promising possibilities for preventing and treating human disease.
While there are many evolutionary questions to ask, Brenda Bass, a distinguished biochemist at University of Utah, Salt Lake City, has set her sights on a particularly profound one: How has innate immunity evolved through the millennia in all living things, including humans? Innate immunity is the immune system’s frontline defense, the first responders that take control of an emerging infectious situation and, if needed, signal for backup.
Exploring the millennia for clues about innate immunity takes a special team, and Bass has assembled a talented one. It includes her Utah colleague Nels Elde, a geneticist; immunologist Dan Stetson, University of Washington, Seattle; and biochemist Jane Jackman, Ohio State University, Columbus.
With a 2020 NIH Director’s Transformative Research Award, this hard-working team will embark on studies looking back at 450 million years of evolution: the point in time when animals diverged to develop very distinct methods of innate immune defense [1]. The team members hope to uncover new possibilities encoded in the innate immune system, especially those that might be latent but still workable. The researchers will then explore whether their finds can be repurposed not only to boost our body’s natural response to external threats but also to internal threats like cancer.
Bass brings a unique perspective to the project. As a postdoc in the 1980s, she stumbled upon a whole new class of enzymes, called ADARs, that edit RNA [2]. Their function was mysterious at the time. It turns out that ADARs specifically edit a molecule called double-stranded RNA (dsRNA). When viruses infect cells in animals, including humans, they make dsRNA, which the innate immune system detects as a sign that a cell has been invaded.
It also turns out that animal cells make their own dsRNA. Over the years, Bass and her lab have identified thousands of dsRNAs made in animal cells—in fact, a significant number of human genes produce dsRNA [3]. Also interesting, ADARs are crucial to marking our own dsRNA as “self” to avoid triggering an immune response when we don’t need it [4].
Bass and others have found that evolution has produced dramatic differences in the biochemical pathways powering the innate immune system. In vertebrate animals, dsRNA leads to release of the immune chemical interferon, a signaling pathway that invertebrate species don’t have. Instead, in response to detecting dsRNA from an invader, and repelling it, worms and other invertebrates trigger a gene-silencing pathway known as RNA interference, or RNAi.
With the new funding, Bass and team plan to mix and match immune strategies from simple and advanced species, across evolutionary time, to craft an entirely new set of immune tools to fight disease. The team will also build new types of targeted immunotherapies based on the principles of innate immunity. Current immunotherapies, which harness a person’s own immune system to fight disease, target infections, autoimmune disorders, and cancer. But they work through our second-line adaptive immune response, which is a biological system unique to vertebrates.
Bass and her team will first hunt for more molecules like ADARs: innate immune checkpoints, as they refer to them. The name comes from a functional resemblance to the better-known adaptive immune checkpoints PD-1 and CTLA-4, which sparked a revolution in cancer immunotherapy. The team will run several screens that sort molecules successful at activating innate immune responses—both in invertebrates and in mammals—hoping to identify a range of durable new immune switches that evolution skipped over but that might be repurposed today.
Another intriguing direction for this research stems from the observation that decreasing normal levels of ADARs in tumors kickstarts innate immune responses that kill cancer cells [5]. Along these lines, the scientists plan to test newly identified immune switches to look for novel ways to fight cancer where existing approaches have not worked.
Evolution is the founding principle for all of biology—organisms learn from what works to improve their ability to survive. In this case, research to re-examine such lessons and apply them for new uses may help transform bygone evolution into a therapeutic revolution!
References:
[1] Evolution of adaptive immunity from transposable elements combined with innate immune systems. Koonin EV, Krupovic M. Nat Rev Genet. 2015 Mar;16(3):184-192.
[2] A developmentally regulated activity that unwinds RNA duplexes. Bass BL, Weintraub H. Cell. 1987 Feb 27;48(4):607-613.
[3] Mapping the dsRNA World. Reich DP, Bass BL. Cold Spring Harb Perspect Biol. 2019 Mar 1;11(3):a035352.
