Cytochrome P450-derived eicosanoids: the neglected pathway in cancer (original) (raw)
1 Introduction
Products of arachidonic acid metabolism, including prostaglandins and leukotrienes are potent mediators of inflammation [1]. These lipid mediators, collectively called eicosanoids, play critical roles in diverse physiological and pathological processes such as pulmonary fibrosis and cancer (Fig. 1). The first two pathways of arachidonic acid metabolism are controlled by the enzyme families cyclooxygenase (COX) and lipoxygenase (LOX). These enzymes are the target of approved drugs for the treatment of pain, inflammation, asthma, and allergies [2]. Both of these pathways produce prostaglandins and leukotrienes, respectively, and have been implicated in cancer [3]. However, a third eicosanoid pathway, in which cytochrome P450 (CYP) enzymes convert arachidonic acid into hydroxyeicosatetraenoic acids (HETEs) or epoxyeicosatrienoic acids (EETs), appears to have a role in tumor growth. COX- and LOX-derived eicosanoids have been intensely studied in tumor biology, while the study of cytochrome P450-derived eicosanoids has focused on inflammation, angiogenesis, and cardiovascular function rather than cancer pathways [1–6].
Fig. 1
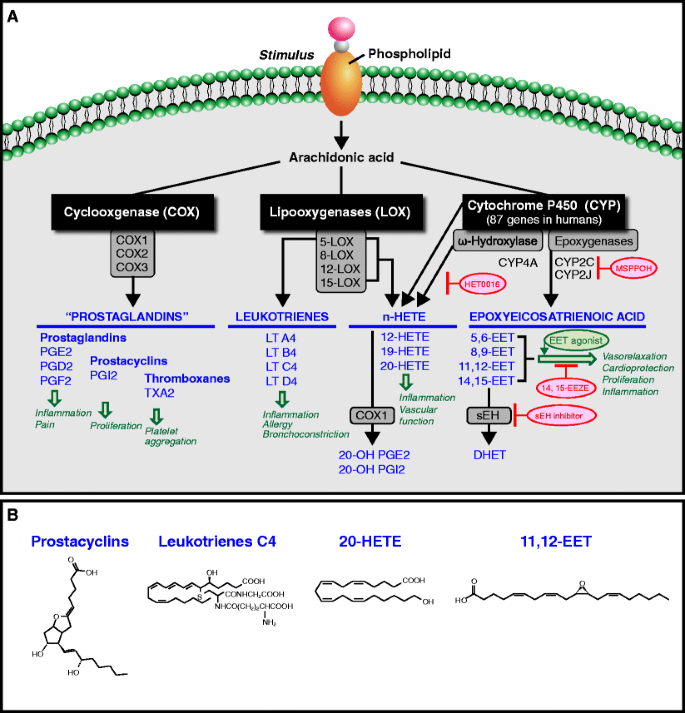
a, b Bioactive eicosanoids derived from the arachidonic acid cascade. Arachidonic acid is metabolized by three pathways—the cyclooxygenase (COX), lipoxygenase (LOX), and cytochrome P450 (CYP) pathways. Schematic overview of major mediators and their metabolites (blue); enzymes (black, boxed) and biological role (green). Inhibitors (red ovals) and agonists (green ovals). HETEs Hydroxyeicosatetraenoic acids, EETs epoxyeicosatrienoic acids, CYP cytochrome P450 enzymes. MS-PPOH is a selective inhibitor of a subset of epoxygenases. HET0016 is a selective inhibitor of the ω-hydroxlase CYP4A. The sEH inhibitor (soluble epoxide hydrolase inhibitor) increases EET levels by acting as an agonist of the EET pathway. 14,15-EEZE is a putative EET receptor antagonist. PGE 2 prostaglandin E2, PGI 2 prostacyclin, LTA 4 leukotriene A4, DHET dihydroxyeicosatrienoic acid, 20-OH PGE 2 20-hydroxy-prostaglandin E2
1.1 Overview of the CYP pathway
Cytochrome P450-dependent metabolism of arachidonic acid occurs in several tissues including liver, kidney, and the cardiovascular system. The CYP enzymes relevant to arachidonic acid metabolism include two distinct pathways: the ω-hydroxylase and epoxygenase pathways. The ω-hydroxylases of the 4A and 4F gene families of cytochrome P450 (CYP4A and CYP4F) convert arachidonic acid to autacoids such as hydroxyeicosatetraenoic acids. 20-hydroxyeicosatetraenoic acid is the principal isoform of this pathway and has shown vasoconstrictory activity [7–9]. The epoxygenase pathway is encoded predominantly by the CYP2C and CYP2J genes and generates epoxyeicosatrienoic acids, which have demonstrated vasodilatory activity [1, 10, 11]. EETs are then metabolized mainly by soluble epoxide hydrolase (sEH) to the dihydroxyeicosatrienoic acids (DHETs), which have traditionally been considered to be less active than EETs [12, 13]. The biology of both the epoxygenase and ω-hydroxylase pathways of cytochrome P450 enzymes has been extensively reviewed [1, 2, 4–6].
1.2 History of the CYP eicosanoids
Based on the pioneering work of Estabrook, both Capdevila and Falck found and characterized a third pathway, microsomal cytochrome P450 arachidonic acid metabolism [14, 15]. In 1981, metabolites separate from the prostanoids and leukotrienes were identified by the oxidative metabolism of arachidonic acid through microsomal cytochrome P450 systems [16–19]. In 1996, EETs were identified by Campbell and colleagues as endothelium-derived substances that hyperpolarize vascular smooth muscle [20]. This discovery sparked interest in the newly developing field of CYP eicosanoids. Within this field, Zeldin and colleagues identified the EET regiospecificity of sEH and were the first to identify and clone the CYP2J2 gene. Over the past decade, the Falck laboratory has synthesized agonists and antagonists of CYP 450 metabolites, including EETs and 20-HETE. However, the rapid metabolism of EETs and other epoxylipids has made it difficult to study the biological relevance of these metabolites. To address this challenge, the Hammock laboratory pioneered a series of sEH inhibitors which further stabilized EETs [2, 21]. sEH inhibitors, which increase EET levels, have been evaluated in the clinic for cardiovascular diseases, such as hypertension [2]. In addition, EET and HETE levels are now quantifiable by liquid chromatography–tandem mass spectrometry [22].
In this review, we survey the largely unexplored field of cytochrome P450 metabolites of arachidonic acid in tumorigenesis. We will focus on their roles in cancer as well as in angiogenesis and inflammation; two interdependent processes in the tumor stroma that play pivotal roles in tumor growth and metastasis.
2 CYP P450 genes, enzymes, and current role in pharmacology
This CYP superfamily is a complex group of enzymes that consist of upwards of 102 putatively functional genes in mice, and as few as 57 in humans [23, 24]. These CYP enzymes differ greatly from mouse to man, presenting challenges in the characterization of CYPs in this field [25, 26].
The best known function of the CYP enzymes is the detoxification of compounds, such as anti-cancer drugs and xenobiotics in the liver. Blocking these enzymes improves the half-life of the cytotoxic drugs—a strategy that is currently under evaluation to improve the efficacy of cancer drug delivery [23, 27, 28]. Conversely, prodrugs activated by cytochrome P450 enzymes are being used to inhibit tumor growth by targeting the tumor cells and tumor-associated endothelial cells [29, 30].
Therapeutic success has already been obtained using cytochrome P450 inhibitors to treat breast cancer [31]. This has prompted investigators to determine whether cytochrome P450 inhibitors can be utilized to treat other hormonally responsive cancers including prostate cancer [29, 32, 33]. While the field of directly targeting cytochrome P450 enzymes in cancer has rapidly expanded, the biological role of CYP-derived lipid autacoids in cancer has been largely neglected.
3 Synthesis and degradation of hydroxyeicosatetraenoic acids and epoxyeicosatrienoic acids
Arachidonic acid is an essential component of mammalian cell membranes and plays a critical role in the synthesis of bioactive eicosanoids [1]. Eicosanoids are generated via the oxidation of the 20-carbon chain present on arachidonic acid or other related fatty acids [2]. During processes, such as inflammation, arachidonic acid is released from the cell membrane through the activation of phospholipase A2 [1]. Arachidonic acid is metabolized by the CYP ω-hydroxylases to 7-, 10-, 12-, 13-, 15-, 16-, 17-, 18-, 19-, and 20-HETEs, the principal metabolite being the pro-inflammatory 20-HETE [4].
The epoxygenase CYP enzymes metabolize arachidonic acid by olefin epoxidation, resulting in four regioisomeric epoxyeicosatrienoic acids (EETs): 5,6-EET, 8,9-EET, 11,12-EET, and 14,15-EET [1]. Each regioisomer can be formed as either an R,S or S,R enantiomer as the epoxide group can attach at each of the double bonds in two separate configurations, resulting in a total of eight EETs (reviewed by Zeldin [1]).
EETs are primarily synthesized in endothelial cells which express isoforms of CYP2C and CYP2J (e.g., CYP2C9 and CYP2J2) [10, 34–37]. EETs are also produced in other cell types such as astrocytes and cardiac myocytes [38–42]. Additionally, monocytic leukocytes have recently been shown to express CYP2J2 and therefore, may generate EETs [43].
