GLUT2, glucose sensing and glucose homeostasis (original) (raw)
Introduction
Glucose is a major source of metabolic energy for most cells of the body and is of critical importance for the brain. Because of its role in cellular energetics and brain function the concentration of glucose in the blood is constantly monitored to adapt cellular and whole body physiology to maintain glycaemia at ∼5 mmol/l. Glucose detection mechanisms are multiple and the term ‘glucose sensing’, as used in this review, refers to any cellular or molecular mechanism whose activity is directly responsive to changes in physiological glucose concentrations. Examples of such glucose-sensing systems include the taste bud cells. These express the sweet taste receptors (T1R2/T1R3) [1], which detect the presence of sugar in the food and initiate nervous responses to control the cephalic phase of insulin secretion, as well as food preference [2, 3]. In hepatocytes, high glucose concentrations activate the transcription factor known as carbohydrate response element-binding protein (ChREBP), a key inducer of glycolysis and lipogenesis [4]. ChREBP is also expressed in beta cells where it regulates expression of glycolytic and lipogenic genes and contributes to glucose-stimulated beta cell proliferation [5–7]. A major function of pancreatic beta cells is to secrete insulin in response to a rise in circulating glucose concentrations, and the signalling pathway involved depends on glucose metabolism [8]. In pancreatic alpha cells, an as yet incompletely defined mechanism, which probably also involves glucose metabolism, links hypoglycaemia to the release of glucagon [9]. Importantly, glucose-responsive neurons have been identified in several brain regions involved in the homeostatic control of feeding, energy expenditure and glucose homeostasis [10, 11]. However, glucose detection in the nervous system is not restricted to neurons, but also involves astrocytes and tanycytes.
A large variety of glucose-sensing mechanisms have been described in the nervous system [12, 13]. These include membrane depolarisation following glucose uptake by the electrogenic Na+-coupled glucose transporter (SGLT)1 [14]; the binding of glucose to SGLT3, which does not transport glucose but can induce a depolarisation signal [15]; the use of beta cell-like glucose-sensing systems, or glucose binding proteins linked to a signalling pathway that closes K+ leak channels, as described in orexin neurons [16]. The glucose transporter GLUT2 is also associated with various glucose-sensing cells, which are activated by either hypoglycaemia or hyperglycaemia, as further discussed below.
GLUT2 structure and function
The family of facilitated diffusion glucose transporters (GLUTs, encoded by the SLC2A genes) comprises 14 isoforms. These have distinct amino acid sequences, substrate specificities, kinetic properties and tissue and cellular localisations [17]. GLUT2 has a uniquely low affinity for glucose (K m ∼ 17 mmol/l), and can also use mannose, galactose and fructose as low affinity substrates; it has, however, a high affinity for glucosamine (K m ∼ 0.8 mmol/l) [18]. GLUT2 is present in the basolateral membrane of enterocytes and of epithelial cells from the kidney proximal convoluted tubule where it functions in the second step of transepithelial glucose transport, a process initiated by the apical sodium–glucose co-transporters SGLT1 or SGLT2. As discussed further below, knocking out Glut2 in the mouse revealed that it is dispensable for glucose absorption in the intestine but required for glucose reabsorption in the kidney. GLUT2 is also the major glucose transporter in the plasma membrane of hepatocytes, and studies on _Glut2_-null mice indicated that it is required for glucose uptake but dispensable for glucose output. In rodent pancreatic beta cells, GLUT2 is the major glucose transporter and its genetic inactivation suppresses glucose uptake and glucose-stimulated insulin secretion (GSIS). GLUT2 is also expressed in the central nervous system.
Thus, although the function of GLUT2 is merely to catalyse the passive transport of glucose across plasma membranes, studies of knockout mice have revealed that this transport activity is important for the control of cellular mechanisms impinging on gene expression, regulation of intracellular metabolic pathways, and induction of hormonal and neuronal signals which together form the basis of an integrated inter-organ communication system to control glucose homeostasis (Fig. 1, Table 1). It is important to note that the mouse phenotypes described below are only observed when both Glut2 alleles are inactivated and that heterozygous knockout mice are indistinguishable from control mice. This is further evidence that glucose transport by GLUT2 is not a rate-controlling step in any of the signalling pathways studied.
Fig. 1
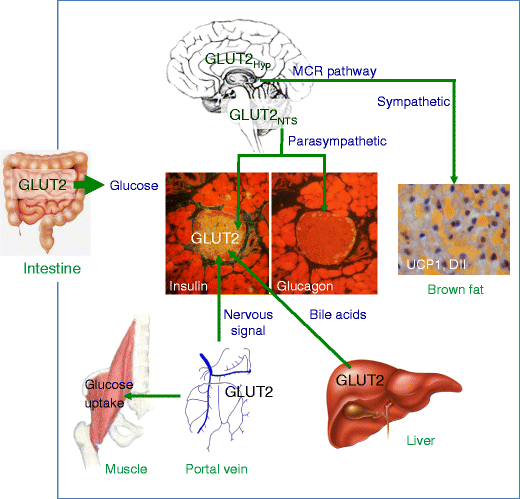
Sites of GLUT2-dependent glucose transport and sensing in inter-organ communication. GLUT2 is present in the basolateral membrane of intestinal epithelial cells, where it allows entry of glucose in the organism. GLUT2 is present in glucose-sensing cells of the hepatoportal vein region. Activation of these glucose sensors induces a nervous signal that stimulates first-phase insulin secretion, as well as other physiological responses. In hepatocytes, GLUT2 is required for equilibration of cytoplasmic glucose with the extracellular milieu in order to allow appropriate regulation of glucose-sensitive genes during the fed-to-fast transition. These glucose-regulated events modulate bile acid production, which can increase beta cell glucose competence through activation of farnesoid X receptor, a nuclear hormone receptor. GLUT2 is expressed in beta cells; in rodents this transporter is essential for normal glucose-stimulated insulin secretion. GLUT2 is expressed in the brain, in the nucleus tractus solitarius (NTS), in few hypothalamic (Hyp) cells, and in neurons that send projections to neuropeptide Y and pro-opiomelanocortin neurons of the melanocortin (MCR) pathway. Nervous system GLUT2-dependent glucose-sensing cells control the regulation by glucose by both parasympathetic and sympathetic nervous systems. Glucose-regulated parasympathetic activity controls beta cell proliferation during the weaning period and glucose-stimulated insulin secretion in adult mice; it also controls glucagon secretion in response to hypoglycaemia. GLUT2-dependent glucose-sensing cells control the sensitivity to leptin of the melanocortin pathway and sympathetic activity in brown fat to control uncoupling protein 1 and deiodinase-2 expression and thermoregulation
Table 1 GLUT2 involvement in cellular functions and integrated physiology
GLUT2 in pancreatic islet beta cells
The first indication that GLUT2 was associated with glucose sensing was the observation that it is the major glucose transporter in rodent islet beta cells [19, 20]. Because of its high K m for glucose and high transport capacity, it allows for fast equilibration of extracellular glucose with that present inside the cell. This gives glucose unrestricted access to glucokinase, the enzyme that catalyses the rate-controlling step in GSIS [21]. In diabetic mice and rats, impaired GSIS is associated with markedly reduced expression of GLUT2 [22, 23]. The loss of GLUT2 expression is regulated at both the mRNA and protein levels. Elevated NEFA suppresses Glut2 mRNA expression [24] and, in diabetes, the reduced Glut2 expression is only observed in beta cells and not in liver; this is due to a tissue-specific activity of the transcription factor hepatic nuclear factor-1α (HNF-1α) [25]. The rate of GLUT2 protein degradation can be increased by elevated glucocorticoid levels [26] and, in mice fed a high-fat diet, a reduction in the expression of _N_-acetylglucosamine transferase-4, a Golgi apparatus enzyme involved in N-linked glycosylation, leads to structural alterations in the transporter N-glycan. This suppresses the interaction of GLUT2 with galectin-9, a lectin that normally anchors the transporter at the cell surface, and reduces GSIS [27–29]. Transplantation of islets from diabetic mice in control animals normalised GLUT2 levels and restored GSIS [30, 31]. Thus, the above data suggest that reduced glucose uptake may become limiting in the control of GSIS in the diabetic state.