[4] To protect and modify double-stranded RNA – the critical roles of ADARs in development, immunity and oncogenesis. Erdmann EA, Mahapatra A, Mukherjee P, Yang B, Hundley HA. Crit Rev Biochem Mol Biol. 2021 Feb;56(1):54-87.
[5] Loss of ADAR1 in tumours overcomes resistance to immune checkpoint blockade. Ishizuka JJ, Manguso RT, Cheruiyot CK, Bi K, Panda A, et al. Nature. 2019 Jan;565(7737):43-48.
Links:
Bass Lab (University of Utah, Salt Lake City)
Elde Lab (University of Utah)
Jackman Lab (Ohio State University, Columbus)
Stetson Lab (University of Washington, Seattle)
Bass/Elde/Jackman/Stetson Project Information (NIH RePORTER)
NIH Director’s Transformative Research Award Program (Common Fund)
NIH Support: Common Fund; National Cancer Institute
Posted In: Creative Minds
Tags: 2020 NIH Director's Transformative Research Award, acquired immunity, ADAR, autoimmune disorders, basic research, cancer, cancer immunotherapy, checkpoint inhibitors, CTLA-4, double-stranded RNA, dsRNA, evolution, evolutionary biology, immunity, immunotherapy, innate immunity, invertebrates, PD-1, RNA, RNA interference, RNAi, vertebrates
Tracking the Evolution of a ‘Variant of Concern’ in Brazil
Posted on April 27th, 2021 by Dr. Francis Collins

By last October, about three out of every four residents of Manaus, Brazil already had been infected with SARS-CoV-2, the virus that causes COVID-19 [1]. And yet, despite hopes of achieving “herd immunity” in this city of 2.2 million in the Amazon region, the virus came roaring back in late 2020 and early 2021 to cause a second wave of illness and death [2]. How is this possible?
The answer offers a lesson in viral evolution, especially when an infectious virus such as SARS-CoV-2 replicates and spreads through a population largely unchecked. In a recent study in the journal Science, researchers tied the city’s resurgence of SARS-CoV-2 to the emergence and rapid spread of a new SARS-CoV-2 “variant of concern” known as P.1 [3]. This variant carries a unique constellation of mutations that allow it not only to sneak past the human immune system and re-infect people, but also to be about twice as transmissible as earlier variants.
To understand how this is possible, consider that each time the coronavirus SARS-CoV-2 makes copies of itself in an infected person, there’s a chance a mistake will be made. Each mistake can produce a new variant that may go on to make more copies of itself. In most cases, those random errors are of little to no consequence. This is evolution in action.
But sometimes a spelling change can occur that benefits the virus. In the special case of patients with suppressed immune systems, the virus can have ample opportunity to accrue an unusually high number of mutations. Variants carrying beneficial mutations can make more copies of themselves than other variants, allowing them to build their numbers and spread to cause more infection.
At this advanced stage of the COVID-19 pandemic, such rapidly spreading new variants remain cause for serious concern. That includes variants such as B.1.351, which originated in South Africa; B.1.1.7 which emerged in the United Kingdom; and now P.1 from Manaus, Brazil.
In the new study, Nuno Faria and Samir Bhatt, Imperial College London, U.K., and Ester Cerdeira Sabino, Universidade de Sao Paulo, Brazil, and their colleagues sequenced SARS-CoV-2 genomes from 184 patient samples collected in Manaus in November and December 2020. The research was conducted under the auspices of the Brazil-UK Centre for Arbovirus Discovery, Diagnosis, Genomics and Epidemiology (CADDE), a project focused on viral genomics and epidemiology for public health.
Those genomic data revealed the P.1 variant had acquired 17 new mutations. Ten were in the spike protein, which is the segment of the virus that binds onto human cells and the target of current COVID-19 vaccines. In fact, the new work reveals that three of these spike protein mutations make it easier for the P.1 spike to bind the human ACE2 receptor, which is SARS-CoV-2’s preferred entry point.