The synthesis of 20-HETE and 12-HETE occurs in vascular smooth muscle cells and fibroblasts respectively through the cytochrome P-450 (CYP450) pathway [4, 44]. 20-HETE synthesis can be controlled by a positive feedback mechanism by activating calcium/calmodulin-dependent protein kinase-induced mitogen-activated protein kinase (MAPK) in smooth muscle cells [45]. This Ras/MAPK pathway then amplifies cytosolic phospholipase A2. This mechanism results in the release of additional arachidonic acid substrates that can then be converted to 20-HETE [45].
Degradation of 20-HETE occurs via multiple pathways. For example, in endothelial cells 20-HETE can be metabolized by cyclooxygenase to 20-hydroxy-prostaglandin G2 and H2 [46]. 20-HETE may also be oxidized by ω-oxidation or β-oxidation [47]. In contrast, the degradation of EETs appears to be more uniform and is exerted mainly by sEH resulting in DHETs [12, 13].
The synthesis pathways for HETEs and EETs are complex exhibiting multiple routes leading to the same compound. HETEs can be generated through the three arachidonic acid metabolic pathways, COX, LOX, and CYP 450 [48]. While EETs are mainly formed by CYP2C and CYP2J, other EET-producing CYPs such as CYP4X1 and CYP2U1 have been characterized [11, 49, 50]. These two cytochrome P450s (CYP4X1 and CYP2U1) metabolize arachidonic acid to 8,9- and 14,15-EETs as well as 19- and 20-HETE, respectively [49, 50].
4 Targets of EETs
The molecular mechanism(s) of EETs is poorly understood, but continues to be fiercely studied. A series of agonists have been developed to help characterize the binding and metabolism of 14,15-EET [51]. While the EET receptor(s) has not yet been identified, intracellular signals by G protein pathways have been implicated [2, 51, 52]. Over 90% of circulating EETs are incorporated into phospholipids of the cell membrane, mainly as low-density lipoproteins [53]. EETs can act as long-chain fatty acids and bind to fatty-acid-binding proteins and nuclear peroxisome-proliferator-activated receptors (PPARγ and PPARα). These actions suggest an intracellular mechanism [52, 54–56]. In fact, all four EETs and their metabolite DHET can stimulate PPAR/RXR heterodimer binding to a peroxisome proliferator response element [56–60].
5 The role of tumor stroma in tumorigenesis: angiogenesis and inflammation
Prior to examining a potential role of 20-HETE and EETs in cancer, we need to review the various cellular components of a tumor that could serve as sources and targets of these lipid autacoids. A paradigm shift has taken place over the past decade in cancer research. The simple notion, unquestioned for decades, was that cancer is a cell-autonomous disease driven by mutations for fast growing and increasingly malignant cell clones. Now it is accepted that tumor growth is a non-cell-autonomous process, requiring support from the “tissue microenvironment” in the “tumor bed” [61–63]. Non-cell-autonomous contribution to tumorigenesis from the “host-tissue”, most clearly epitomized by tumor vasculature, is crucial for tumor expansion and progression [64]. This hypothesis, developed by Folkman in 1971, stated that tumor growth requires neovascularization, and that such “tumor angiogenesis” is induced by tumor-derived soluble factors [65]. Since then, the contributions of non-cancerous cells to the growth of tumors has extended beyond endothelial cells to pericytes, inflammatory cells, immune cells, fibroblasts, myofibroblasts, and adipocytes; for example, carcinoma-associated fibroblasts can promote the growth of invasive breast cancer [66]. Non-local cells, including bone-marrow-derived macrophages, neutrophils, mast cells, and mesenchymal stem cells are also recruited, contributing to the invasiveness and metastatic ability of neoplastic epithelial cells [67].
Both angiogenesis and inflammation are interdependent stromal processes that exert substantial influence on tumor growth and metastasis. Pro-inflammatory enzymes and cytokines act to promote tumors; increased infiltration of macrophages and neutrophils can increase angiogenesis and correlates with a poor prognosis [68, 69]. In other cases, inflammatory infiltration of lymphocytic/monocytic cells can actually inhibit tumor growth [70]. Conversely, blocking inflammation can be associated with the stimulation of cancer [3, 71, 72]. Both inhibition and activation of the nuclear factor-kappaB (NF-κB) protein complex can promote carcinogenesis [73–75]. Inflammation in the tumor bed can then either stimulate or inhibit tumor growth [72, 76, 77]. Thus, pharmacological modulation of inflammation in cancer treatments must be evaluated with the notion that inflammation may be a double-edged sword in tumor growth.
6 Lipid autacoids in cancer
It has been recognized that tumor growth is a complex process involving many cell types. The intercellular communication that takes place between these cells is conducted by an array of soluble factors such as: proteinaceous growth factors and chemokines, vascular endothelial growth factor (VEGF), FGF-2, TGF-β, TNF-α, interleukin (IL)-1, and oxygen radicals [78].
Little attention has been paid to small molecule mediators, such as lipid autacoids, whose role in cancer has only recently emerged. Given that a tumor consists of both cancerous and non-cancerous cells, the role of autacoids in tumor growth can be separated into their direct effects on neoplastic growth and their effects on inflammation, angiogenesis, and stromal cells.
The pro-inflammatory prostaglandins and leukotrienes directly induce epithelial tumor cell proliferation, survival, migration, and invasion in an autocrine and paracrine manner [3]. Lipid autacoids, such as prostaglandin E2 (PGE2) and leukotriene B4 (LTB4), stimulate both epithelial cells and stromal cells to produce VEGF and FGF-2. These angiogenic growth factors induce COX2 and in turn produce PGE2 and PGI2 in endothelial cells [3, 79]. Other studies have linked eicosanoids to stroma inflammation in epithelial ovarian cancer [80]. Levels of eicosanoid metabolites, such as PGE2, 5-HETE, and 12-HETE, increase progressively in patients with benign pelvic disease to those with epithelial ovarian cancer. This demonstrates the involvement of lipid autacoids in the inflammatory environment of cancer [80]. However, the role of lipid autacoids derived from the third eicosanoid pathway of arachidonic acid remains poorly characterized in cancer.
7 HETEs effects on inflammation and the vasculature
Lipoxygenase-derived HETEs inhibit apoptosis, stimulate angiogenesis, and enhance proliferation and migration of cancer cells [48]. 20-HETE, the principal metabolite of the ω-hydroxylation pathway, is a pro-inflammatory mediator that markedly stimulates the production of inflammatory cytokines/chemokines in endothelial cells, including IL-8, IL-13, IL-4, and prostaglandin E2 [81]. 20-HETE stimulates NF-κB activation and MAPK/ERK pathways, which suggests that HETE’s pro-inflammatory effect may be mediated by the central inflammatory pathway of NF-κB [81].
In addition to its pro-inflammatory activity, 20-HETE has pro-angiogenic activity including the stimulation of endothelial cell proliferation, migration, and cell survival [82–85]. 20-HETE has an important role in VEGF-dependent angiogenesis [86] (reviewed in [85]). While VEGF seems to be the primary mediator of 20-HETE–induced endothelial cell proliferation, inhibition with a VEGF antibody does not completely abrogate the mitogenic effect of 20-HETE [82]. This suggests other pathways are involved in 20-HETE-mediated angiogenesis [82].
The pro-angiogenic factor fibroblast growth factor-2 (FGF-2) can activate cytosolic phospholipase A2 (the enzyme which releases arachidonic acid from cell membranes) in endothelial cells [87]. FGF-2 increases arachidonic acid production, potentially stimulating CYP4A and production of 20-HETE [85]. The overexpression of CYP4A1, which increases 20-HETE production, results in increased neovessel formation [88].
HET0016, a selective inhibitor of CYP4A, suppresses the formation of 20-HETE at a concentration <10 nM, and has no effect on epoxygenase, cyclooxygenase, or lipoxygenase activity at concentrations up to 1 μM [4, 89]. HET0016 inhibits VEGF-induced endothelial cell proliferation in vitro and corneal neovascularization in vivo when administered locally with pellets containing VEGF [84]. When administered locally into the cornea, HET0016 inhibited tumor-induced (U251 glioblastoma cells) angiogenesis by 70% [84]. Furthermore, the administration of the stable 20-HETE agonist, 20-hydroxyeicosa-6(Z) 15(Z)-dienoic acid (WIT003), induced mitogenesis in endothelial cells and corneal neovascularization in vivo [84]. These studies provide experimental evidence that inhibiting 20-HETE may offer a strategy to reduce pathological angiogenesis not only in tumors but in angiogenic diseases such as diabetic retinopathy, macular degeneration and chronic inflammatory diseases, such as psoriasis [84]. However, these studies did not determine whether 20-HETE was produced by the cornea or endothelial cells and, therefore, further studies are needed [90].