Whether GLUT2 has unique properties that are required for glucose sensing has been tested in genetic rescue experiments. Glut2 −/− mice die at around the weaning period [32] and transgenic expression of Glut1 or Glut2 in their beta cells (RIP_Glut1;Glut2_ −/− or RIP_Glut2;Glut2_ −/− mice) restores normal GSIS and glucose-stimulated insulin biosynthesis [33]; a minimum of ∼20% of the normal GLUT2 expression level was found to be sufficient to normalise GSIS. Thus, the total transport capacity rather than the specific isoform of glucose transporter expressed at the beta cell surface is necessary for normal GSIS. This is consistent with the observation that human beta cells express several glucose transporters: the low K m glucose transporters GLUT1 and GLUT3, and GLUT2, which is expressed at a relatively low level (see also the discussion of Fanconi–Bickel patients in the ‘GLUT2 in human pathophysiology’ section).
A role for GLUT2 in glucose sensing, independent from its transport activity, has been proposed based on the study of liver cells transfected with a cDNA construct encoding a green fluorescent protein fused to the large intracellular loop of GLUT2 (amino acids 237–301). These studies showed that the fusion protein is translocated into the nucleus when the cells are exposed to high glucose concentrations, leading to impaired induction of glucose-regulated genes such as Glut2 and that encoding l-pyruvate kinase [34]. In subsequent experiments, transgenic mice were produced that expressed the same fusion protein under the control of the actin promoter to achieve ubiquitous expression of the transgene [35]. These mice showed a large diversity of phenotypes associated with deregulation of glucose metabolism [35] and feeding behaviour [36]. However, even though the transgenic mice show a clear and intriguing phenotype, this is induced by overexpression of a soluble fusion protein that can migrate to the nucleus. It is not clear how these observations can support a role for GLUT2, a membrane protein that has so far never been reported to be present in the nucleus, as a glucose sensor that directly controls biological processes in the nucleus. Nevertheless, a role for GLUT2 in transducing an intracellular glucose-dependent signal, independent from its glucose transport activity, cannot be excluded.
GLUT2 is required for GSIS in rodent islets; GLUT2 is dispensable in human beta cells since they also express GLUT1 and GLUT3.
GLUT2 and the hepatoportal glucose sensor
The finding that GLUT2 is required for glucose signalling in beta cells prompted the assessment of an association of the transporter with other glucose-sensing systems. A first indication that this was the case came from studies assessing the regulation of glucagon secretion in response to hypo- or hyperglycaemia. Initial observations with Glut2 knockout mice revealed strong hyperglucagonaemia [32]. Using RIP_Glut1;Glut2_ −/− mice it was observed that the stimulation of glucagon secretion by hypoglycaemia or its suppression by hyperglycaemia were lost [37]. As GLUT2 is not expressed in pancreatic alpha cells, this indicated that glucose sensing by extra-pancreatic GLUT2-expressing cells were involved in glucagon secretion. In addition, since the hyperglucagonaemia of the knockout mice could be normalised by ganglionic blockade, this suggested that these extrapancreatic GLUT2-dependent glucose sensors control autonomic nervous activity.
The hepatoportal vein is richly innervated by vagal afferents [38, 39], which are activated following intestinal glucose absorption [40, 41], leading to several physiological responses: suppression of glucagon secretion [42, 43], feeding termination [44] and stimulation of glucose uptake by some muscles and brown fat [45]. Studies of RIP_Glut1;Glut2_ −/− mice showed that the signal generated by glucose infusion into the portal vein and that stimulates glucose utilisation by peripheral tissues required GLUT2 expression [45, 46]. Studies of mice with inactivation of GLUT4 or of the insulin receptor specifically in muscles indicated that the signal generated by the hepatoportal sensor was distinct from insulin, required GLUT4 expression, and was suppressed by expression of a dominant negative form of 5′ AMP-activated protein kinase in muscle [47]. Inactivation of this sensor also suppressed first-phase insulin secretion following i.p. glucose injection; however, insulin secretion from perifused islets isolated from the same mice displayed normal first-phase insulin secretion [48]. This therefore shows that in vivo, the control of first-phase insulin secretion depends on an indirect signal activated by glucose entering the hepatoportal vein area, and probably transmitted to beta cells by the nervous system.
Hepatic GLUT2, bile acids and beta cell glucose competence
GLUT2 is the major glucose transporter of hepatocytes in rodents and humans [19, 49]. The generally accepted role of this transporter is to take up glucose during the absorptive phase and to release it in the blood during fasting. In hepatocytes, glucose can be stored as glycogen, degraded through the glycolytic pathway or converted to fatty acids by the lipogenic pathway. The release of glucose in the circulation follows degradation of glycogen or gluconeogenesis.
Glucose also modifies cellular metabolism by allosteric and transcriptional regulation. Glucose inhibits glycogenolysis by activating glycogen phosphorylase kinase, which inactivates glycogen phosphorylase, the rate-limiting enzyme in glycogen breakdown; glucose 6-phosphate, on the other hand, is an allosteric activator of glycogen synthase [50]. Thus, increased absorption and phosphorylation of glucose stimulates glycogen deposition by allosteric stimulation of glycogen synthesis and inhibition of its degradation. A fraction of the absorbed glucose is metabolised through the pentose shunt pathway, which produces xylulose 5-phosphate, an activator of the protein phosphatase that dephosphorylates ChREBP [4, 51]. This induces translocation of ChREBP into the nucleus, where it activates the transcription of the gene encoding l-pyruvate kinase, a key regulator of glycolysis, and lipogenic genes such as those encoding acetyl-CoA carboxylase and fatty acid synthase. These allosteric and transcriptional responses are important in the absorptive phase to control glucose uptake, storage and conversion into fat.
Investigation of the impact of Glut2 inactivation in hepatocytes led to three surprising findings. First, Glut2 inactivation in hepatocytes of adult mice using a tamoxifen-dependent recombination system (LG2KO mice) [52] suppressed hepatic glucose uptake but did not modify glucose homeostasis in the fed, fasted, or fasted and refed states. Measurement of tissue glucose uptake in LG2KO mice showed increased uptake in the tibialis anterior and extensor digitorum longus muscles. Thus, glucose fluxes were redirected to other tissues to preserve normal control of glycaemia in the absorptive state.
