Direct evidence of attempted beta cell regeneration in an 89-year-old patient with recent-onset type 1 diabetes (original) (raw)
Abstract
Aims/hypothesis
We investigated whether there was evidence of attempted beta cell regeneration in the pancreas obtained from a patient with recent-onset type 1 diabetes, and if so by what mechanism this occurred.
Subjects, materials and methods
We examined pancreas tissue from a lean 89-year-old patient (BMI 18.0 kg/m2) with recent-onset type 1 diabetes who had had a distal pancreatectomy to remove a low-grade pancreatic intraepithelial neoplasia.
Results
In the tumour-free tissue, the fractional beta cell area was 0.54±0.2% of pancreas area (about one-third of that in non-diabetic humans). CD3-positive T lymphocytes and macrophages had infiltrated the majority of the islets. Subclassification of the T cell population revealed a predominance of CD8-positive cells over CD4-positive cells. Beta cell apoptosis (terminal deoxynucleotidyl transferase-mediated dUTP-biotin nick-end labelling [TUNEL] staining) was greatly increased, consistent with ongoing immune-mediated beta cell destruction. There was also a marked increase (more than ∼100-fold) in the frequency of beta cell replication (0.69±0.15% Ki67-positive beta cells) in all blocks examined.
Conclusions/interpretation
The present report provides direct evidence of attempted beta cell regeneration through the mechanism of beta cell replication in a case of newly diagnosed type 1 diabetes, and affirms that beta cell apoptosis is an important mechanism for beta cell loss in type 1 diabetes.
Similar content being viewed by others
Introduction
Type 1 diabetes is believed to be due to immune-mediated beta cell loss, leading to insufficient insulin secretion [1–3]. It has long been recognised that a few beta cells can still be identified in pancreas tissue from people with longstanding type 1 diabetes [4–6]. We recently reported that beta cells in longstanding type 1 diabetes have increased apoptosis and are associated with a low-grade immune infiltrate [7]. These data affirm that increased beta cell apoptosis is present in humans with type 1 diabetes, as previously reported in non-obese diabetic (NOD) mice [8]. The presence of beta cells with ongoing beta cell apoptosis up to 60 years after the onset of type 1 diabetes also provided indirect evidence of ongoing beta cell formation in patients with longstanding type 1 diabetes [7]. These studies were limited because, among other reasons, the pancreas tissue was obtained at autopsy and there were insufficient beta cells to establish whether the source of the putative new beta cell formation was beta cell replication.
The present report is unique inasmuch as ∼50% of the pancreas became available via surgery from an 89-year-old man with recent-onset type 1 diabetes. This permitted us (1) to establish in freshly fixed tissue that beta cell apoptosis is indeed an important mechanism of beta cell destruction in humans with type 1 diabetes, and (2) to establish directly the presence of attempted beta cell regeneration through the mechanism of a more than 100-fold increase in the frequency of beta cell replication.
Subject, materials and methods
Case history
An 89-year-old male presented to a community hospital complaining of knee pain and failure to thrive. A review of his systems was significant for an unexplained weight loss of 9 kg over the past 3 months. At the time of presentation, his height was 1.75 m, his weight was 55 kg and his BMI was 18.0 kg/m2. His family history included a sister who developed diabetes late in life. His only medication prior to hospitalisation was aspirin (81 mg/day). The physical examination was normal except for patches of vitiligo on his chest. Thyroid function tests revealed a mildly elevated concentration of thyroid-stimulating hormone (5.07 μIU/ml; normal range 0.46–4.68 μIU/ml), normal free T4 (13.4 pmol/l [1.04 ng/dl]; normal range 10.3–28.2 pmol/l [0.78–2.19 ng/dl]), mildly decreased total T3 (1.37 pmol/l [89 ng/dl]; normal range 1.5–2.6 pmol/l [97–169 ng/dl]), and positive thyroid peroxidase antibodies (975 IU/ml), consistent with Hashimoto’s thyroiditis. Laboratory studies showed a random blood glucose of 19.4 mmol/l (349 mg/dl), an HbA1c concentration of 10.8% (normal range 4.4–5.9%), glucosuria 4+ on urinalysis, and high titres of islet autoantibodies (GAD 65 antibodies, 65 U/ml; normal range <0.5 U/ml; ICA-512, 49 U/ml; normal range <1 U/ml). The plasma C-peptide measured when the plasma glucose was 12.7 mmol/l was undetectable (limit of detection 0.5 ng/ml [1.5 nmol/l], reference range at normal fasting glucose 0.36–1.52 nmol/l [1.1–4.6 ng/ml]). Computed tomography (CT) of the abdomen was performed because of the weight loss. This CT scan demonstrated a mass of 3.1×1.8 cm at the distal body and tail of the pancreas. He was referred to UCLA, where he was started on insulin and levothyroxine. Subsequently, the patient underwent an exploratory laparotomy with an en bloc distal pancreatectomy (8×4×1.8 cm of the distal pancreas) with splenectomy.