In the systemic circulation, 20-HETE produced by vascular smooth muscle cells acts as a vasoconstrictor [4]. However, in pulmonary arteries, 20-HETE contributes to VEGF-induced relaxation of the lungs [91]. VEGF, a nitric oxide (NO)-dependent dilator of systemic arteries, plays a key role in maintaining the integrity of the pulmonary vasculature [91].
8 20-HETE effects in cancer
In 2008, U251 glioblastoma cells were genetically altered (transfected with rat CYP4A1 cDNA) to increase the formation of 20-HETE [92]. This stimulated proliferation in culture. When these transfected U251 glioblastoma cells were implanted into the brain of rats, a tenfold increase in tumor volume was observed when compared to animals receiving mock-transfected U251 cells [92].
Conversely, Guo et al. demonstrated that HET0016 significantly inhibited human U251 glioblastoma cell proliferation in a dose-dependent manner [90]. HET0016 inhibited the phosphorylation of the epidermal growth factor receptor (EGFR) and the subsequent phosphorylation of p42/p44 MAPK [90]. While U251 cells expressed CYP4A11 mRNA and protein, HPLC and mass spectrometry analysis of U251 cell extracts revealed that they did not appear to synthesize 20-HETE [90]. Thus, HET0016 has other effects independent of suppressing 20-HETE. Subsequently, the same group demonstrated that 9L gliosarcoma proliferation and tumor growth in rats are suppressed by HET0016 [93]. Systemic administration of HET0016 inhibited the tumor growth of 9L gliosarcomas by 80%, and tumor angiogenesis by roughly 50%. In a separate study, HET0016 and a 20-HETE antagonist (WIT002) both inhibited the proliferation of a renal adenocarcinoma. This cell type expressed CYP4F isoforms and produced 20-HETE [94].
Little is known about 20-HETE in cancer patients. In one study, 12-HETE and 20-HETE concentrations were shown to be elevated in the urine of patients with benign prostatic hypertrophy and prostate cancer patients as compared to normal subjects [95]. Further analysis did not establish a correlation between the concentrations of HETEs and prostatic specific antigen level, gland size, or tumor grade [95].
9 EETs and angiogenesis
EETs are mainly secreted by endothelial cells and play critical roles in cellular proliferation, migration, and inflammation; their major target is blood vessels [6, 37]. EETs may act in an autocrine fashion on the endothelium inducing vasodilatory and anti-inflammatory effects in blood vessels [96]. As a result of these effects, EETs lower blood pressure and protect the myocardium and brain from ischemia [56, 97–99].
The initial finding that linked EETs to angiogenesis was shown by an increase in proliferation of cerebral capillary endothelial cells by astrocyte conditioned media [40]. In contrast, an inhibitor of cytochrome P450, 17-octadecynoic acid (17-ODYA), suppressed the formation of capillary tubes in a co-culture of astrocytes and endothelial cells. Both EETs secreted by astrocytes and synthetic EETs stimulated endothelial cell proliferation, tube formation, and angiogenesis in a matrigel plug in vivo [40, 100, 101].
Angiogenesis is critically dependent on endothelial cell migration [102]. The development of synthetic EETs has provided insight into the angiogenic functions and pathways of the various EETs. For instance, EETs have been shown to promote endothelial cell migration via endothelial NO synthase, MEK/MAPK, and PI3K [103]. Another assay to evaluate angiogenesis is the chick chorioallantoic membrane assay, which uses the chorioallantoic membrane (CAM) of a chicken embryo [104]. Michaelis et al. employed this assay to demonstrate that 11,12-EET stimulates vessel formation [105]. Importantly, this CAM-mediated angiogenesis was suppressed by either an EGF receptor-neutralizing antibody or an inhibitor of the EGF receptor. Thus, 11,12-EET may stimulate angiogenesis through the activation of the EGF receptor [105].
Several other pathways have been implicated in 11,12-EET- and 14,15-EET-mediated angiogenesis. Sphingosine kinase-1 (SK1) is one important mediator of 11,12-EET-induced angiogenic effects [106]. The expression of a dominant-negative SK1 or knockdown of SK1 by siRNA, inhibited 11,12-EET-induced endothelial cell proliferation, migration, tube formation, and matrigel plug vessel formation [106]. In other studies, EphB4 is a critical component of the CYP2C9-activated signaling cascade [107]. Both CYP2C9 overexpression or the administration of 11,12-EET showed increased expression of EphB4 in endothelial cells. The availability of these synthetic EETs has made it possible to evaluate another regioisomer, 14,15-EET. 14,15-EET was shown to induce angiogenesis via several pathways including: Src, phospatidylinositol-3-kinase/Akt (PI3K/Akt) signaling in parallel with mTOR-S6K1 activation and Src-dependent STAT-3-mediated VEGF expression [108, 109].
Other groups have studied CYP 450-derived metabolites, utilizing the strategy of overexpressing CYP epoxygenases. In lieu of EETs, this system inhibited endothelial cell apoptosis through activation of the PI3K/Akt pathway [110]. The overexpression of CYP epoxygenases, including CYP2J2, also increased muscle capillary density in a rat ischemic hind limb model [103]. Thus, CYP 450-derived metabolites may stimulate the development of collateral circulation in ischemic tissue [103].
While most investigators have focused on 11,12-EET and 14,15-EET, Pozzi et al. identified 5,6- and 8,9-EET as pro-angiogenic lipids [36]. These regioisomers increased blood vessel density and formed functionally intact vessels in a subcutaneous sponge model in mice. This neovascularization was enhanced by the co-administration of an epoxide hydrolase inhibitor, which elevates the levels of EETs [36]. This study corroborates the critical role that EETs plays in angiogenesis.
It is known that hypoxia stimulates angiogenesis via transcriptional VEGF induction, a response that is mediated by the hypoxia-inducible factor-1α (HIF-1α) [111]. It was shown by the Fleming laboratory that hypoxia also stimulates CYP 2C8 and 2C9 expression [5, 112, 113]. Consistently, the CYP inhibitor (MS-PPOH) and the putative EET receptor antagonist (14,15-EEZE), inhibited hypoxia-induced endothelial tube formation [112]. Furthermore, the angiogenic effect of EETs is partially dependent on HIF-1α-mediated VEGF induction [114]. This may have implications in cancer beyond angiogenesis, since HIF-1α can provide a growth and survival advantage to tumor cells, especially under metabolic stress [72].
The effects of EETs and VEGF regulation are closely intertwined. EETs can enhance the effects of VEGF-induced angiogenesis [115]. In turn, VEGF can increase CYP2C promoter activity in endothelial cells and induce the expression of CYP2C8, resulting in increased intracellular EET levels [115]. The putative EET receptor antagonist, 14,15-EEZE, inhibits VEGF-induced endothelial cell tube formation. However, 14,15-EEZE does not affect VEGF-induced phosphorylation of its receptor or FGF-2-stimulated tube formation [115]. In a parallel study, CYP2C44 epoxygenase appears to be an important component in the VEGF signaling pathway [116]. For example, in cultured lung endothelial cells that express VEGF-inducible CYP2C44 epoxygenase, resulting in increased levels of 11,12- and 14,15-EET, angiogenesis was stimulated in vitro. Taken together, these studies suggest that the pro-angiogenic activity of EETs is mediated at least, in part, by VEGF [115, 116].
10 CYP 450 epoxygenases and cancer
While the pro-angiogenic activity of EETs has extensively been investigated [36, 103, 117], the role of EETs in cancer remains poorly characterized. Although two decades ago, 14,15-EET was shown to stimulate mesangial and renal epithelial cell proliferation [118, 119], only in the last 5 years has evidence, supporting cytochrome P450 epoxygenases as a potential tumor-promoting enzyme, begun to emerge [120]. The role of CYP2J2 epoxygenase in cancer was first shown by Jiang et al. In this study, CYP2J2 was upregulated in 77% of human carcinoma tissues and eight different human carcinoma cell lines [120]. Furthermore, the transfection of tumor cells with cytochrome epoxygenase 2J2 enhanced tumor formation [120]. Subsequent studies, in which CYP epoxygenase levels were manipulated, by either overexpression of CYP2J2 or antisense in the xenotransplanted tumor cell, suggest EETs may play a role in cancer metastasis [121].
EETs also appear to be important for cancer cell survival. Specific CYP2J2 inhibitors suppress human tumor cell proliferation [122]. These inhibitors activate caspase-3, which leads to reduced tumor cell adhesion, migration, invasion, and suppressed murine xenograft tumor growth [122]. It is often difficult to distinguish a direct effect on the tumor cells or the stromal processes. It is likely that both mechanisms synergize to account for the potential pro-tumorigenic activity of EETs.
There are few pharmacological studies using drugs which can non-specifically affect EETs. In one study conducted by Pozzi et al., mice treated with PPARα ligands exhibited a reduction of tumor growth, vascularization, and plasma EETs [123]. In a separate study, two mechanistically different synthetic inhibitors of cytochrome P450, 17-ODYA, and miconazole significantly reduced tumor size and capillary formation in intracranial glial tumors, and prolonged survival of treated rats [124]. Interestingly, these inhibitors had no effect on EETs in the tumor tissue suggesting that the tumor endothelium may be the target of these CYP inhibitors [124]. It has recently been reported that EET antagonists inhibit prostate carcinoma cell motility [125]. This may represent a novel mechanism of EET antagonists acting directly on the tumor cell [125].