Second, hepatic glucose production was quantitatively normal in fasted LG2KO mice. This was surprising because the last steps in glucose release from hepatocytes are classically described as involving the transport of glucose 6-phosphate into the endoplasmic reticulum where the active site of the glucose 6-phosphatase is located. Glucose 6-phosphate is hydrolysed into glucose and phosphate, and glucose is transported back into the cytosol to be exported from the cell by GLUT2. Our data indicated that absence of GLUT2 suppressed glucose uptake but, unexpectedly, did not impair glucose output. This revealed the existence of a second pathway for glucose output that does not require transport of glucose back into the cytoplasm. Investigation of this export mechanism suggested that glucose release was dependent on a membrane traffic-based pathway originating in the endoplasmic reticulum [53–55]. Although not yet fully characterised, this second pathway could offer a major regulatory advantage. Indeed, compartmentalising glucose away from the cytoplasm when hepatic glucose production is needed, i.e. in the fasted state, would prevent the redirection of glucose to glycogen storage, glycolysis and lipogenesis. In the fasted state, GLUT2 expression in the plasma membrane is nevertheless required for normal hepatic glucose metabolism. Indeed, since part of the glucose generated from glucose 6-phosphate in the endoplasmic reticulum can be transported back into the cytoplasm, it needs to be equilibrated with the extracellular glucose. Preventing this equilibration in LG2KO hepatocytes leads to the accumulation of glucose and glucose 6-phosphate in the cytoplasm, which causes a permanent increase in nuclear ChREBP and a paradoxical upregulation of the gene encoding l-pyruvate kinase and the lipogenic genes in the fasted state [52, 53]. The presence of GLUT2 in hepatocytes is therefore required for the normal control by glucose of gene expression under physiological conditions.
The third surprise was that, even though Glut2 gene inactivation in adult mouse liver had no immediate impact on whole body glucose homeostasis, LG2KO mice progressively developed glucose intolerance as a result of a defect in GSIS [52]. Thus, altered glucose metabolism in hepatocytes induced progressive beta cell dysfunction. However, beta cell mass and insulin content were normal. To identify possible signals from the liver that control beta cell function, we performed gene expression analysis of livers of fasted and fed control and LG2KO mice. Gene set enrichment analysis revealed that, besides modifications of glycolytic and lipogenic gene expression, most cholesterol biosynthesis genes were downregulated. However, this was not associated with changes in plasma cholesterol in either the fed or fasted stated. As cholesterol is also the precursor of bile acids, we measured their concentrations in faeces and plasma. Faecal bile acids were reduced by 30% in LG2KO mice as compared with control mice, and the plasma bile acid concentration was also lower in the mutant mice. When islets from control mice were incubated for 24 h in the presence of bile acids, GSIS measured in subsequent incubations was strongly increased, and this effect was not seen in islets from mice with genetic inactivation of Fxr, which encodes a nuclear receptor for bile acids. An acute role of bile acids on GSIS, through _Fxr_-dependent effects on KATP channels has also been reported [56]. Thus, bile acids may form a functional link between hepatic glucose metabolism and beta cell function.
In hepatocytes, GLUT2 is required for glucose uptake but is dispensable for glucose output, which takes place through a separate, probably membrane traffic-based release pathway. GLUT2 is nevertheless required to equilibrate intracellular and extracellular glucose concentrations to ensure proper control of glucose-sensitive gene expression in the liver. Failure of this process, as seen in LG2KO mice, reveals a new role of hepatocytes in linking glucose sensing to the long-term preservation of beta cell glucose competence by a mechanism likely involving bile acids.
GLUT2 in the nervous system
GLUT2 is expressed in the central nervous system of humans [57, 58], rodents and zebrafish [59]. In rats, a careful immunohistochemical mapping of GLUT2 expression at the light and electron microscopy levels revealed its presence in most brain structures, and expression was found in neurons, astrocytes, endothelial cells and tanycytes, which are specialised astrocytes lining the lower part of the third ventricle [60–62]. In mice, using a genetic reporter system in which a fluorescent protein is expressed under the control of the Glut2 promoter (Glut2-Cre transgenic mice crossed with Rosa26tdTomato mice), the distribution of GLUT2 expressing cells in the brain was found to be very similar to that of the rat [63].
Glucose sensing, feeding and thermogenesis
Initial investigations of the role of rat brain GLUT2 in glucose homeostasis and feeding control used intracarotid [64] or intracerebroventricular (i.c.v.) [65] injections of specific antisense oligonucleotides to silence Glut2 expression. In the first study this led to reduced body weight with no change in feeding and a reduced insulin response to intracarotid glucose administration. In the second one, Glut2 silencing reduced feeding and body weight gain. Using RIP_Glut1;Glut2_ −/− mice the feeding response to i.c.v. (as well as to i.p.) glucose or 2-deoxy-d-glucose (2-DG) injections was lost and this was associated with impaired regulation of the orexinergic (neuropeptide Y [NPY], agouti-related peptide [AGRP]) and anorexigenic (pro-opiomelanocortin [POMC], cocaine and amphetamine regulated transcript [CART]) neuropeptides of the melanocortin pathway during the fast-to-refed transition or following i.c.v. glucose injection [66]. These data indicated that central GLUT2-dependent glucose sensing regulates feeding through control of the melanocortin pathway.
Thermoregulation in RIP_Glut1;Glut2_ −/− mice is abnormal due to a defect in central glucose sensing. This was shown by the much lower hypothermic response induced by i.c.v. 2-DG injections in RIP_Glut1;Glut2_ −/− as compared with control mice [63]. This was secondary to reduced activation of brown fat uncoupling protein 1 and deiodinase-2 expression by the sympathetic nervous system and was associated with fasting-induced torpor in the _Glut2-_null mice. Abnormal sympathetic tone was caused, at least in part, by the reduced sensitivity to leptin of arcuate nucleus neurons, further supporting a role for central GLUT2-dependent glucose sensing in the control of the melanocortin pathway. Analysis of mice that express the red fluorescent protein tdTomato in _Glut2_-expressing cells revealed that NPY and POMC neurons do not express this transporter. However, they are in contact with nerve terminals from _Glut2_-expressing neurons, which probably have their soma outside the arcuate nucleus, suggesting an indirect control by glucose of the melanocortin pathway.
GLUT2 is required for glucose sensing by cells of the central nervous system. Absence of central GLUT2-dependent glucose sensing impairs the normal sensitivity of the melanocortin pathway to leptin and is associated with impaired control of feeding and thermoregulation.
Nervous glucose sensing and the regulation of islet cell mass and function
Analysis of mice with genetic inactivation of Glut2 in the nervous system (NG2KO mice), generated by crossing Glut2 flox mice with a strain of nestin-Cre mice that do not show any metabolic or growth abnormalities [67], provided a new view of the role of nervous glucose sensing in autonomic regulation of the endocrine pancreas [68]. Direct recording of nerve activity showed that the firing rate of the parasympathetic nerve was lower in NG2KO mice than in control mice and was not increased by i.p. glucose injections. Sympathetic nerve activity was similar in the basal state in NG2KO and control mice but was not suppressed by glucose in NG2KO mice. Thus, Glut2 is required for normal control by glucose of both branches of the autonomic nervous system.
Absence of parasympathetic nerve regulation by glucose had two consequences. First, the proliferation rate of beta cells during the weaning period was reduced by half in NG2KO mice vs control mice and this lower rate of proliferation led to a 30% reduction in adult beta cell mass. However, the difference in proliferation rates was not seen when the mice were weaned on a high-fat, carbohydrate-free diet. Thus, during the weaning period, when there is a high rate of beta cell proliferation [69] and when the diet changes from a lipid-rich milk to a carbohydrate-rich chow [70], GLUT2-dependent nervous glucose sensing and parasympathetic activity play a critical role in stimulating beta cell proliferation to achieve normal adult beta cell mass.