The first P.1 variant case was detected by genomic surveillance on December 6, 2020, after which it spread rapidly. Through further evolutionary analysis, the team estimates that P.1 must have emerged, undetected for a brief time, in mid-November 2020.
To understand better how the P.1 variant led to such an explosion of new COVID-19 cases, the researchers developed a mathematical model that integrated the genomic data with mortality data. The model suggests that P.1 may be 1.7 to 2.4 times more transmissible than earlier variants. They also estimate that a person previously infected with a variant other than P.1 will have only 54 percent to 79 percent protection against a subsequent infection with P.1.
The researchers also observed an increase in mortality following the emergence of the P.1 variant. However, it’s not yet clear if that’s an indication P.1 is inherently more deadly than earlier variants. It’s possible the increased mortality is related primarily to the extra stress on the healthcare system in Manaus from treating so many people with COVID-19.
These findings are yet another reminder of the importance of genomic surveillance and international data sharing for detecting and characterizing emerging SARS-CoV-2 variants quickly. It’s worth noting that at about the same time this variant was detected in Brazil, it also was reported in four individuals who had traveled to Brazil from Japan. The P.1 variant continues to spread rapidly across Brazil. It has also been detected in more than 37 countries [4], including the United States, where it now accounts for more than 1 percent of new cases [5].
No doubt you are wondering what this means for vaccines, such as the Pfizer and Moderna mRNA vaccines, that have been used to immunize (at least one dose) over 140 million people in the United States. Here the news is encouraging. Serum from individuals who received the Pfizer vaccine had titers of neutralizing antibodies that were only slightly reduced for P.1 compared to the original SARS-CoV-2 virus [6]. Therefore, the vaccine is predicted to be highly protective. This is another example of a vaccine providing more protection than a natural infection.
The United States has made truly remarkable progress in combating COVID-19, but we must heed this lesson from Manaus: this terrible pandemic isn’t over just yet. While the P.1 variant remains at low levels here for now, the “U.K. variant” B.1.1.7 continues to spread rapidly and now is the most prevalent variant circulating in the U.S., accounting for 44 percent of new cases [6]. Fortunately, the mRNA vaccines also work well against B.1.1.7.
We must continue to do absolutely everything possible, individually and collectively, to prevent these new SARS-CoV-2 variants from slowing or even canceling the progress made over the last year. We need to remain vigilant for just a while longer, while encouraging our friends, neighbors, and loved ones to get vaccinated.
References:
[1] Three-quarters attack rate of SARS-CoV-2 in the Brazilian Amazon during a largely unmitigated epidemic. Buss, L. F., C. A. Prete, Jr., C. M. M. Abrahim, A. C. Dye, V. H. Nascimento, N. R. Faria and E. C. Sabino et al. (2021). Science 371(6526): 288-292.
[2] Resurgence of COVID-19 in Manaus, Brazil, despite high seroprevalence. Sabino EC, Buss LF, Carvalho MPS, Prete Jr CCA, Crispim MAE, Fraiji NA, Pereira RHM, Paraga KV, Peixoto PS, Kraemer MUG, Oikawa MJ, Salomon T, Cucunuba ZM, Castro MC, Santos AAAS, Nascimento VH, Pereira HS, Ferguson NM, Pybus OG, Kucharski A, Busch MP, Dye C, Faria NR Lancet. 2021 Feb 6;397(10273):452-455.
[3] Genomics and epidemiology of the P.1 SARS-CoV-2 lineage in Manaus, Brazil. Faria NR, Mellan TA, Whittaker C, Claro IM, Fraiji NA, Carvalho MDPSS, Pybus OG, Flaxman S, Bhatt S, Sabino EC et al. Science. 2021 Apr 14:eabh2644.
[4] GRINCH Global Report Investigating novel coronavirus haplotypes. PANGO Lineages.
[5] COVID Data Tracker. Variant Proportions. Centers for Disease Control and Prevention.