Several CYP epoxygenases have been detected in tumor cells in vitro and in vivo, supporting the potential role of EETs in cancer (Fig. 2). For instance, CYP2C8, CYP2C9, and CYP2J2 were recently shown to be expressed in three prostate carcinoma cell lines (PC3, DU-145, and LNCaP) [125]. In these studies no consistent correlation between mRNA expression, protein expression, or EET concentrations was found [125]. Another epoxygenase, CYP2C11, was shown by Zagorac et al. to be upregulated in cerebral brain tumors of rats [124]. In addition to tumor cells, CYP epoxygenases are also expressed in the tumor stroma. For example, CYP2C44 epoxygenase is expressed in the tumor vessels of a xenograft model of human non-small cell lung cancer in mice [36]. Furthermore, in human renal tumors, CYP2C9 epoxygenase was recently found to be selectively expressed in the vasculature [126]. These findings open the possibility that EETs may act as a trophic factor for both tumor stroma and parenchyma.
Fig. 2
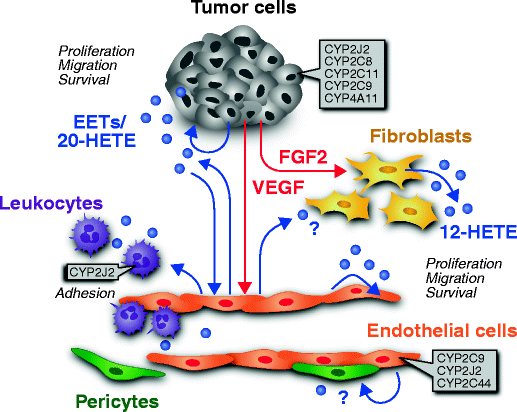
Cytochrome P450 epoxygenase expression in tumor cell compartments and their potential role in cell–cell communication in the tumor stroma. Note that both tumor cells and endothelial cell can produce EETs (blue circles), establishing an autocatalytic loop. EETs may act in an autocrine or paracrine fashion
In earlier clinical studies, 14,15-EET levels were detected in the urine samples of patients with benign prostatic hypertrophy and prostate cancer in comparison to normal volunteers [95]. Interestingly, after the removal of the prostate gland in prostate cancer patients, the urinary concentration of 14,15-EET did not decrease. This data suggests that the origin of the 14,15-EET was not the prostate gland but another source [95].
Whether the levels of CYP epoxygenases are dependent on isoenzyme or tumor type remains up for debate. Enayetallah et al. analyzed three cytochrome P450 epoxygenases (CYP2C8, CYP2C9, and CYP2J2) and soluble epoxide hydrolase in human malignant neoplasms [127]. CYP2C9 was the most abundantly expressed epoxygenase in several human malignant neoplasms [127]. In contrast CYP2J2 staining was not detected in pancreatic or prostate adenocarcinoma. Furthermore, CYP2J2 expression was detected in less than 50% of lung squamous cell carcinoma samples and in less than 15% of lung adenocarcinoma samples [127]. These results showing that CYP2J2 can be suppressed in tumors were confirmed by Leclerc et al. [128]. It has been suggested that this decrease in arachidonic acid epoxidation in certain tumors may allow arachidonic acid to be metabolized to the other eicosanoids [128]. Consequently, the inconsistent overexpression and underexpression of CYP epoxygenases in tumors makes it difficult to understand the biological significance of these enzymes in cancer. This variability must be examined in the wider context of the complex metabolic pathways of lipid autacoids.
In addition to the study of CYP epoxygenases, the expression of sEH, the main metabolizing enzyme of EETs, has been investigated in cancer. The loss of sEH has been reported in hepatocellular carcinoma and hepatoma cells [129, 130]. Enayetallah et al. further confirmed that sEH can be downregulated in renal and hepatic tumors, in principle increasing the levels of EETs in the tumor tissue [127]. These studies support a potential role for EETs in cancer due to downregulation of its metabolizing enzyme. Mirroring the expression of CYP epoxygenases in tumors, the expression of sEH can also be upregulated. sEH expression was increased in seminoma, cholangiocarcinoma, and advanced ovarian cancer when compared to normal tissues or early stage cancer [127]. As in the case of CYP, there is no consistent finding in the expression of sEH in tumors. According to the rationale that the elevation of EETs in tissues promotes tumor growth, sEH would be expected to be downregulated in tumors—which has been observed only in certain tumors, to date. More studies will be needed to reveal whether a potential increase of EETs is associated with a particular tumor type or cancer stage. Whether another biochemical component can compensate for a lack of EET increase in tumors remains unknown.
There is another aspect to consider when discussing CYP expression in cancer. Since cytochrome P450 (CYP) 2C8 metabolizes drugs, such as paclitaxel (Taxol), tamoxifen, and other chemotherapeutic agents, genetic polymorphisms in the CYP2C8/9 gene may affect cancer patient survival [131, 132]. Some polymorphisms in human CYP2C8 have been identified and shown to decrease the metabolism of paclitaxel and arachidonic acid [133]. Recent studies have implicated that genetic polymorphisms in CYP2C8 and CYP2C9 influence disease-free survival in breast cancer patients [132]. Furthermore, a genetic polymorphism of CYP2C19 is associated with increased susceptibility to biliary tract cancer [134].
11 Outlook
Several types of drugs, originally designed to target inflammation and cardiovascular diseases, have now been discovered to play an important role in cancer biology. This appears to be the case with the cytochrome P450-derived metabolites of arachidonic acid, EETs and HETEs. These lipid autacoids may be involved in cancer in a few different ways: either in cell-autonomous tumor survival and growth or in modulating stromal processes, such as angiogenesis and inflammation that can support tumor progression. Recently, classical mediators of inflammation, such as prostaglandins, have received new attention as potential targets in cancer treatment. The EET and HETE pathways should be evaluated as potential targets in cancer therapy, directed both against tumor cells and their surrounding stroma. Today, the role of cytochrome P450 metabolites in cancer is still poorly characterized, in part because of their biochemical complexity. The increasing availability of research tools, such as novel synthetic agonists, antagonists, and enzyme inhibitors, now offer a reasonable platform for dissecting the role of EETs and HETEs family autacoids in cancer.
References
- Zeldin, D. C. (2001). Epoxygenase pathways of arachidonic acid metabolism. The Journal of Biological Chemistry, 276, 36059–36062.
CAS PubMed Google Scholar - Imig, J. D., & Hammock, B. D. (2009). Soluble epoxide hydrolase as a therapeutic target for cardiovascular diseases. Nature Reviews. Drug Discovery, 8, 794–805.
CAS PubMed Google Scholar - Wang, D., & Dubois, R. N. (2010). Eicosanoids and cancer. Nature Reviews. Cancer, 10, 181–193.
CAS PubMed Google Scholar - Roman, R. J. (2002). P-450 metabolites of arachidonic acid in the control of cardiovascular function. Physiological Reviews, 82, 131–185.
CAS PubMed Google Scholar - Fleming, I. (2007). Epoxyeicosatrienoic acids, cell signaling and angiogenesis. Prostaglandins & Other Lipid Mediators, 82, 60–67.
CAS Google Scholar - Spector, A. A., & Norris, A. W. (2007). Action of epoxyeicosatrienoic acids on cellular function. American Journal of Physiology. Cell Physiology, 292, C996–C1012.
CAS PubMed Google Scholar - Hardwick, J. P., Song, B. J., Huberman, E., & Gonzalez, F. J. (1987). Isolation, complementary DNA sequence, and regulation of rat hepatic lauric acid omega-hydroxylase (cytochrome P-450LA omega). Identification of a new cytochrome P-450 gene family. The Journal of Biological Chemistry, 262, 801–810.
CAS PubMed Google Scholar - Powell, P. K., Wolf, I., Jin, R., & Lasker, J. M. (1998). Metabolism of arachidonic acid to 20-hydroxy-5, 8, 11, 14-eicosatetraenoic acid by P450 enzymes in human liver: Involvement of CYP4F2 and CYP4A11. The Journal of Pharmacology and Experimental Therapeutics, 285, 1327–1336.
CAS PubMed Google Scholar - Miyata, N., & Roman, R. J. (2005). Role of 20-hydroxyeicosatetraenoic acid (20-HETE) in vascular system. Journal of Smooth Muscle Research, 41, 175–193.
PubMed Google Scholar - Fisslthaler, B., Popp, R., Kiss, L., Potente, M., Harder, D. R., et al. (1999). Cytochrome P450 2 C is an EDHF synthase in coronary arteries. Nature, 401, 493–497.
CAS PubMed Google Scholar - Kaspera, R., & Totah, R. A. (2009). Epoxyeicosatrienoic acids: Formation, metabolism and potential role in tissue physiology and pathophysiology. Expert Opinion on Drug Metabolism & Toxicology, 5, 757–771.