Pancreas resection and tissue treatment
Resected pancreas tissue was obtained for histological evaluation after written informed consent had been obtained from the patient, in compliance with institutional review board guidelines. Specimens from five areas of the pancreas were fixed in formaldehyde and embedded in paraffin for subsequent analysis as described previously [7]. Sequential 5 μm sections from each block were stained as follows: (1) haematoxylin and eosin for light microscopy; (2) insulin (peroxidase staining) and haematoxylin for light microscopy; (3) insulin, terminal deoxynucleotidyl transferase-mediated dUTP-biotin nick-end labelling (TUNEL) and 4,6-diamidino-2-phenylindole (DAPI) (immunofluorescence); (4) insulin, Ki67 and DAPI (immunofluorescence); (5) insulin, CD3, CD68 (immunofluorescence); (6) insulin, CD4 and DAPI (immunofluorescence); (7) insulin and CD8 (immunofluorescence); and (8) glucagon (peroxidase staining) and haematoxylin for light microscopy.
For immunohistochemistry the following primary antibodies were used: guinea-pig anti-insulin, 1:400 (Dako, Glostrup, Denmark; lot no. 50381573); mouse anti-Ki67, 1:200 (MIB-1; Dako; lot no. 00014101); rabbit-anti glucagon, 1:500 (Immunostar; lot no. 13627); rabbit anti-gastrin, 1:100 (Novocastra, Newcastle upon Tyne, UK, lot no. 402203); rabbit anti-CD3, 1:100 (Dako; lot no. 9110); mouse anti-CD68, 1:100 (Dako; lot no. 5445); mouse anti-CD4, 1:50 (Acris Antibodies, Hiddenhausen, Germany; lot no. 310305); mouse anti-CD8, 1:50 (Acris; lot no. 527C).
Secondary antibodies labelled with Cy3, fluorescein isothiocyanate and aminomethylcoumarin acetate were obtained from Jackson Laboratories (West Grove, PA, USA) and used at dilutions of 1:100. For TUNEL staining, a commercially available kit was used (In Situ Cell Death Detection Kit; Roche Diagnostics, Indianapolis, IN, USA) was used.
Morphometric analysis
To measure the fractional alpha and beta cell areas, each pancreatic section was imaged at 40× magnification (4× objective), and the ratios of alpha cell area or beta cell area to the exocrine area were quantified digitally as previously described [7] using Image Pro Plus software (Media Cybernetics, Silver Springs, MD, USA).
To quantify beta cell apoptosis, the total number of TUNEL-positive beta cells per section was counted and expressed in relation to the respective beta cell area. For quantification of beta cell replication, ten random islets per section were imaged, and the number of cells co-staining for insulin and Ki67 was quantified in each islet and expressed as the percentage of the total number of beta cells. In this way a total of 3,030 beta cells were evaluated. The extent of T lymphocyte infiltration was evaluated by counting the number of CD4- and CD8-positive cells in or adjacent to ten random islets per block. Adjacency to an islet was defined as less than five nuclei distant in the plane of section from an islet. An islet was classified as being infiltrated if five or more T cells were present inside or adjacent, within the plane of section of the islet.
Results
Histological evaluation of the pancreas tumour revealed a low-grade pancreatic intraepithelial neoplasia (PanIN 1), the pre-invasive form of ductal pancreatic cancer (Fig. 1). The tumour was negative for the endocrine markers insulin, glucagon and gastrin, except for occasional islets entrapped between tumour stroma. This was therefore a pre-invasive form of pancreatic cancer and not an endocrine tumour.