The second consequence of the lack of glucose regulation of the autonomic nervous system was a loss of first-phase insulin secretion. In vivo, this early secretion response depends on nervous glucose sensing, in particular activation of the hepatoportal glucose sensor. This response was lost in NG2KO mice, whereas first-phase insulin secretion from isolated and perifused islets from young mice was normal. Thus, the defect in achieving normal adult beta cell mass together with the loss of first-phase insulin secretion led to the progressive development of glucose intolerance. This became evident at around 24 weeks of age and was caused by a defect in GSIS observed in vivo as well as in isolated islets. These physiological defects were accelerated by high-fat-diet feeding and were also associated with the development of hyperglucagonaemia.
Nervous GLUT2-dependent glucose sensing is required for the normal control by glucose of the sympathetic and parasympathetic nervous activity. Absence of this sensing mechanism prevents normal beta cell expansion in the postnatal period and achievement of normal adult beta cell mass. Over time this leads to development of glucose intolerance due to a defect in GSIS.
Optogenetic control of _Glut2_-expressing neurons to stimulate glucagon secretion
In order to better define the role of _Glut2_-expressing neurons in glucose regulation, we studied a small group of such cells present in the nucleus tractus solitarius (NTS). This brainstem structure is at the crossroad of afferent signals coming from different segments of the absorptive track, it can directly sense changes in nutrient and hormone concentrations, and the neurons forming this structure send projections not only to neighbouring nuclei such as the area postrema (AP) and the dorsal motor nucleus of the vagus (also referred to as the DMNX or DMV) but also to various forebrain regions [71–73]. The DMNX is of particular interest since it is formed by the somas of the vagus nerve neurons and NTS neurons send numerous projections to the DMNX to control vagal activity [74].
Mice expressing a fluorescent protein in _Glut2_-expressing neurons (Glut2Cre;Rosa26tdTomato) were used to prepare acute brainstem slices for patch-clamp analysis. NTS _Glut2_-expressing neurons were shown to be activated by hypoglycaemia by a signalling pathway that involves glucose metabolism, stimulation of AMP kinase and the control of K+ leak channels [75]. The Glut2-Cre mice were then crossed with Rosa26ChR2 [76] mice to express channelrhodopsin-2, a light-sensitive cation channel [77], in _Glut2_-expressing neurons. Patch-clamp analysis demonstrated that pulses of blue light could induce firing of the NTS _Glut2_-expressing neurons. Stimulation by light in living mice with simultaneous recording of vagal activity showed that activation of the NTS _Glut2_-expressing neurons increased vagal nerve firing and this led to a strong stimulation of glucagon secretion [75]. Thus, _Glut2_-expressing neurons from the NTS are part of a neuronal circuit that links hypoglycaemia detection to the counter-regulatory response. These observations are also in agreement with the fact that when hypoglycaemia develops, vagal activity is increased first and sympathetic activity increases only at deeper hypoglycaemic levels [78]. They are also relevant in the context of the insulin treatment of type 1 and type 2 diabetes. Indeed, insulin-treated patients are at risk of developing hypoglycaemia, a risk that increases following antecedent hypoglycaemic episodes [79]. This condition, referred to as hypoglycaemia-associated autonomic failure, is due to impaired hypoglycaemia detection. The identification of an NTS glucose-sensing system involved in glucagon secretion provides a new pathway to investigate the molecular mechanisms of counter-regulation control and defects in these.
_Glut2_-expressing neurons of the NTS are glucose-inhibited (GI) neurons that are activated by hypoglycaemia and control vagal nerve activity and glucagon secretion.
GLUT2 in human pathophysiology
GLUT2 mutations
The role of GLUT2 in human physiology and pathogenic processes can be inferred from genetic studies. First, inactivating mutations in the GLUT2 gene cause Fanconi–Bickel syndrome, a condition associated with hepatomegaly, growth retardation and renal Fanconi syndrome [80, 81]. The liver and kidney phenotypes of the Fanconi–Bickel patients are very similar to those of Glut2 knockout mice [82]. There is a similar increase in liver weight and increased glycogen accumulation, but a preserved hyperglycaemic response to glucagon injection, indicating that glycogen degradation can be followed by hepatic glucose release in the absence of GLUT2, both in humans and mice. Patients with Fanconi–Bickel syndrome have aminoaciduria, hyperphosphaturia and hypercalciuria, which are also observed in the _Glut2_-null mice [83]. In the mouse, exaggerated secretion of phosphate is linked to the suppressed expression in the proximal tubule of the type 2 sodium–phosphate co-transporter (NPT2C). Thus, defects in glucose reabsorption in the proximal tubule leads to secondary changes in other transporters expression, which may explain the observed renal syndrome.
Impaired GSIS in adult Fanconi–Bickel patients have only been reported in a few cases [84–86], but inactivating mutations in GLUT2 have been found to cause transient neonatal diabetes mellitus [87]. As this condition disappears after approximately 18 months, this indicates that there is a transient requirement for GLUT2 for the control of insulin secretion in the first months of life. However, transient neonatal diabetes was not reported in all Fanconi–Bickel patients. This may be because such cases went undiagnosed. Alternatively, defects caused by GLUT2 mutations may be compensated for by the expression of other genes. For instance, human beta cells express the low K m glucose transporters GLUT1 and GLUT3 in addition to GLUT2, thereby providing multiple means of efficiently taking up glucose to ensure that glucokinase remains the rate-controlling step in GSIS.
Another, attractive possible explanation for the impact of the mutation is that GLUT2 expression in the nervous system has similar functions in humans as in mice. If this were the case, impaired GLUT2 expression in glucose-sensing cells of the nervous system would prevent the normal expansion of beta cell mass in the postnatal period and prevent first-phase insulin secretion, leading to hyperglycaemia. Glycaemia is, however, normalised at a later stage of development, when other signals, independent from autonomic nervous activity, are activated to increase beta cell mass, leading to a sufficient insulin secretion capacity.
Interestingly, a role for GLUT2 expression in the control of food preference has been suggested by studies of two cohorts of individuals with the common Thr110Ile variant of GLUT2 [88]; these individuals show a significant preference for high-sugar-containing food. A similar preference for glucose solutions has been observed in cohorts of NG2KO mice (G. Labouèbe, B. Thorens, unpublished observations). However, when expressed in Xenopus oocytes the GLUT2 Thr110Ile variant is normally transported to the cell surface and its glucose transport activity is normal [89]. Thus, in humans, the genetic mutation that codes for the Thr110Ile variant may also be involved in mechanisms regulating GLUT2 expression at a pre-translational level; alternatively, it may suppress a putative signalling mechanism that is distinct from GLUT2 sugar transport activity and is dependent on Thr110.
Inactivating mutations in GLUT2 in humans cause Fanconi–Bickel syndrome, characterised by hepatomegaly, growth retardation and renal syndrome. Patients with Fanconi–Bickel syndrome have been observed to have transient neonatal diabetes mellitus, indicating a link between GLUT2 and insulin secretion in human neonates.