CAS Google Scholar - Zeldin, D. C., Kobayashi, J., Falck, J. R., Winder, B. S., Hammock, B. D., et al. (1993). Regio- and enantiofacial selectivity of epoxyeicosatrienoic acid hydration by cytosolic epoxide hydrolase. The Journal of Biological Chemistry, 268, 6402–6407.
CAS PubMed Google Scholar - Yu, Z., Xu, F., Huse, L. M., Morisseau, C., Draper, A. J., et al. (2000). Soluble epoxide hydrolase regulates hydrolysis of vasoactive epoxyeicosatrienoic acids. Circulation Research, 87, 992–998.
CAS PubMed Google Scholar - Chacos, N., Capdevila, J., Falck, J. R., Manna, S., Martin-Wixtrom, C., et al. (1983). The reaction of arachidonic acid epoxides (epoxyeicosatrienoic acids) with a cytosolic epoxide hydrolase. Archives of Biochemistry and Biophysics, 223, 639–648.
CAS PubMed Google Scholar - Capdevila, J. H., Harris, R. C., & Falck, J. R. (2002). Microsomal cytochrome P450 and eicosanoid metabolism. Cellular and Molecular Life Sciences, 59, 780–789.
CAS PubMed Google Scholar - Capdevila, J., Chacos, N., Werringloer, J., Prough, R. A., & Estabrook, R. W. (1981). Liver microsomal cytochrome P-450 and the oxidative metabolism of arachidonic acid. Proceedings of the National Academy of Sciences of the United States of America, 78, 5362–5366.
CAS PubMed Google Scholar - Capdevila, J., Marnett, L. J., Chacos, N., Prough, R. A., & Estabrook, R. W. (1982). Cytochrome P-450-dependent oxygenation of arachidonic acid to hydroxyicosatetraenoic acids. Proceedings of the National Academy of Sciences of the United States of America, 79, 767–770.
CAS PubMed Google Scholar - Morrison, A. R., & Pascoe, N. (1981). Metabolism of arachidonate through NADPH-dependent oxygenase of renal cortex. Proceedings of the National Academy of Sciences of the United States of America, 78, 7375–7378.
CAS PubMed Google Scholar - Oliw, E. H., Lawson, J. A., Brash, A. R., & Oates, J. A. (1981). Arachidonic acid metabolism in rabbit renal cortex. Formation of two novel dihydroxyeicosatrienoic acids. The Journal of Biological Chemistry, 256, 9924–9931.
CAS PubMed Google Scholar - Campbell, W. B., Gebremedhin, D., Pratt, P. F., & Harder, D. R. (1996). Identification of epoxyeicosatrienoic acids as endothelium-derived hyperpolarizing factors. Circulation Research, 78, 415–423.
CAS PubMed Google Scholar - Morisseau, C., Goodrow, M. H., Dowdy, D., Zheng, J., Greene, J. F., et al. (1999). Potent urea and carbamate inhibitors of soluble epoxide hydrolases. Proceedings of the National Academy of Sciences of the United States of America, 96, 8849–8854.
CAS PubMed Google Scholar - Newman, J. W., Watanabe, T., & Hammock, B. D. (2002). The simultaneous quantification of cytochrome P450 dependent linoleate and arachidonate metabolites in urine by HPLC-MS/MS. Journal of Lipid Research, 43, 1563–1578.
CAS PubMed Google Scholar - Nelson, D. R., Zeldin, D. C., Hoffman, S. M., Maltais, L. J., Wain, H. M., et al. (2004). Comparison of cytochrome P450 (CYP) genes from the mouse and human genomes, including nomenclature recommendations for genes, pseudogenes and alternative-splice variants. Pharmacogenetics, 14, 1–18.
CAS PubMed Google Scholar - Nebert, D. W., & Russell, D. W. (2002). Clinical importance of the cytochromes P450. Lancet, 360, 1155–1162.
CAS PubMed Google Scholar - Wang, H., Zhao, Y., Bradbury, J. A., Graves, J. P., Foley, J., et al. (2004). Cloning, expression, and characterization of three new mouse cytochrome p450 enzymes and partial characterization of their fatty acid oxidation activities. Molecular Pharmacology, 65, 1148–1158.
CAS PubMed Google Scholar - Finta, C., & Zaphiropoulos, P. G. (2000). The human CYP2C locus: A prototype for intergenic and exon repetition splicing events. Genomics, 63, 433–438.
CAS PubMed Google Scholar - Waxman, D. J., Chen, L., Hecht, J. E., & Jounaidi, Y. (1999). Cytochrome P450-based cancer gene therapy: Recent advances and future prospects. Drug Metabolism Reviews, 31, 503–522.
CAS PubMed Google Scholar - Nebert, D. W., & Dalton, T. P. (2006). The role of cytochrome P450 enzymes in endogenous signalling pathways and environmental carcinogenesis. Nature Reviews. Cancer, 6, 947–960.
CAS PubMed Google Scholar - Swanson, H. I., Njar, V. C., Yu, Z., Castro, D. J., Gonzalez, F. J., et al. (2010). Targeting drug-metabolizing enzymes for effective chemoprevention and chemotherapy. Drug Metabolism and Disposition, 38, 539–544.
CAS PubMed Google Scholar - Lu, H., Chen, C. S., & Waxman, D. J. (2009). Potentiation of methoxymorpholinyl doxorubicin antitumor activity by P450 3A4 gene transfer. Cancer Gene Therapy, 16, 393–404.
PubMed Google Scholar - Jordan, V. C., & Brodie, A. M. (2007). Development and evolution of therapies targeted to the estrogen receptor for the treatment and prevention of breast cancer. Steroids, 72, 7–25.
CAS PubMed Google Scholar - Bruno, R. D., & Njar, V. C. (2007). Targeting cytochrome P450 enzymes: A new approach in anti-cancer drug development. Bioorganic & Medicinal Chemistry, 15, 5047–5060.
CAS Google Scholar - Moreira, V. M., Salvador, J. A., Vasaitis, T. S., & Njar, V. C. (2008). CYP17 inhibitors for prostate cancer treatment—An update. Current Medicinal Chemistry, 15, 868–899.
CAS PubMed Google Scholar - Node, K., Huo, Y., Ruan, X., Yang, B., Spiecker, M., et al. (1999). Anti-inflammatory properties of cytochrome P450 epoxygenase-derived eicosanoids. Science, 285, 1276–1279.
CAS PubMed Google Scholar - Rosolowsky, M., & Campbell, W. B. (1996). Synthesis of hydroxyeicosatetraenoic (HETEs) and epoxyeicosatrienoic acids (EETs) by cultured bovine coronary artery endothelial cells. Biochimica et Biophysica Acta, 1299, 267–277.
PubMed Google Scholar - Pozzi, A., Macias-Perez, I., Abair, T., Wei, S., Su, Y., et al. (2005). Characterization of 5, 6- and 8, 9-epoxyeicosatrienoic acids (5, 6- and 8, 9-EET) as potent in vivo angiogenic lipids. The Journal of Biological Chemistry, 280, 27138–27146.
CAS PubMed Google Scholar - Fleming, I. (2007). DiscrEET regulators of homeostasis: Epoxyeicosatrienoic acids, cytochrome P450 epoxygenases and vascular inflammation. Trends in Pharmacological Sciences, 28, 448–452.
CAS PubMed Google Scholar - Alkayed, N. J., Narayanan, J., Gebremedhin, D., Medhora, M., Roman, R. J., et al. (1996). Molecular characterization of an arachidonic acid epoxygenase in rat brain astrocytes. Stroke, 27, 971–979.
CAS PubMed Google Scholar - Amruthesh, S. C., Boerschel, M. F., McKinney, J. S., Willoughby, K. A., & Ellis, E. F. (1993). Metabolism of arachidonic acid to epoxyeicosatrienoic acids, hydroxyeicosatetraenoic acids, and prostaglandins in cultured rat hippocampal astrocytes. Journal of Neurochemistry, 61, 150–159.
CAS PubMed Google Scholar - Munzenmaier, D. H., & Harder, D. R. (2000). Cerebral microvascular endothelial cell tube formation: Role of astrocytic epoxyeicosatrienoic acid release. American Journal of Physiology. Heart and Circulatory Physiology, 278, H1163–H1167.
CAS PubMed Google Scholar - Wu, S., Moomaw, C. R., Tomer, K. B., Falck, J. R., & Zeldin, D. C. (1996). Molecular cloning and expression of CYP2J2, a human cytochrome P450 arachidonic acid epoxygenase highly expressed in heart. The Journal of Biological Chemistry, 271, 3460–3468.
CAS PubMed Google Scholar - Wu, S., Chen, W., Murphy, E., Gabel, S., Tomer, K. B., et al. (1997). Molecular cloning, expression, and functional significance of a cytochrome P450 highly expressed in rat heart myocytes. The Journal of Biological Chemistry, 272, 12551–12559.
CAS PubMed Google Scholar - Nakayama, K., Nitto, T., Inoue, T., & Node, K. (2008). Expression of the cytochrome P450 epoxygenase CYP2J2 in human monocytic leukocytes. Life Sciences, 83, 339–345.