Fig. 1
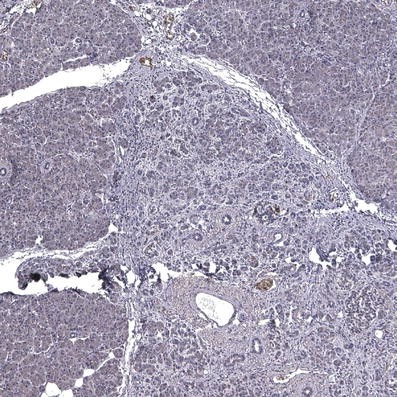
Pancreatic section from an 89-year-old patient presenting with recent-onset type 1 diabetes, stained for insulin and haematoxylin, showing the low-grade pancreatic intraepithelial neoplasia and adjacent normal pancreatic tissue. The image was acquired at 10× objective magnification
Staining of the tumour-free pancreas sections for CD3 and CD68 revealed a high-grade islet infiltration with T lymphocytes and macrophages in all five pancreas blocks examined (Figs. 2 and 3), consistent with the clinical diagnosis of recent-onset type 1 diabetes. The extent of islet infiltration was much more pronounced than in the cases of longstanding type 1 diabetes studied previously by our group [7], and appeared similar to that typically found at diabetes onset in NOD mice [9]. Interestingly, the appearance of this infiltration was quite heterogeneous, some islets being completely unaffected and others showing a marked invasion of T lymphocytes. The T lymphocytes were primarily organised as a peri-islet infiltration, with ∼2 to 3 times more T cells present around the islets than inside the islet structure.
Fig. 2
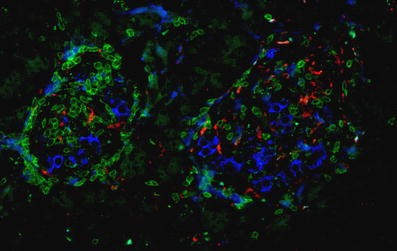
Pancreatic section from an 89-year-old patient presenting with recent-onset type 1 diabetes, stained for insulin (blue), CD3 (green) and CD68 (red), showing lymphocytic infiltration of the islets. The image was acquired at 20× objective magnification
Fig. 3
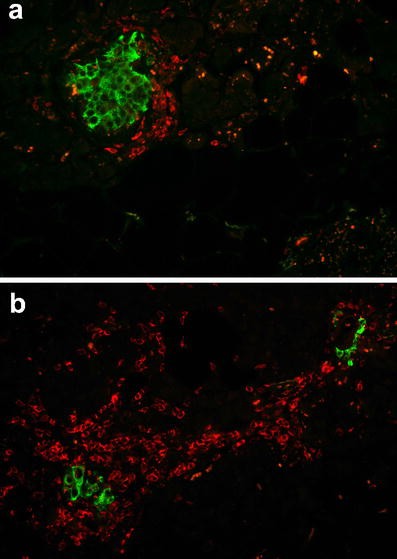
Pancreatic sections from an 89-year-old patient presenting with recent-onset type 1 diabetes, stained for (a) insulin (green) and CD4 (red), and (b) for insulin (green) and CD8 (red). The images were acquired at 20× objective magnification
Further characterisation of the T cell population showed both CD4- and CD8-positive cells; the latter were most frequent (Figs. 2 and 3). The average number of CD4-positive cells (in a single plane of section) was 2.8±1.2 per islet (range 0–50 cells/islet) within the islets and 5.8±1.2 per islet (range 0–28 cells/islet) around islets. The respective numbers of CD8-positive lymphocytes per single plane of section were 3.13±0.64 per islet (range 0–17 cells/islet) within islets and 11.48±1.64 per islet (range 0–57 cells/islet) around the islets. An islet infiltration was present in 36% of the islets based on CD4-positive T cells, and in 64% of the islets based on CD8-positive T lymphocytes.
The fractional beta cell area in the tumour-free pancreas sections of the patients was 0.54±0.2% of the total pancreatic area, corresponding to a deficit of ∼70–80% in fractional beta cell area compared with lean non-diabetic individuals [10, 11]. Again, there was a large degree of heterogeneity between different pancreas blocks (range 0.014–1.12%). In contrast, the fractional alpha cell area was within normal limits (0.56±0.08%, range 0.30–0.78%) [11].
Beta cell apoptosis, as measured by the frequency of TUNEL staining, was clearly increased in the pancreas of the patient (212±184 TUNEL-positive beta cells/cm2 beta cell area; range 4.6–945 cells/cm2) (Fig. 4). Beta cell apoptosis occurred in a focal pattern, with sometimes up to four beta cells within one islet staining positive for TUNEL and other islets being completely free of apoptotic cells.