Genome-wide association studies
Genome-wide association studies have found that variants in GLUT2 (including the Thr110Ile variant encoded by the rs5400 single nucleotide polymorphism) are associated with impaired fasting glucose [90, 91], type 2 diabetes and an increased risk of transition from impaired fasting glycaemia to diabetes [90, 92–94]. When GLUT2 SNPs are investigated in association with other phenotypes, such as intensity of exercise, the presence of the major allele predicts that low physical activity increases the risk of developing type 2 diabetes by about threefold as compared with high physical activity; the presence of the minor allele has no predictive value [93]. A search for genetic regions associated with plasma lipid profiles and adjusted for diet and physical activity found that the GLUT2 locus has a major influence on serum cholesterol levels [95]. Another study found that out of 46 genetic variants examined, only the minor risk allele of GLUT2 was significantly associated with a risk of cardiovascular diseases [96].
These genetic studies therefore indicate that GLUT2 is involved in regulatory mechanisms that control impaired fasting glucose, the risk to transition to type 2 diabetes, preference for sugar-containing foods, as well as cholesterol levels and risk of cardiovascular disease. Because GLUT2 is only one of the glucose transporters present in beta cells, it is unlikely that slight changes in expression could have an impact on insulin secretion. Studies in mice showed that Glut2 expression in the nervous system controls glucose-regulated autonomic nervous activity, which could impact liver, adipocytes and heart function. It is tempting to speculate that GLUT2 has a dominant role in glucose sensing in the nervous system in humans and that its deregulation precedes the development of type 2 diabetes.
Genome-wide association studies have reported that GLUT2 variants are associated with fasting hyperglycaemia and an increased risk of type 2 diabetes.
Summary and perspectives
Studies of the facilitated diffusion glucose transporters have revealed a multitude of novel regulatory systems controlling glucose homeostasis [17, 97]. The investigations of GLUT2 function reported here shed light on some unique regulatory mechanisms, in particular related to blood glucose monitoring and the control of pancreatic hormone secretion, activity of the autonomic nervous system, and feeding and thermoregulation. The results of mouse physiological studies and human genetic-physiological studies to date suggest that, in humans, the association of GLUT2 mutations with transient neonatal diabetes, fasting hyperglycaemia, and risk of type 2 diabetes may be best explained by defects in neuronal glucose sensing, which induce loss of first-phase insulin secretion, deregulated feeding behaviour, hyperglucagonaemia, and progressive development of glucose intolerance and impaired GSIS. This suggests that loss of nervous glucose sensing may actually precede the establishment of the alpha and beta cell defects that are characteristic of overt type 2 diabetes. Thus, it would be highly interesting to test the loss of nervous glucose sensing in humans. The investigations could include assessment of the cephalic phase of insulin secretion, which relies on nervous glucose sensing. If this response shows a conserved pattern in healthy individuals, a decreased response may indicate a prediabetic state. Designing specific clinical investigations to test this hypothesis could be an important goal of future research.
A still unresolved question is why is a high K m glucose transporter required in glucose-sensing cells in the brain when the parenchymal glucose concentration is approximately one-third of that in the blood [11], i.e. much lower than the K m of GLUT2 for glucose? And why is GLUT2 associated with both GI and glucose-excited (GE) neurons? Related to the first question, it is still unknown whether the high K m for glucose represents an advantage for glucose sensing in the brain or whether GLUT2 has signalling properties that are independent of its transport capacity. Concerning the role of GLUT2 in both GI and GE neurons, we previously proposed a model to explain the observation that plasma glucagon levels in _Glut2_-null mice are elevated in the basal state and no longer increased by hypoglycaemia or suppressed by hyperglycaemia [37]. This model proposes that during hypoglycaemia, GI neurons generate a positive signal to stimulate autonomic nervous activity and glucagon secretion; in the absence of GLUT2 this positive signal can no longer be suppressed when euglycaemia is restored. On the other hand, when glycaemia increases GE, neurons generate an inhibitory signal that suppresses autonomic activation of alpha cells; in the absence of GLUT2, this signal cannot be generated. Thus, Glut2 knockout leads to permanently elevated stimulation of glucagon secretion [98]. Our studies on NTS _Glut2_-expressing neurons have shown that these are indeed activated by hypoglycaemia to control parasympathetic activity and glucagon secretion [75]. On the other hand, _Glut2_-expressing neurons of the basolateral medulla are activated by glucose injections as revealed by c-fos immunostaining [63]. Whether they are GE neurons that negatively control glucagon secretion will, however, require direct analysis using electrophysiological and optogenetic techniques. These studies will be important to further validate our hypothesis that GE and GI _Glut2_-expressing neurons functionally interact to fine-tune autonomic nervous activity and glucagon secretion. The identification of these neurons will also pave the way to a new understanding of the mechanisms leading to hypoglycaemia-associated autonomic failure.
Finally, because knocking out Glut2 in the nervous system leads to suppressed regulation of parasympathetic activity by glucose and impaired control of beta cell mass and function [68], it will be important to identify the glucose-sensing neurons that control this autonomic activity. Identification of these glucose sensing cells may lead to alternative ways of improving insulin secretion capacity in diabetic patients.
References
- Chandrashekar J, Hoon MA, Ryba NJ, Zuker CS (2006) The receptors and cells for mammalian taste. Nature 444:288–294
Article CAS PubMed Google Scholar - de Araujo IE, Oliveira-Maia AJ, Sotnikova TD et al (2008) Food reward in the absence of taste receptor signaling. Neuron 57:930–941
Article PubMed Google Scholar - Berthoud HR, Bereiter DA, Trimble ER, Siegel EG, Jeanrenaud B (1981) Cephalic phase, reflex insulin secretion. Neuroanatomical and physiological characterization. Diabetologia 20(Suppl):393–401
Article CAS PubMed Google Scholar - Dentin R, Denechaud PD, Benhamed F, Girard J, Postic C (2006) Hepatic gene regulation by glucose and polyunsaturated fatty acids: a role for ChREBP. J Nutr 136:1145–1149