CAS PubMed Google Scholar - Nieves, D., & Moreno, J. J. (2006). Hydroxyeicosatetraenoic acids released through the cytochrome P-450 pathway regulate 3 T6 fibroblast growth. Journal of Lipid Research, 47, 2681–2689.
CAS PubMed Google Scholar - Muthalif, M. M., Benter, I. F., Karzoun, N., Fatima, S., Harper, J., et al. (1998). 20-Hydroxyeicosatetraenoic acid mediates calcium/calmodulin-dependent protein kinase II-induced mitogen-activated protein kinase activation in vascular smooth muscle cells. Proceedings of the National Academy of Sciences of the United States of America, 95, 12701–12706.
CAS PubMed Google Scholar - Schwartzman, M. L., Falck, J. R., Yadagiri, P., & Escalante, B. (1989). Metabolism of 20-hydroxyeicosatetraenoic acid by cyclooxygenase. Formation and identification of novel endothelium-dependent vasoconstrictor metabolites. The Journal of Biological Chemistry, 264, 11658–11662.
CAS PubMed Google Scholar - Kaduce, T. L., Fang, X., Harmon, S. D., Oltman, C. L., Dellsperger, K. C., et al. (2004). 20-hydroxyeicosatetraenoic acid (20-HETE) metabolism in coronary endothelial cells. The Journal of Biological Chemistry, 279, 2648–2656.
CAS PubMed Google Scholar - Moreno, J. J. (2009). New aspects of the role of hydroxyeicosatetraenoic acids in cell growth and cancer development. Biochemical Pharmacology, 77, 1–10.
CAS PubMed Google Scholar - Stark, K., Dostalek, M., & Guengerich, F. P. (2008). Expression and purification of orphan cytochrome P450 4×1 and oxidation of anandamide. The FEBS Journal, 275, 3706–3717.
CAS PubMed Google Scholar - Chuang, S. S., Helvig, C., Taimi, M., Ramshaw, H. A., Collop, A. H., et al. (2004). CYP2U1, a novel human thymus- and brain-specific cytochrome P450, catalyzes omega- and (omega-1)-hydroxylation of fatty acids. The Journal of Biological Chemistry, 279, 6305–6314.
CAS PubMed Google Scholar - Yang, W., Holmes, B. B., Gopal, V. R., Kishore, R. V., Sangras, B., et al. (2007). Characterization of 14, 15-epoxyeicosatrienoyl-sulfonamides as 14, 15-epoxyeicosatrienoic acid agonists: Use for studies of metabolism and ligand binding. The Journal of Pharmacology and Experimental Therapeutics, 321, 1023–1031.
CAS PubMed Google Scholar - Spector, A. A. (2009). Arachidonic acid cytochrome P450 epoxygenase pathway. Journal of Lipid Research, 50(Suppl), S52–S56.
PubMed Google Scholar - Karara, A., Wei, S., Spady, D., Swift, L., Capdevila, J. H., et al. (1992). Arachidonic acid epoxygenase: Structural characterization and quantification of epoxyeicosatrienoates in plasma. Biochemical and Biophysical Research Communications, 182, 1320–1325.
CAS PubMed Google Scholar - Spector, A. A., Fang, X., Snyder, G. D., & Weintraub, N. L. (2004). Epoxyeicosatrienoic acids (EETs): Metabolism and biochemical function. Progress in Lipid Research, 43, 55–90.
CAS PubMed Google Scholar - Widstrom, R. L., Norris, A. W., Van Der Veer, J., & Spector, A. A. (2003). Fatty acid-binding proteins inhibit hydration of epoxyeicosatrienoic acids by soluble epoxide hydrolase. Biochemistry, 42, 11762–11767.
CAS PubMed Google Scholar - Liu, Y., Zhang, Y., Schmelzer, K., Lee, T. S., Fang, X., et al. (2005). The antiinflammatory effect of laminar flow: The role of PPARgamma, epoxyeicosatrienoic acids, and soluble epoxide hydrolase. Proceedings of the National Academy of Sciences of the United States of America, 102, 16747–16752.
CAS PubMed Google Scholar - Deng, Y., Theken, K. N., & Lee, C. R. (2010). Cytochrome P450 epoxygenases, soluble epoxide hydrolase, and the regulation of cardiovascular inflammation. Journal of Molecular and Cellular Cardiology, 48, 331–341.
CAS PubMed Google Scholar - Cowart, L. A., Wei, S., Hsu, M. H., Johnson, E. F., Krishna, M. U., et al. (2002). The CYP4A isoforms hydroxylate epoxyeicosatrienoic acids to form high affinity peroxisome proliferator-activated receptor ligands. The Journal of Biological Chemistry, 277, 35105–35112.
CAS PubMed Google Scholar - Fang, X., Hu, S., Watanabe, T., Weintraub, N. L., Snyder, G. D., et al. (2005). Activation of peroxisome proliferator-activated receptor alpha by substituted urea-derived soluble epoxide hydrolase inhibitors. The Journal of Pharmacology and Experimental Therapeutics, 314, 260–270.
CAS PubMed Google Scholar - Fang, X., Hu, S., Xu, B., Snyder, G. D., Harmon, S., et al. (2006). 14, 15-Dihydroxyeicosatrienoic acid activates peroxisome proliferator-activated receptor-alpha. American Journal of Physiology. Heart and Circulatory Physiology, 290, H55–H63.
CAS PubMed Google Scholar - Folkman, J. (1990). What is the evidence that tumors are angiogenesis-dependent? Journal of the National Cancer Institute, 82, 4–6.
CAS PubMed Google Scholar - McAllister, S. S., & Weinberg, R. A. (2010). Tumor-host interactions: A far-reaching relationship. Journal of Clinical Oncology, 28, 4022–4028.
PubMed Google Scholar - Panigrahy, D., Huang, S., Kieran, M. W., & Kaipainen, A. (2005). PPARgamma as a therapeutic target for tumor angiogenesis and metastasis. Cancer Biology & Therapy, 4, 687–693.
CAS Google Scholar - Bhowmick, N. A., Neilson, E. G., & Moses, H. L. (2004). Stromal fibroblasts in cancer initiation and progression. Nature, 432, 332–337.
CAS PubMed Google Scholar - Folkman, J. (1971). Tumor angiogenesis: Therapeutic implications. The New England Journal of Medicine, 285, 1182–1186.
CAS PubMed Google Scholar - Orimo, A., Gupta, P. B., Sgroi, D. C., Arenzana-Seisdedos, F., Delaunay, T., et al. (2005). Stromal fibroblasts present in invasive human breast carcinomas promote tumor growth and angiogenesis through elevated SDF-1/CXCL12 secretion. Cell, 121, 335–348.
CAS PubMed Google Scholar - Joyce, J. A., & Pollard, J. W. (2009). Microenvironmental regulation of metastasis. Nature Reviews. Cancer, 9, 239–252.
CAS PubMed Google Scholar - Lin, E. Y., & Pollard, J. W. (2004). Role of infiltrated leucocytes in tumour growth and spread. British Journal of Cancer, 90, 2053–2058.
CAS PubMed Google Scholar - de Visser, K. E., Eichten, A., & Coussens, L. M. (2006). Paradoxical roles of the immune system during cancer development. Nature Reviews. Cancer, 6, 24–37.
PubMed Google Scholar - Zhang, L., Conejo-Garcia, J. R., Katsaros, D., Gimotty, P. A., Massobrio, M., et al. (2003). Intratumoral T cells, recurrence, and survival in epithelial ovarian cancer. The New England Journal of Medicine, 348, 203–213.
CAS PubMed Google Scholar - Clevers, H. (2004). At the crossroads of inflammation and cancer. Cell, 118, 671–674.
CAS PubMed Google Scholar - Aggarwal, B. B., Shishodia, S., Sandur, S. K., Pandey, M. K., & Sethi, G. (2006). Inflammation and cancer: How hot is the link? Biochemical Pharmacology, 72, 1605–1621.
CAS PubMed Google Scholar - Seitz, C. S., Lin, Q., Deng, H., & Khavari, P. A. (1998). Alterations in NF-kappaB function in transgenic epithelial tissue demonstrate a growth inhibitory role for NF-kappaB. Proceedings of the National Academy of Sciences of the United States of America, 95, 2307–2312.
CAS PubMed Google Scholar - Dajee, M., Lazarov, M., Zhang, J. Y., Cai, T., Green, C. L., et al. (2003). NF-kappaB blockade and oncogenic Ras trigger invasive human epidermal neoplasia. Nature, 421, 639–643.
CAS PubMed Google Scholar - Karin, M. (2009). NF-kappaB as a critical link between inflammation and cancer. Cold Spring Harbor Perspectives in Biology, 1, a000141.
PubMed Google Scholar - Kaipainen, A., Kieran, M. W., Huang, S., Butterfield, C., Bielenberg, D., et al. (2007). PPARalpha deficiency in inflammatory cells suppresses tumor growth. PLoS ONE, 2, e260.
PubMed Google Scholar - Panigrahy, D., Kaipainen, A., Kieran, M.W., Huang, S. (2008). PPARs: A Double-Edged Sword in Cancer Therapy? PPAR Res 2008: 350351.