[6] Antibody evasion by the P.1 strain of SARS-CoV-2. Dejnirattisai W, Zhou D, Supasa P, Liu C, Mongkolsapaya J, Ren J, Stuart DI, Screaton GR, et al. Cell. 2021 Mar 30:S0092-8674(21)00428-1.
Links:
COVID-19 Research (NIH)
Brazil-UK Centre for Arbovirus Discovery, Diagnosis, Genomics and Epidemiology (CADDE)
Nuno Faria (Imperial College, London, U.K.)
Samir Bhatt (Imperial College)
Ester Cerdeira Sabino (Universidade de Sao Paulo, Brazil)
NIH Support: National Institute of Allergy and Infectious Diseases
Posted In: News
Tags: ACE2, B.1.1.7, B.1.351, Brazil, CADDE, coronavirus, COVID-19, COVID-19 infections, COVID-19 variants, evolution, genomic epidemiology, genomic surveillance, genomics, herd immunity, Manaus, Moderna, mRNA vaccine, novel coronavirus, P.1, pandemic, Pfizer/BioNTech vaccine, public health, SARS-CoV-2, South Africa, spike protein, U.K. variant, vaccines, variant of concern, viral evolution
South Africa Study Shows Power of Genomic Surveillance Amid COVID-19 Pandemic
Posted on February 18th, 2021 by Dr. Francis Collins
Credit: iStock/Thomas Faull
Considerable research is underway around the world to monitor the spread of new variants of SARS-CoV-2, the coronavirus that causes COVID-19. That includes the variant B.1.351 (also known as 501Y.V2), which emerged in South Africa towards the end of 2020 [1, 2]. Public health officials in South Africa have been busy tracing the spread of this genomic variant and others across their country. And a new analysis of such data reveals that dozens of distinct coronavirus variants were already circulating in South Africa well before the appearance of B.1.351.
A study of more than 1,300 near-whole genome sequences of SARS-CoV-2, published recently in the journal Nature Medicine, shows there were in fact at least 42 SARS-CoV-2 variants spreading in South Africa within the pandemic’s first six months in that country [3]. Among them were 16 variants that had never before been described. Most of the single-letter changes carried by these variants didn’t change the virus in important ways and didn’t rise to significant frequency. But the findings come as another critical reminder of the value of genomic surveillance to track the spread of SARS-CoV-2 to identify any potentially worrisome new variants and to inform measures to get this devastating pandemic under control.
SARS-CoV-2 was first detected in South Africa on March 5, 2020, in a traveler returning from Italy. By November 2020, despite considerable efforts to slow the spread, more than 785,000 people in South Africa were infected, accounting for about half of all reported COVID-19 cases on the African continent.
Recognizing the importance of genomic surveillance, researchers led by Houriiyah Tegally and Tulio de Oliveira, University of KwaZulu-Natal, Durban, South Africa, wasted no time in producing 1,365 near-complete SARS-CoV-2 genomes by mid-September, near the end of the coronavirus’s first peak in the country. Those samples had been collected in hundreds of clinics over the course of the pandemic in eight of South Africa’s nine provinces, offering a broad picture of the spread and emergence of new variants across the country.
The data revealed three main variants, dubbed B.1.1.54, B.1.1.56, and C.1, that were responsible for 42 percent of all the infections in South Africa’s first wave. Of the 16 newly described variants, most carried single-letter changes that haven’t been identified in other countries.
The majority of changes were what scientists refer to as “synonymous,” meaning that they don’t change the structure or function of any of the virus’s essential proteins. The exception is the newly identified C.1, which includes 16 single-letter changes compared to the original sequence from Wuhan, China. One of those 16 changes swaps a single amino acid for another on SARS-CoV-2’s spike protein. That’s notable because the spike protein is a key target of antibodies and also is essential to the virus’s ability to infect human cells.
In fact, four of the most prevalent variants in South Africa all carry this same mutation. The researchers also saw three other changes that would alter the spike protein in different ways, although the significance of these for viral spread and our efforts to stop it isn’t yet clear.