CAS PubMed Google Scholar - Metukuri MR, Zhang P, Basantani MK et al (2012) ChREBP mediates glucose-stimulated pancreatic beta-cell proliferation. Diabetes 61:2004–2015
Article CAS PubMed Central PubMed Google Scholar - Iizuka K, Bruick RK, Liang G, Horton JD, Uyeda K (2004) Deficiency of carbohydrate response element-binding protein (ChREBP) reduces lipogenesis as well as glycolysis. Proc Natl Acad Sci U S A 101:7281–7286
Article CAS PubMed Central PubMed Google Scholar - Wang H, Wollheim CB (2002) ChREBP rather than USF2 regulates glucose stimulation of endogenous L-pyruvate kinase expression in insulin-secreting cells. J Biol Chem 277:32746–32752
Article CAS PubMed Google Scholar - Prentki M, Matschinsky FM, Madiraju SR (2013) Metabolic signaling in fuel-induced insulin secretion. Cell Metab 18:162–185
Article CAS PubMed Google Scholar - Zhang Q, Ramracheya R, Lahmann C et al (2013) Role of KATP channels in glucose-regulated glucagon secretion and impaired counterregulation in type 2 diabetes. Cell Metab 18:871–882
Article CAS PubMed Central PubMed Google Scholar - Marty N, Dallaporta M, Thorens B (2007) Brain glucose sensing, counterregulation, and energy homeostasis. Physiology (Bethesda) 22:241–251
Article CAS Google Scholar - Routh VH (2002) Glucose-sensing neurons: are they physiologically relevant? Physiol Behav 76:403–413
Article CAS PubMed Google Scholar - Thorens B (2012) Sensing of glucose in the brain. Handb Exp Pharmacol 209:277–294
Article CAS PubMed Google Scholar - Gonzalez JA, Reimann F, Burdakov D (2009) Dissociation between sensing and metabolism of glucose in sugar sensing neurones. J Physiol 587:41–48
Article CAS PubMed Central PubMed Google Scholar - O’Malley D, Reimann F, Simpson AK, Gribble FM (2006) Sodium-coupled glucose cotransporters contribute to hypothalamic glucose sensing. Diabetes 55:3381–3386
Article PubMed Central PubMed Google Scholar - Diez-Sampedro A, Hirayama BA, Osswald C et al (2003) A glucose sensor hiding in a family of transporters. Proc Natl Acad Sci U S A 100:11753–11758
Article CAS PubMed Central PubMed Google Scholar - Gonzalez JA, Jensen LT, Fugger L, Burdakov D (2008) Metabolism-independent sugar sensing in central orexin neurons. Diabetes 57:2569–2576
Article CAS PubMed Central PubMed Google Scholar - Mueckler M, Thorens B (2013) The SLC2 (GLUT) family of membrane transporters. Mol Asp Med 34:121–138
Article CAS Google Scholar - Uldry M, Ibberson M, Hosokawa M, Thorens B (2002) GLUT2 is a high affinity glucosamine transporter. FEBS Lett 524:199–203
Article CAS PubMed Google Scholar - Thorens B, Sarkar HK, Kaback HR, Lodish HF (1988) Cloning and functional expression in bacteria of a novel glucose transporter present in liver, intestine, kidney, and beta-pancreatic islet cells. Cell 55:281–290
Article CAS PubMed Google Scholar - Johnson JH, Newgard CB, Milburn JL, Lodish HF, Thorens B (1990) The high K m glucose transporter of islets of Langerhans is functionally similar to the low affinity transporter of liver and has identical primary sequence. J Biol Chem 265:6548–6551
CAS PubMed Google Scholar - Matschinsky FM (1996) A lesson in metabolic regulation inspired by the glucokinase sensor paradigm. Diabetes 45:223–241
Article CAS PubMed Google Scholar - Thorens B, Weir GC, Leahy JL, Lodish HF, Bonner-Weir S (1990) Reduced expression of the liver/beta-cell glucose transporter isoform in glucose-insensitive pancreatic beta cells of diabetic rats. Proc Natl Acad Sci U S A 87:6492–6496
Article CAS PubMed Central PubMed Google Scholar - Orci L, Unger RH, Ravazzola M et al (1990) Reduced b-cell glucose transporter in new onset diabetic BB rats. J Clin Investig 86:1615–1622
Article CAS PubMed Central PubMed Google Scholar - Gremlich S, Bonny C, Waeber G, Thorens B (1997) Fatty acids decrease IDX-1 expression in rat pancreatic islets and reduce GLUT2, glucokinase, insulin, and somatostatin levels. J Biol Chem 272:30261–30269
Article CAS PubMed Google Scholar - Parrizas M, Maestro MA, Boj SF et al (2001) Hepatic nuclear factor 1-alpha directs nucleosomal hyperacetylation to its tissue-specific transcriptional targets. Mol Cell Biol 21:3234–3243
Article CAS PubMed Central PubMed Google Scholar - Gremlich S, Roduit R, Thorens B (1997) Dexamethasone induces posttranslational degradation of GLUT2 and inhibition of insulin secretion in isolated pancreatic beta cells. Comparison with the effects of fatty acids. J Biol Chem 272:3216–3222
Article CAS PubMed Google Scholar - Ohtsubo K, Takamatsu S, Gao C, Korekane H, Kurosawa TM, Taniguchi N (2013) N-Glycosylation modulates the membrane sub-domain distribution and activity of glucose transporter 2 in pancreatic beta cells. Biochem Biophys Res Commun 434:346–351
Article CAS PubMed Google Scholar - Ohtsubo K, Chen MZ, Olefsky JM, Marth JD (2011) Pathway to diabetes through attenuation of pancreatic beta cell glycosylation and glucose transport. Nat Med 17:1067–1075
Article CAS PubMed Central PubMed Google Scholar - Ohtsubo K, Takamatsu S, Minowa MT, Yoshida A, Takeuchi M, Marth JD (2005) Dietary and genetic control of glucose transporter 2 glycosylation promotes insulin secretion in suppressing diabetes. Cell 123:1307–1321
Article CAS PubMed Google Scholar - Thorens B, Wu Y-J, Leahy JL, Weir GC (1992) The loss of GLUT2 expression by glucose-unresponsive beta cells of db/db mice is reversible and is induced by the diabetic environment. J Clin Investig 90:77–85
Article CAS PubMed Central PubMed Google Scholar - Ogawa Y, Noma Y, Davalli AM et al (1995) Loss of glucose-induced insulin secretion and GLUT2 expression in transplanted beta-cells. Diabetes 44:75–79
Article CAS PubMed Google Scholar - Guillam M-T, Hümmler E, Schaerer E et al (1997) Early diabetes and abnormal postnatal pancreatic islet development in mice lacking GLUT2. Nat Genet 17:327–330
Article CAS PubMed Google Scholar - Thorens B, Guillam MT, Beermann F, Burcelin R, Jaquet M (2000) Transgenic reexpression of GLUT1 or GLUT2 in pancreatic beta cells rescues GLUT2-null mice from early death and restores normal glucose-stimulated insulin secretion. J Biol Chem 275:23751–23758
Article CAS PubMed Google Scholar - Guillemain G, Loizeau M, Pincon-Raymond M, Girard J, Leturque A (2000) The large intracytoplasmic loop of the glucose transporter GLUT2 is involved in glucose signaling in hepatic cells. J Cell Sci 113:841–847
CAS PubMed Google Scholar - Stolarczyk E, Le Gall M, Even P et al (2007) Loss of sugar detection by GLUT2 affects glucose homeostasis in mice. PLoS One 2:e1288
Article PubMed Central PubMed Google Scholar - Stolarczyk E, Guissard C, Michau A et al (2010) Detection of extracellular glucose by GLUT2 contributes to hypothalamic control of food intake. Am J Physiol Endocrinol Metab 298:E1078–E1087
Article CAS PubMed Google Scholar - Burcelin R, Thorens B (2001) Evidence that extrapancreatic GLUT2-dependent glucose sensors control glucagon secretion. Diabetes 50:1282–1289
Article CAS PubMed Google Scholar - Berthoud H-R, Neuhuber WL (2000) Functional and chemical anatomy of the afferent vagal system. Auton Neurosci Basic Clin 85:1–17