Fig. 4
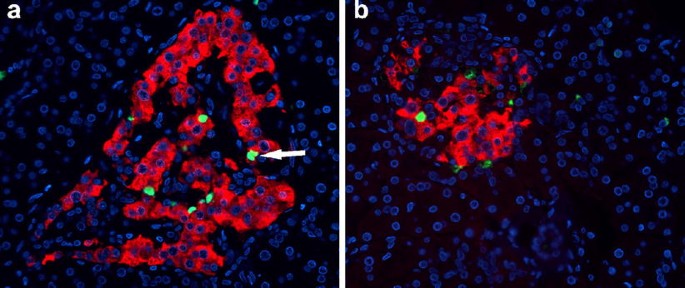
Two pancreatic sections (a, b) from an 89-year-old patient presenting with recent-onset type 1 diabetes, stained for insulin (red), TUNEL (green) and DAPI (blue), showing multiple apoptotic beta cells. The arrow (a) points to a doublet of apoptotic beta cells, indicative of postmitotic apoptosis. The images were acquired at 20× objective magnification
Ki67 staining demonstrated a high frequency of beta cell replication in all pancreas sections examined (Fig. 5). On average, 0.69±0.15% (range 0.32–1.1%) of beta cells were positive for Ki67. There was no apparent increase in the frequency of periductal islets or insulin-positive cells in duct cells compared with non-diabetic controls.
Fig. 5
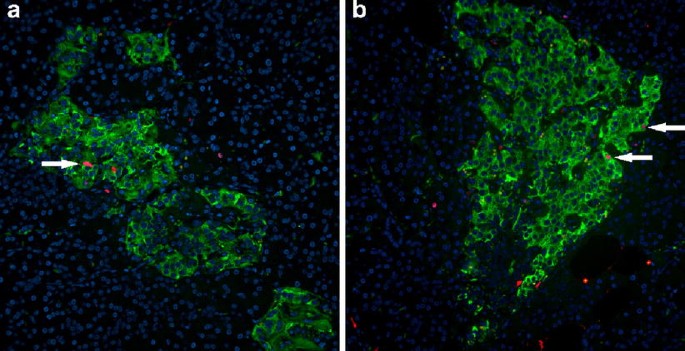
Two pancreatic sections (a, b) from an 89-year-old patient presenting with recent-onset type 1 diabetes, stained for insulin (green), Ki67 (red) and DAPI (blue), showing multiple replicating beta cells (arrows). The images were acquired at 20× objective magnification
Discussion
The present report provides direct evidence of attempted beta cell regeneration through the mechanism of beta cell replication in a case of newly diagnosed type 1 diabetes, and affirms that beta cell apoptosis is an important mechanism for beta cell loss in type 1 diabetes.
The late age of onset of diabetes sometimes raises questions about the diagnosis of type 1 diabetes. However, the incidence of type 1 diabetes remains remarkably constant throughout life [12]. Also, given the extensive T cell insulitis, the high titres of islet autoantibodies, the obvious susceptibility to developing autoimmune disorders (indicated by the presence of Hashimoto’s thyroiditis and vitiligo), the absence of detectable C-peptide secretion from the pancreas remnant and the low BMI of the patient, there is little doubt that this patient did indeed have type 1 diabetes.
Beta cell replication in adult human pancreas is rare (∼1 positive cell in 20 islets, corresponding to a frequency of ∼0.001%) [7, 10, 13–15]. However, the present report reveals a more than 100-fold increased frequency (0.69% Ki67-positive beta cells) in an 89-year-old man with recent-onset type 1 diabetes.
This finding now provides direct evidence that attempted beta cell regeneration in humans with type 1 diabetes does indeed occur [7, 16]. The fact that this was present in a man 89 years of age shows that there is a lifelong capacity to induce beta cell replication in humans. This finding is also consistent with findings in mice using lineage tracing [17] and cyclin D2 deletion [18], which identified beta cell replication as the primary mechanism for postnatal beta cell expansion.
Despite the marked increase in beta cell replication, the number of beta cells was decreased to ∼20–30% of normal in this case [10, 11]. The mechanism subserving this was presumably the increased beta cell apoptosis, now confirmed in freshly fixed pancreas tissue. The relatively slow evolution of type 1 diabetes (up to 10 years from detection of islet antibodies to the onset of hyperglycaemia [1]) may reflect the concurrent processes of beta cell regeneration and increased beta cell apoptosis. Clinical studies showing a continued increased capacity for stimulated insulin secretion during childhood in the presence of islet-related autoantibodies provide indirect evidence for simultaneous beta cell destruction and attempted growth and/or regeneration [19]. That beta cell apoptosis eventually overcomes attempted beta cell replication may be explained by the fact that replicating beta cells are more vulnerable to cytokine-induced apoptosis [20].