Importantly, the data show that the bulk of introductions to South Africa happened early on, before lockdown and travel restrictions were implemented in late March. Subsequently, much of the spread within South Africa stemmed from hospital outbreaks. For example, an outbreak of the C.1 variant in the North West Province in April ultimately led this variant to become the most geographically widespread in South Africa by the end of August. Meanwhile, an earlier identified South African-specific variant, B.1.106, first identified in April, vanished altogether after outbreaks were controlled in KwaZulu-Natal Province, where the researchers reside.
Genomic surveillance has remarkable power for understanding the evolution of SARS-CoV-2 and tracking the dynamics of its transmission. Tegally and de Oliveira’s team notes that this type of intensive genomic surveillance now can be used on a large scale across Africa and around the world to identify new variants of SARS-CoV-2 and to develop timely measures to control the spread of the virus. They’re now working with the African CDC to expand genomic surveillance across Africa [4].
Such genomic surveillance was crucial in the subsequent identification of the B.1.351 variant in South Africa that we’ve been hearing so much about, with its potential to evade our current treatments and vaccines. By picking up on such concerning mutations early through genomic surveillance and understanding how the virus is spreading over time and space, the hope is we’ll be better informed and more adept in our efforts to get this pandemic under control.
References:
[1] Emerging SARS-CoV-2 variants. Centers for Disease Control and Prevention.
[2] Emergence and rapid spread of a new severe acute respiratory syndrome-related coronavirus 2 (SARS-CoV-2) lineage with multiple spike mutations in South Africa. Tegally H, Wilkinson E, Giovanetti M, Iranzadeh A, Bhiman J, Williamson C, de Oliveira T, et al. medRxiv 2020 Dec 22.
[3] Sixteen novel lineages of SARS-CoV-2 in South Africa. Tegally H, Wilkinson E, Lessells RJ, Giandhari J, Pillay S, Msomi N, Mlisana K, Bhiman JN, von Gottberg A, Walaza S, Fonseca V, Allam M, Ismail A, Glass AJ, Engelbrecht S, Van Zyl G, Preiser W, Williamson C, Petruccione F, Sigal A, Gazy I, Hardie D, Hsiao NY, Martin D, York D, Goedhals D, San EJ, Giovanetti M, Lourenço J, Alcantara LCJ, de Oliveira T. Nat Med. 2021 Feb 2.
[4] Accelerating genomics-based surveillance for COVID-19 response in Africa. Tessema SK, Inzaule SC, Christoffels A, Kebede Y, de Oliveira T, Ouma AEO, Happi CT, Nkengasong JN.Lancet Microbe. 2020 Aug 18.
Links:
COVID-19 Research (NIH)
Houriiyah Tegally (University of KwaZulu-Natal, Durban, South Africa)
Tulio de Oliveira (University of KwaZulu-Natal)
Posted In: News
Tags: Africa, B.1.1.54, B.1.1.56, B.1.106, B.1.351, C.1, coronavirus, COVID-19, evolution, genomic sequencing, genomic surveillance, novel coronavirus, pandemic, SARS-CoV-19 variants, SARS-CoV-2, SARS-CoV-2 transmission, South Africa, South African variant, spike protein, variants
Study Shows Genes Unique to Humans Tied to Bigger Brains
Posted on June 5th, 2018 by Dr. Francis Collins
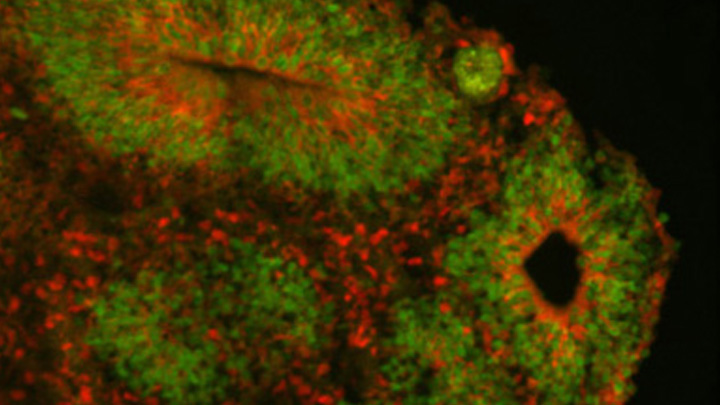
Caption: Cortical organoid, showing radial glial stem cells (green) and cortical neurons (red).