Article CAS Google Scholar - Berthoud H-R, Kressel M, Neuhuber WL (1992) An anterograde tracing study of the vagal innervation of rat liver, portal vein and biliary system. Anat Embryol 186:431–442
Article CAS PubMed Google Scholar - Niijima A (1984) The effect of d-glucose on the firing rate of glucose-sensitive vagal afferents in the liver in comparison with the effect of 2-deoxy-d-glucose. J Auton Nerv Syst 10:279–285
Article CAS PubMed Google Scholar - Niijima A (1982) Glucose-sensitive afferent nerve fibres in the hepatic branch of the vagus nerve in the guinea-pig. J Physiol 332:315–323
Article CAS PubMed Central PubMed Google Scholar - Donovan CM, Hamilton-Wessler M, Halter JB, Bergman RN (1994) Primacy of liver glucosensors in the sympathetic response to progressive hypoglycemia. Proc Natl Acad Sci U S A 91:2863–2867
Article CAS PubMed Central PubMed Google Scholar - Hamilton-Wessler M, Bergman RN, Halter JB, Watanabe RM, Donovan CM (1994) The role of liver glucosensors in the integrated sympathetic response induced by deep hypoglycemia in dogs. Diabetes 43:1052–1060
Article CAS PubMed Google Scholar - Russek M (1963) Participation of hepatic glucoreceptors in the control of intake of food. Nature 197:79–80
Article CAS PubMed Google Scholar - Burcelin R, Dolci W, Thorens B (2000) Portal glucose infusion in the mouse induces hypoglycemia. Evidence that the hepatoportal glucose sensor stimulates glucose utilization. Diabetes 49:1635–1642
Article CAS PubMed Google Scholar - Burcelin R, Dolci W, Thorens B (2000) Glucose sensing by the hepatoportal sensor is GLUT2-dependent. In vivo analysis in GLUT2-null mice. Diabetes 49:1643–1648
Article CAS PubMed Google Scholar - Burcelin R, Crivelli V, Perrin C et al (2003) GLUT4, AMP kinase, but not the insulin receptor, are required for hepatoportal glucose sensor-stimulated muscle glucose utilization. J Clin Invest 111:1555–1562
Article CAS PubMed Central PubMed Google Scholar - Preitner F, Ibberson M, Franklin I et al (2004) Gluco-incretins control insulin secretion at multiple levels as revealed in mice lacking GLP-1 and GIP receptors. J Clin Invest 113:635–645
Article CAS PubMed Central PubMed Google Scholar - Fukumoto H, Seino S, Imura H et al (1988) Sequence, tissue distribution, and chromosomal localization of mRNA encoding a human glucose transporter-like protein. Proc Natl Acad Sci U S A 85:5434–5438
Article CAS PubMed Central PubMed Google Scholar - Carabaza A, Ciudad CJ, Baque S, Guinovart JJ (1992) Glucose has to be phosphorylated to activate glycogen synthase, but not to inactivate glycogen phosphorylase in hepatocytes. FEBS Lett 296:211–214
Article CAS PubMed Google Scholar - Kabashima T, Kawaguchi T, Wadzinski BE, Uyeda K (2003) Xylulose 5-phosphate mediates glucose-induced lipogenesis by xylulose 5-phosphate-activated protein phosphatase in rat liver. Proc Natl Acad Sci U S A 100:5107–5112
Article CAS PubMed Central PubMed Google Scholar - Seyer P, Vallois D, Poitry-Yamate C et al (2013) Hepatic glucose sensing is required to preserve beta cell glucose competence. J Clin Invest 123:1662–1676
Article CAS PubMed Central PubMed Google Scholar - Burcelin R, del Carmen MM, Guillam MT, Thorens B (2000) Liver hyperplasia and paradoxical regulation of glycogen metabolism and glucose-sensitive gene expression in GLUT2-null hepatocytes. Further evidence for the existence of a membrane-based glucose release pathway. J Biol Chem 275:10930–10936
Article CAS PubMed Google Scholar - Guillam MT, Burcelin R, Thorens B (1998) Normal hepatic glucose production in the absence of GLUT2 reveals an alternative pathway for glucose release from hepatocytes. Proc Natl Acad Sci U S A 95:12317–12321
Article CAS PubMed Central PubMed Google Scholar - Hosokawa M, Thorens B (2002) Glucose release from GLUT2-null hepatocytes: characterization of a major and a minor pathway. Am J Physiol Endocrinol Metab 282:E794–E801
CAS PubMed Google Scholar - Düfer M, Hörth K, Wagner R et al (2012) Bile acids acutely stimulate insulin secretion of mouse beta-cells via farnesoid X receptor activation and ATP-sensitive potassium channel inhibition. Diabetes 61:1479–1489
Article PubMed Central PubMed Google Scholar - Roncero I, Alvarez E, Chowen JA et al (2004) Expression of glucose transporter isoform GLUT-2 and glucokinase genes in human brain. J Neurochem 88:1203–1210
Article CAS PubMed Google Scholar - Liu Y, Liu F, Grundke-Iqbal I, Iqbal K, Gong CX (2009) Brain glucose transporters, O-GlcNAcylation and phosphorylation of tau in diabetes and Alzheimer’s disease. J Neurochem 111:242–249
Article CAS PubMed Central PubMed Google Scholar - Castillo J, Crespo D, Capilla E et al (2009) Evolutionary structural and functional conservation of an ortholog of the GLUT2 glucose transporter gene (SLC2A2) in zebrafish. Am J Physiol Regul Integr Comp Physiol 297:R1570–R1581
Article CAS PubMed Google Scholar - Arluison M, Quignon M, Thorens B, Leloup C, Penicaud L (2004) Immunocytochemical localization of the glucose transporter 2 (GLUT2) in the adult rat brain. II. Electron microscopic study. J Chem Neuroanat 28:137–146
Article CAS PubMed Google Scholar - Arluison M, Quignon M, Nguyen P, Thorens B, Leloup C, Penicaud L (2004) Distribution and anatomical localization of the glucose transporter 2 (GLUT2) in the adult rat brain–an immunohistochemical study. J Chem Neuroanat 28:117–136
Article CAS PubMed Google Scholar - Garcia M, Millan C, Balmaceda-Aguilera C et al (2003) Hypothalamic ependymal-glial cells express the glucose transporter GLUT2, a protein involved in glucose sensing. J Neurochem 86:709–724
Article PubMed Google Scholar - Mounien L, Marty N, Tarussio D et al (2010) Glut2-dependent glucose-sensing controls thermoregulation by enhancing the leptin sensitivity of NPY and POMC neurons. FASEB J 24:1747–1758
Article CAS PubMed Google Scholar - Leloup C, Orosco M, Serradas P, Nicolaidis S, Penicaud L (1998) Specific inhibition of GLUT2 in arcuate nucleus by antisense oligonucleotides suppresses nervous control of insulin secretion. Brain Res Mol Brain Res 57:275–280
Article CAS PubMed Google Scholar - Wan HZ, Hulsey MG, Martin RJ (1998) Intracerebroventricular administration of antisense oligodeoxynucleotide against GLUT2 glucose transporter mRNA reduces food intake, body weight change and glucoprivic feeding response in rats. J Nutr 128:287–291
CAS PubMed Google Scholar - Bady I, Marty N, Dallaporta M et al (2006) Evidence from glut2-null mice that glucose is a critical physiological regulator of feeding. Diabetes 55:988–995
Article CAS PubMed Google Scholar - Trumpp A, Depew MJ, Rubenstein JL, Bishop JM, Martin GR (1999) Cre-mediated gene inactivation demonstrates that FGF8 is required for cell survival and patterning of the first branchial arch. Genes Dev 13:3136–3148
Article CAS PubMed Central PubMed Google Scholar - Tarussio D, Metref S, Seyer P et al (2014) Nervous glucose sensing regulates postnatal beta cell proliferation and glucose homeostasis. J Clin Invest 124:413–424