- Ono, M. (2008). Molecular links between tumor angiogenesis and inflammation: Inflammatory stimuli of macrophages and cancer cells as targets for therapeutic strategy. Cancer Science, 99, 1501–1506.
CAS PubMed Google Scholar - Salcedo, R., Zhang, X., Young, H. A., Michael, N., Wasserman, K., et al. (2003). Angiogenic effects of prostaglandin E2 are mediated by up-regulation of CXCR4 on human microvascular endothelial cells. Blood, 102, 1966–1977.
CAS PubMed Google Scholar - Freedman, R. S., Wang, E., Voiculescu, S., Patenia, R., Bassett, R. L., Jr., et al. (2007). Comparative analysis of peritoneum and tumor eicosanoids and pathways in advanced ovarian cancer. Clinical Cancer Research, 13, 5736–5744.
CAS PubMed Google Scholar - Ishizuka, T., Cheng, J., Singh, H., Vitto, M. D., Manthati, V. L., et al. (2008). 20-Hydroxyeicosatetraenoic acid stimulates nuclear factor-kappaB activation and the production of inflammatory cytokines in human endothelial cells. The Journal of Pharmacology and Experimental Therapeutics, 324, 103–110.
CAS PubMed Google Scholar - Guo, A. M., Arbab, A. S., Falck, J. R., Chen, P., Edwards, P. A., et al. (2007). Activation of vascular endothelial growth factor through reactive oxygen species mediates 20-hydroxyeicosatetraenoic acid-induced endothelial cell proliferation. The Journal of Pharmacology and Experimental Therapeutics, 321, 18–27.
CAS PubMed Google Scholar - Dhanasekaran, A., Bodiga, S., Gruenloh, S., Gao, Y., Dunn, L., et al. (2009). 20-HETE increases survival and decreases apoptosis in pulmonary arteries and pulmonary artery endothelial cells. American Journal of Physiology. Heart and Circulatory Physiology, 296, H777–H786.
CAS PubMed Google Scholar - Chen, P., Guo, M., Wygle, D., Edwards, P. A., Falck, J. R., et al. (2005). Inhibitors of cytochrome P450 4A suppress angiogenic responses. The American Journal of Pathology, 166, 615–624.
CAS PubMed Google Scholar - Ljubimov, A. V., & Grant, M. B. (2005). P450 in the angiogenesis affair: The unusual suspect. The American Journal of Pathology, 166, 341–344.
CAS PubMed Google Scholar - Amaral, S. L., Maier, K. G., Schippers, D. N., Roman, R. J., & Greene, A. S. (2003). CYP4A metabolites of arachidonic acid and VEGF are mediators of skeletal muscle angiogenesis. American Journal of Physiology. Heart and Circulatory Physiology, 284, H1528–H1535.
CAS PubMed Google Scholar - Sa, G., Murugesan, G., Jaye, M., Ivashchenko, Y., & Fox, P. L. (1995). Activation of cytosolic phospholipase A2 by basic fibroblast growth factor via a p42 mitogen-activated protein kinase-dependent phosphorylation pathway in endothelial cells. The Journal of Biological Chemistry, 270, 2360–2366.
CAS PubMed Google Scholar - Jiang, M., Mezentsev, A., Kemp, R., Byun, K., Falck, J. R., et al. (2004). Smooth muscle-specific expression of CYP4A1 induces endothelial sprouting in renal arterial microvessels. Circulation Research, 94, 167–174.
CAS PubMed Google Scholar - Miyata, N., Taniguchi, K., Seki, T., Ishimoto, T., Sato-Watanabe, M., et al. (2001). HET0016, a potent and selective inhibitor of 20-HETE synthesizing enzyme. British Journal of Pharmacology, 133, 325–329.
CAS PubMed Google Scholar - Guo, M., Roman, R. J., Falck, J. R., Edwards, P. A., & Scicli, A. G. (2005). Human U251 glioma cell proliferation is suppressed by HET0016 [N-hydroxy-N’-(4-butyl-2-methylphenyl)formamidine], a selective inhibitor of CYP4A. The Journal of Pharmacology and Experimental Therapeutics, 315, 526–533.
CAS PubMed Google Scholar - Jacobs, E. R., Zhu, D., Gruenloh, S., Lopez, B., & Medhora, M. (2006). VEGF-induced relaxation of pulmonary arteries is mediated by endothelial cytochrome P-450 hydroxylase. American Journal of Physiology. Lung Cellular and Molecular Physiology, 291, L369–L377.
CAS PubMed Google Scholar - Guo, A. M., Sheng, J., Scicli, G. M., Arbab, A. S., Lehman, N. L., et al. (2008). Expression of CYP4A1 in U251 human glioma cell induces hyperproliferative phenotype in vitro and rapidly growing tumors in vivo. The Journal of Pharmacology and Experimental Therapeutics, 327, 10–19.
CAS PubMed Google Scholar - Guo, M., Roman, R. J., Fenstermacher, J. D., Brown, S. L., Falck, J. R., et al. (2006). 9 L gliosarcoma cell proliferation and tumor growth in rats are suppressed by N-hydroxy-N’-(4-butyl-2-methylphenol) formamidine (HET0016), a selective inhibitor of CYP4A. The Journal of Pharmacology and Experimental Therapeutics, 317, 97–108.
CAS PubMed Google Scholar - Alexanian, A., Rufanova, V. A., Miller, B., Flasch, A., Roman, R. J., et al. (2009). Down-regulation of 20-HETE synthesis and signaling inhibits renal adenocarcinoma cell proliferation and tumor growth. Anticancer Research, 29, 3819–3824.
CAS PubMed Google Scholar - Nithipatikom, K., Isbell, M. A., See, W. A., & Campbell, W. B. (2006). Elevated 12- and 20-hydroxyeicosatetraenoic acid in urine of patients with prostatic diseases. Cancer Letters, 233, 219–225.
CAS PubMed Google Scholar - Fleming, I. (2008). Vascular cytochrome p450 enzymes: Physiology and pathophysiology. Trends in Cardiovascular Medicine, 18, 20–25.
CAS PubMed Google Scholar - Gross, G. J., Falck, J. R., Gross, E. R., Isbell, M., Moore, J., et al. (2005). Cytochrome P450 and arachidonic acid metabolites: Role in myocardial ischemia/reperfusion injury revisited. Cardiovascular Research, 68, 18–25.
CAS PubMed Google Scholar - Chaudhary, K. R., Batchu, S. N., Das, D., Suresh, M. R., Falck, J. R., et al. (2009). Role of B-type natriuretic peptide in epoxyeicosatrienoic acid-mediated improved post-ischaemic recovery of heart contractile function. Cardiovascular Research, 83, 362–370.
CAS PubMed Google Scholar - Simpkins, A. N., Rudic, R. D., Schreihofer, D. A., Roy, S., Manhiani, M., et al. (2009). Soluble epoxide inhibition is protective against cerebral ischemia via vascular and neural protection. The American Journal of Pathology, 174, 2086–2095.
CAS PubMed Google Scholar - Zhang, C., & Harder, D. R. (2002). Cerebral capillary endothelial cell mitogenesis and morphogenesis induced by astrocytic epoxyeicosatrienoic acid. Stroke, 33, 2957–2964.
CAS PubMed Google Scholar - Medhora, M., Daniels, J., Mundey, K., Fisslthaler, B., Busse, R., et al. (2003). Epoxygenase-driven angiogenesis in human lung microvascular endothelial cells. American Journal of Physiology. Heart and Circulatory Physiology, 284, H215–H224.
CAS PubMed Google Scholar - Ausprunk, D. H., & Folkman, J. (1977). Migration and proliferation of endothelial cells in preformed and newly formed blood vessels during tumor angiogenesis. Microvascular Research, 14, 53–61.
CAS PubMed Google Scholar - Wang, Y., Wei, X., Xiao, X., Hui, R., Card, J. W., et al. (2005). Arachidonic acid epoxygenase metabolites stimulate endothelial cell growth and angiogenesis via mitogen-activated protein kinase and phosphatidylinositol 3-kinase/Akt signaling pathways. The Journal of Pharmacology and Experimental Therapeutics, 314, 522–532.
CAS PubMed Google Scholar - Ribatti, D., Vacca, A., Roncali, L., & Dammacco, F. (2000). The chick embryo chorioallantoic membrane as a model for in vivo research on antiangiogenesis. Current Pharmaceutical Biotechnology, 1, 73–82.
CAS PubMed Google Scholar - Michaelis, U. R., Fisslthaler, B., Medhora, M., Harder, D., Fleming, I., et al. (2003). Cytochrome P450 2 C9-derived epoxyeicosatrienoic acids induce angiogenesis via cross-talk with the epidermal growth factor receptor (EGFR). The FASEB Journal, 17, 770–772.
CAS PubMed Google Scholar - Yan, G., Chen, S., You, B., & Sun, J. (2008). Activation of sphingosine kinase-1 mediates induction of endothelial cell proliferation and angiogenesis by epoxyeicosatrienoic acids. Cardiovascular Research, 78, 308–314.