Credit: Sofie Salama, University of California, Santa Cruz
In seeking the biological answer to the question of what it means to be human, the brain’s cerebral cortex is a good place to start. This densely folded, outer layer of grey matter, which is vastly larger in Homo sapiens than in other primates, plays an essential role in human consciousness, language, and reasoning.
Now, an NIH-funded team has pinpointed a key set of genes—found only in humans—that may help explain why our species possesses such a large cerebral cortex. Experimental evidence shows these genes prolong the development of stem cells that generate neurons in the cerebral cortex, which in turn enables the human brain to produce more mature cortical neurons and, thus, build a bigger cerebral cortex than our fellow primates.
That sounds like a great advantage for humans! But there’s a downside. Researchers found the same genomic changes that facilitated the expansion of the human cortex may also render our species more susceptible to certain rare neurodevelopmental disorders.
Posted In: News
Tags: autism, Autism Spectrum Disorder, brain, cerebral cortex, cortical neurons, CRISPR/Cas9, DNA sequencing, duplication, evolution, gene-editing technology, genes, genomics, human genome, Human Genome Project, humans, macrocephaly, microcephaly, microdeletion, neurodevelopmental disorders, neurons, neuroscience, Notch, organoids, primates, radial glial stem cells, schizophrenia, signaling genes, stem cells
Finding Brain Circuits Tied to Alertness
Posted on November 14th, 2017 by Dr. Francis Collins
Everybody knows that it’s important to stay alert behind the wheel or while out walking on the bike path. But our ability to react appropriately to sudden dangers is influenced by whether we feel momentarily tired, distracted, or anxious. How is it that the brain can transition through such different states of consciousness while performing the same routine task, even as its basic structure and internal wiring remain unchanged?
A team of NIH-funded researchers may have found an important clue in zebrafish, a popular organism for studying how the brain works. Using a powerful new method that allowed them to find and track brain circuits tied to alertness, the researchers discovered that this mental state doesn’t work like an on/off switch. Rather, alertness involves several distinct brain circuits working together to bring the brain to attention. As shown in the video above that was taken at cellular resolution, different types of neurons (green) secrete different kinds of chemical messengers across the zebrafish brain to affect the transition to alertness. The messengers shown are: serotonin (red), acetylcholine (blue-green), and dopamine and norepinephrine (yellow).
What’s also fascinating is the researchers found that many of the same neuronal cell types and brain circuits are essential to alertness in zebrafish and mice, despite the two organisms being only distantly related. That suggests these circuits are conserved through evolution as an early fight-or-flight survival behavior essential to life, and they are therefore likely to be important for controlling alertness in people too. If correct, it would tell us where to look in the brain to learn about alertness not only while doing routine stuff but possibly for understanding dysfunctional brain states, ranging from depression to post-traumatic stress disorder (PTSD).
Posted In: Health, Science, technology
Tags: acetylcholine, alertness, brain, brain circuits, brain imaging, brain states, Danio rerio, depression, dopamine, evolution, evolutionary biology, locus coeruleus, mice, model organism, Multi-MAP, neurology, neuromodulation, neurotransmitter, norepinephrine, optogenetics, PTSD, serotonin, zebrafish
Happy New Year: Looking Back at 2016 Research Highlights
Posted on January 4th, 2017 by Dr. Francis Collins
 Happy New Year! While everyone was busy getting ready for the holidays, the journal Science announced its annual compendium of scientific Breakthroughs of the Year. If you missed it, the winner for 2016 was the detection of gravitational waves—tiny ripples in the fabric of spacetime created by the collision of two black holes 1.3 billion years ago! It’s an incredible discovery, and one that Albert Einstein predicted a century ago.