Article CAS PubMed Central PubMed Google Scholar - Finegood DT, Scaglia L, Bonner-Weir S (1995) Dynamics of beta-cell mass in the growing rat pancreas. Estimation with a simple mathematical model. Diabetes 44:249–256
Article CAS PubMed Google Scholar - Girard J, Ferré P, Pégorier J-P, Duée P-H (1992) Adaptation of glucose and fatty acid metabolism during perinatal period and suckling-weaning transition. Physiol Rev 72:507–562
CAS PubMed Google Scholar - Dallaporta M, Himmi T, Perrin J, Orsini JC (1999) Solitary tract nucleus sensitivity to moderate changes in glucose level. Neuroreport 10:2657–2660
Article CAS PubMed Google Scholar - Balfour RH, Hansen AM, Trapp S (2006) Neuronal responses to transient hypoglycaemia in the dorsal vagal complex of the rat brainstem. J Physiol 570:469–484
Article CAS PubMed Central PubMed Google Scholar - Grill HJ, Hayes MR (2012) Hindbrain neurons as an essential hub in the neuroanatomically distributed control of energy balance. Cell Metab 16:296–309
Article CAS PubMed Google Scholar - Babic T, Browning KN, Travagli RA (2011) Differential organization of excitatory and inhibitory synapses within the rat dorsal vagal complex. Am J Physiol Gastrointest Liver Physiol 300:G21–G32
Article CAS PubMed Central PubMed Google Scholar - Lamy CM, Sanno H, Labouèbe G et al (2014) Hypoglycemia-activated GLUT2 neurons of the nucleus tractus solitarius stimulate vagal activity and glucagon secretion. Cell Metab 19:527–538
Article CAS PubMed Google Scholar - Madisen L, Mao T, Koch H et al (2012) A toolbox of Cre-dependent optogenetic transgenic mice for light-induced activation and silencing. Nat Neurosci 15:793–802
Article CAS PubMed Central PubMed Google Scholar - Fenno L, Yizhar O, Deisseroth K (2011) The development and application of optogenetics. Annu Rev Neurosci 34:389–412
Article CAS PubMed Google Scholar - Taborsky GJ Jr, Mundinger TO (2012) Minireview: the role of the autonomic nervous system in mediating the glucagon response to hypoglycemia. Endocrinology 153:1055–1062
Article CAS PubMed Central PubMed Google Scholar - Cryer PE, Davis SN, Shamoon H (2003) Hypoglycemia in diabetes. Diabetes Care 26:1902–1912
Article CAS PubMed Google Scholar - Fanconi G, Bickel H (1949) Die chronische Aminoacidurie bei der Glykogenose und der Cystinkrankheit. Helv Paediatr Acta 4:359–396, article in German
CAS PubMed Google Scholar - Santer R, Schneppenheim R, Suter D, Schaub J, Steinmann B (1998) Fanconi-Bickel syndrome - the original patient and his natural history, historical steps leading to the primary defect, and a review of the literature. Eur J Pediatr 157:783–797
Article CAS PubMed Google Scholar - Foretz M, Thorens B (2003) The facilitative glucose transporter 2: pathophysiological role in mouse and human. In: Broër S, Wagner CA (eds) Membrane transporter diseases. Kluwer Academic/Plenum Publishers, New York, pp 175–190
Chapter Google Scholar - Mannstadt M, Magen D, Segawa H et al (2012) Fanconi-Bickel syndrome and autosomal recessive proximal tubulopathy with hypercalciuria (ARPTH) Are allelic variants caused by GLUT2 mutations. J Clin Endocrinol Metab 97:E1978–E1986
Article CAS PubMed Central PubMed Google Scholar - Manz F, Bickel H, Brodehl J et al (1987) Fanconi-Bickel syndrome. Pediatr Nephrol 1:509–518
Article CAS PubMed Google Scholar - Chesney RW, Kaplan BS, Colle E et al (1980) Abnormalities of carbohydrate metabolism in idiopathic Fanconi syndrome. Pediatr Res 14:209–215
Article CAS PubMed Google Scholar - Garty R, Cooper M, Tabachnik E (1974) The Fanconi syndrome associated with hepatic glycogenosis and abnormal metabolism of galactose. J Pediatr 85:821–823
Article CAS PubMed Google Scholar - Sansbury FH, Flanagan SE, Houghton JA et al (2012) SLC2A2 mutations can cause neonatal diabetes, suggesting GLUT2 may have a role in human insulin secretion. Diabetologia 55:2381–2385
Article CAS PubMed Google Scholar - Eny KM, Wolever TM, Fontaine-Bisson B, El-Sohemy A (2008) Genetic variant in the glucose transporter type 2 is associated with higher intakes of sugars in two distinct populations. Physiol Genomics 33:355–360
Article CAS PubMed Google Scholar - Mueckler M, Kruse M, Strube M, Riggs AC, Chiu KC, Permutt MA (1994) A mutation in the GLUT2 glucose transporter gene of a diabetic patient abolishes transport activity. J Biol Chem 269:17765–17767
CAS PubMed Google Scholar - Dupuis J, Langenberg C, Prokopenko I et al (2010) New genetic loci implicated in fasting glucose homeostasis and their impact on type 2 diabetes risk. Nat Genet 42:105–116
Article CAS PubMed Central PubMed Google Scholar - Barker A, Sharp SJ, Timpson NJ et al (2011) Association of genetic loci with glucose levels in childhood and adolescence: a meta-analysis of over 6,000 children. Diabetes 60:1805–1812
Article CAS PubMed Central PubMed Google Scholar - Gaulton KJ, Willer CJ, Li Y et al (2008) Comprehensive association study of type 2 diabetes and related quantitative traits with 222 candidate genes. Diabetes 57:3136–3144
Article CAS PubMed Central PubMed Google Scholar - Kilpelainen TO, Lakka TA, Laaksonen DE et al (2007) Physical activity modifies the effect of SNPs in the SLC2A2 (GLUT2) and ABCC8 (SUR1) genes on the risk of developing type 2 diabetes. Physiol Genomics 31:264–272
Article CAS PubMed Google Scholar - Laukkanen O, Lindstrom J, Eriksson J et al (2005) Polymorphisms in the SLC2A2 (GLUT2) gene are associated with the conversion from impaired glucose tolerance to type 2 diabetes: the Finnish Diabetes Prevention Study. Diabetes 54:2256–2260
Article CAS PubMed Google Scholar - Igl W, Johansson A, Wilson JF et al (2010) Modeling of environmental effects in genome-wide association studies identifies SLC2A2 and HP as novel loci influencing serum cholesterol levels. PLoS Genet 6:e1000798
Article PubMed Central PubMed Google Scholar - Borglykke A, Grarup N, Sparsø T et al (2012) Genetic variant SLC2A2 [corrected] is associated with risk of cardiovascular disease - assessing the individual and cumulative effect of 46 type 2 diabetes related genetic variants. PLoS One 7:e50418
Article CAS PubMed Central PubMed Google Scholar - Thorens B, Mueckler M (2010) Glucose transporters in the 21st Century. Am J Physiol Endocrinol Metab 298:E141–E145
Article CAS PubMed Central PubMed Google Scholar - Thorens B (2001) GLUT2 in pancreatic and extra-pancreatic gluco-detection (review). Mol Membr Biol 18:265–273
Article CAS PubMed Google Scholar - Stumpel F, Burcelin R, Jungermann K, Thorens B (2001) Normal kinetics of intestinal glucose absorption in the absence of GLUT2: evidence for a transport pathway requiring glucose phosphorylation and transfer into the endoplasmic reticulum. Proc Natl Acad Sci U S A 98:11330–11335
Article CAS PubMed Central PubMed Google Scholar - Marty N, Dallaporta M, Foretz M et al (2005) Regulation of glucagon secretion by glucose transporter type 2 (glut2) and astrocyte-dependent glucose sensors. J Clin Invest 115:3545–3553
Article CAS PubMed Central PubMed Google Scholar