CAS PubMed Google Scholar - Webler, A. C., Popp, R., Korff, T., Michaelis, U. R., Urbich, C., et al. (2008). Cytochrome P450 2 C9-induced angiogenesis is dependent on EphB4. Arteriosclerosis, Thrombosis, and Vascular Biology, 28, 1123–1129.
CAS PubMed Google Scholar - Zhang, B., Cao, H., & Rao, G. N. (2006). Fibroblast growth factor-2 is a downstream mediator of phosphatidylinositol 3-kinase-Akt signaling in 14, 15-epoxyeicosatrienoic acid-induced angiogenesis. The Journal of Biological Chemistry, 281, 905–914.
CAS PubMed Google Scholar - Cheranov, S. Y., Karpurapu, M., Wang, D., Zhang, B., Venema, R. C., et al. (2008). An essential role for SRC-activated STAT-3 in 14, 15-EET-induced VEGF expression and angiogenesis. Blood, 111, 5581–5591.
CAS PubMed Google Scholar - Yang, S., Lin, L., Chen, J. X., Lee, C. R., Seubert, J. M., et al. (2007). Cytochrome P-450 epoxygenases protect endothelial cells from apoptosis induced by tumor necrosis factor-alpha via MAPK and PI3K/Akt signaling pathways. American Journal of Physiology. Heart and Circulatory Physiology, 293, H142–H151.
CAS PubMed Google Scholar - Tsuzuki, Y., Fukumura, D., Oosthuyse, B., Koike, C., Carmeliet, P., et al. (2000). Vascular endothelial growth factor (VEGF) modulation by targeting hypoxia-inducible factor-1alpha– > hypoxia response element– > VEGF cascade differentially regulates vascular response and growth rate in tumors. Cancer Research, 60, 6248–6252.
CAS PubMed Google Scholar - Michaelis, U. R., Fisslthaler, B., Barbosa-Sicard, E., Falck, J. R., Fleming, I., et al. (2005). Cytochrome P450 epoxygenases 2 C8 and 2 C9 are implicated in hypoxia-induced endothelial cell migration and angiogenesis. Journal of Cell Science, 118, 5489–5498.
CAS PubMed Google Scholar - Earley, S., Pastuszyn, A., & Walker, B. R. (2003). Cytochrome p-450 epoxygenase products contribute to attenuated vasoconstriction after chronic hypoxia. American Journal of Physiology. Heart and Circulatory Physiology, 285, H127–H136.
CAS PubMed Google Scholar - Suzuki, S., Oguro, A., Osada-Oka, M., Funae, Y., & Imaoka, S. (2008). Epoxyeicosatrienoic acids and/or their metabolites promote hypoxic response of cells. Journal of Pharmacological Sciences, 108, 79–88.
CAS PubMed Google Scholar - Webler, A. C., Michaelis, U. R., Popp, R., Barbosa-Sicard, E., Murugan, A., et al. (2008). Epoxyeicosatrienoic acids are part of the VEGF-activated signaling cascade leading to angiogenesis. American Journal of Physiology. Cell Physiology, 295, C1292–C1301.
CAS PubMed Google Scholar - Yang, S., Wei, S., Pozzi, A., & Capdevila, J. H. (2009). The arachidonic acid epoxygenase is a component of the signaling mechanisms responsible for VEGF-stimulated angiogenesis. Archives of Biochemistry and Biophysics, 489, 82–91.
CAS PubMed Google Scholar - Dunn, L. K., Gruenloh, S. K., Dunn, B. E., Reddy, D. S., Falck, J. R., et al. (2005). Chick chorioallantoic membrane as an in vivo model to study vasoreactivity: Characterization of development-dependent hyperemia induced by epoxyeicosatrienoic acids (EETs). The Anatomical Record. Part A: Discoveries in Molecular, Cellular, and Evolutionary Biology, 285, 771–780.
Google Scholar - Harris, R. C., Homma, T., Jacobson, H. R., & Capdevila, J. (1990). Epoxyeicosatrienoic acids activate Na+/H+exchange and are mitogenic in cultured rat glomerular mesangial cells. Journal of Cellular Physiology, 144, 429–437.
CAS PubMed Google Scholar - Sellmayer, A., Uedelhoven, W. M., Weber, P. C., & Bonventre, J. V. (1991). Endogenous non-cyclooxygenase metabolites of arachidonic acid modulate growth and mRNA levels of immediate-early response genes in rat mesangial cells. The Journal of Biological Chemistry, 266, 3800–3807.
CAS PubMed Google Scholar - Jiang, J. G., Chen, C. L., Card, J. W., Yang, S., Chen, J. X., et al. (2005). Cytochrome P450 2 J2 promotes the neoplastic phenotype of carcinoma cells and is up-regulated in human tumors. Cancer Research, 65, 4707–4715.
CAS PubMed Google Scholar - Jiang, J. G., Ning, Y. G., Chen, C., Ma, D., Liu, Z. J., et al. (2007). Cytochrome p450 epoxygenase promotes human cancer metastasis. Cancer Research, 67, 6665–6674.
CAS PubMed Google Scholar - Chen, C., Li, G., Liao, W., Wu, J., Liu, L., et al. (2009). Selective inhibitors of CYP2J2 related to terfenadine exhibit strong activity against human cancers in vitro and in vivo. The Journal of Pharmacology and Experimental Therapeutics, 329, 908–918.
CAS PubMed Google Scholar - Pozzi, A., Ibanez, M. R., Gatica, A. E., Yang, S., Wei, S., et al. (2007). Peroxisomal proliferator-activated receptor-alpha-dependent inhibition of endothelial cell proliferation and tumorigenesis. The Journal of Biological Chemistry, 282, 17685–17695.
CAS PubMed Google Scholar - Zagorac, D., Jakovcevic, D., Gebremedhin, D., & Harder, D. R. (2008). Antiangiogenic effect of inhibitors of cytochrome P450 on rats with glioblastoma multiforme. Journal of Cerebral Blood Flow and Metabolism, 28, 1431–1439.
CAS PubMed Google Scholar - Nithipatikom, K., Brody, D.M., Tang, A.T., Manthati, V.L., Falck, J.R., et al. (2010). Inhibition of carcinoma cell motility by epoxyeicosatrienoic acid (EET) antagonists. Cancer Sci.
- Pozzi, A., Popescu, V., Yang, S., Mei, S., Shi, M., et al. (2010). The anti-tumorigenic properties of peroxisomal proliferator-activated receptor alpha are arachidonic acid epoxygenase-mediated. The Journal of Biological Chemistry, 285, 12840–12850.
CAS PubMed Google Scholar - Enayetallah, A. E., French, R. A., & Grant, D. F. (2006). Distribution of soluble epoxide hydrolase, cytochrome P450 2 C8, 2 C9 and 2 J2 in human malignant neoplasms. Journal of Molecular Histology, 37, 133–141.
CAS PubMed Google Scholar - Leclerc, J., Tournel, G., Courcot-Ngoubo Ngangue, E., Pottier, N., Lafitte, J. J., et al. (2010). Profiling gene expression of whole cytochrome P450 superfamily in human bronchial and peripheral lung tissues: Differential expression in non-small cell lung cancers. Biochimie, 92, 292–306.
CAS PubMed Google Scholar - Yang, M. D., Wu, C. C., Chiou, S. H., Chiu, C. F., Lin, T. Y., et al. (2003). Reduction of dihydrodiol dehydrogenase expression in resected hepatocellular carcinoma. Oncology Reports, 10, 271–276.
CAS PubMed Google Scholar - Roques, M., Bagrel, D., Magdalou, J., & Siest, G. (1991). Expression of arylhydrocarbon hydroxylase, epoxide hydrolases, glutathione S-transferase and UDP-glucuronosyltransferases in H5-6 hepatoma cells. General Pharmacology, 22, 677–684.
CAS PubMed Google Scholar - Rahman, A., Korzekwa, K. R., Grogan, J., Gonzalez, F. J., & Harris, J. W. (1994). Selective biotransformation of taxol to 6 alpha-hydroxytaxol by human cytochrome P450 2C8. Cancer Research, 54, 5543–5546.
CAS PubMed Google Scholar - Jernstrom, H., Bageman, E., Rose, C., Jonsson, P. E., & Ingvar, C. (2009). CYP2C8 and CYP2C9 polymorphisms in relation to tumour characteristics and early breast cancer related events among 652 breast cancer patients. British Journal of Cancer, 101, 1817–1823.
CAS PubMed Google Scholar - Dai, D., Zeldin, D. C., Blaisdell, J. A., Chanas, B., Coulter, S. J., et al. (2001). Polymorphisms in human CYP2C8 decrease metabolism of the anticancer drug paclitaxel and arachidonic acid. Pharmacogenetics, 11, 597–607.
CAS PubMed Google Scholar - Isomura, Y., Yamaji, Y., Ohta, M., Seto, M., Asaoka, Y., et al. (2010). A genetic polymorphism of CYP2C19 is associated with susceptibility to biliary tract cancer. J Gastroenterol.