Happy New Year! While everyone was busy getting ready for the holidays, the journal Science announced its annual compendium of scientific Breakthroughs of the Year. If you missed it, the winner for 2016 was the detection of gravitational waves—tiny ripples in the fabric of spacetime created by the collision of two black holes 1.3 billion years ago! It’s an incredible discovery, and one that Albert Einstein predicted a century ago.
Among the nine other advances that made the first cut for Breakthrough of the Year, several involved the biomedical sciences. As I’ve done in previous years (here and here), I’ll kick off this New Year by taking a quick look of some of the breakthroughs that directly involved NIH support:
Tags: 2016, Africa, aging, All of Us, astronaut, atherosclerosis, Breakthroughs of 2016, Breakthroughs of the Year, chronic kidney disease, custom-designed proteins, designer proteins, DNA analysis, DNA sequencing, embryos, evolution, genomic analysis, genomics, hemagglutinin, human development, human embyos, human evolution, human migration, International Space Station, kidney dysfunction, longevity, nanopore sequencing, osteoarthritis, Out of Africa, portable laboratories, precision medicine, Precision Medicine Initiative, proteins, pulmonary fibrosis, Science's Breakthroughs of the Year, senescent cells, senolytic drugs, Simons Genome Diversity Project, universal flu vaccine, Zika vaccine
Talking Music and Science with Yo-Yo Ma
Posted on December 8th, 2016 by Dr. Francis Collins
It’s not every day that an amateur guitar picker gets to play a duet with an internationally renowned classical cellist. But that was my thrill this week as I joined Yo-Yo Ma in a creative interpretation of the traditional song, “How Can I Keep from Singing?” Our short jam session capped off Mr. Ma’s appearance as this year’s J. Edward Rall Cultural Lecture.
The event, which counts The Dalai Lama, Maya Angelou, and Atul Gawande among its distinguished alumni, this year took the form of a conversation on the intersection of music and science—and earned a standing ovation from a packed house of researchers, patients, and staff here on the National Institutes of Health (NIH) campus in Bethesda, MD.
Posted In: Science, Tribute, Video
Tags: brain, cello, cerebral cortex, chamber music, classical music, dopamine, evolution, How Can I Keep from Singing, J. Edward Rall Cultural Lecture, music, neuroscience, neuroscience of music, Science, world music, Yo-Yo Ma
Out of Africa: DNA Analysis Points to a Single Major Exodus
Posted on September 27th, 2016 by Dr. Francis Collins

Credit: NASA
If you go back far enough, the ancestors of all people trace to Africa. That much is clear. We are all Africans. But there’s been considerable room for debate about exactly when and how many times modern humans made their way out of Africa to take up residence in distant locations throughout the world. It’s also unclear what evolutionary or other factors might have driven our human ancestors to set off on such a perilous and uncertain journey (or journeys) in the first place.
By analyzing 787 newly sequenced complete human genomes representing more than 280 diverse and understudied populations, three new studies—two of which received NIH funding—now help to fill in some of those missing pages of our evolutionary history. The genomic evidence suggests that the earliest human inhabitants of Eurasia came from Africa and began to diverge genetically at least 50,000 years ago. While the new studies differ somewhat in their conclusions, the findings also lend support to the notion that our modern human ancestors dispersed out of Africa primarily in a single migratory event. If an earlier and ultimately failed voyage occurred, it left little trace in the genomes of people alive today.
Posted In: Science
Tags: Aborigines, Africa, anthropology, Australia, DNA, DNA analysis, Euroasia, evolution, evolutionary biology, genome, genomic history, genomics, human ancestry, human evolution, human genetics, human migration, hunter gatherer, molecular anthropology, New Guinea, Out of Africa, Papuans, population genetics, Sahul, Simons Genome Diversity Project