An interesting question prompted by the present case report concerns the mechanism underlying the increased beta cell replication. Hyperglycaemia may have been responsible, although the absence of increased beta cell replication in patients with type 2 diabetes is inconsistent with this [10]. Alternatively, the islet infiltration itself may contribute to the induction of beta cell replication. Cytokines, such as IL-1β, induce beta cell replication in isolated human islets at certain concentrations [21]. Also, the onset of diabetes in NOD mice diabetes is preceded by increased beta cell proliferation [22]. Another possible explanation for increased beta cell replication in the present case is the secretion of cytokines by the pancreatic tumour. However, this seems unlikely for several reasons. Pancreatic cancers are common and are characterised by islet atrophy rather than beta cell expansion [23, 24]. In the present case there was no relationship between the proximity of islets to the tumour and the extent of beta cell replication. It is also important to point out that this tumour was not an endocrine tumour, and specifically not a tumour of the multiple endocrine neoplasia type.
While the present study directly documents beta cell regeneration by beta cell replication, it does not exclude the possibility that there are other means of new beta cell formation (so-called neogenesis). However, the absence of any perceptible increase in periductal islets or intraductal insulin-positive cells in this case, with an increase in beta cell replication greater than 100-fold, implies that beta cell regeneration in humans is accomplished predominately by beta cell replication.
There was marked heterogeneity of immune infiltrate and the extent of beta cell loss. The fractional beta cell area varied by ∼80-fold between different blocks of pancreas tissue from the same patient. While some islets were completely free of a T cell infiltration and beta cell apoptosis, other islets showed marked insulitis accompanied by high frequencies of beta cell apoptosis. This finding is in agreement with previous studies in NOD mice [25] and in humans [26]. Since both peri-islet and intra-islet infiltration were often confined to a single pole of an islet, it is possible that some islets that appeared infiltration-free were negative because the plane of section missed the infiltration. Nonetheless, this would not explain the remarkable variability in fractional beta cell area observed in different sections.
The finding of attempted beta cell regeneration in an 89-year-old patient with recent-onset type 1 diabetes has interesting clinical implications. If beta cells maintain their capacity to undergo replication through the ninth decade of life, regeneration of beta cells would appear to be possible provided sufficient beta cells remain [27]. However, the deficit in beta cells despite an increase of more than 100-fold in beta cell replication emphasises the futility of seeking to foster beta cell replication as a strategy to reverse diabetes without first attending to the underlying mechanisms leading to increased beta cell apoptosis. The most crucial requirement to permit successful islet regeneration in type 1 diabetes will be to inhibit the continued autoimmune destruction of beta cells. In NOD mice, endogenous regeneration of beta cells has been reported after treatment with CD3 antibodies [9] and infusions of spleen cells [28–30]. It is noteworthy that promoting beta cell replication was not required in any of these studies. There has also been some initial success in preserving insulin secretion in patients with recent-onset type 1 diabetes treated with CD3 antibodies [31].
The present report provides direct evidence of attempted beta cell regeneration through the mechanism of beta cell replication in type 1 diabetes and reveals that human beta cells retain the capacity to replicate throughout life. We conclude that selective inhibition of immune-mediated beta cell apoptosis in type 1 diabetes to permit endogenous beta cell regeneration is a plausible approach to the reversal of type 1 diabetes, provided sufficient beta cells are present at the onset of therapy.
Abbreviations
CT:
computed tomography
DAPI:
4,6-diamidino-2-phenylindole
NOD:
non-obese diabetic (mice)
TUNEL:
terminal deoxynucleotidyl transferase-mediated dUTP-biotin nick-end labelling
References
- Atkinson MA, Eisenbarth GS (2001) Type 1 diabetes: new perspectives on disease pathogenesis and treatment. Lancet 358:221–229
Article PubMed CAS Google Scholar - Kloppel G, Drenck CR, Oberholzer M, Heitz PU (1984) Morphometric evidence for a striking B-cell reduction at the clinical onset of type 1 diabetes. Virchows Arch A Pathol Anat Histopathol 403:441–452
Article PubMed CAS Google Scholar - Gale EA (2001) The discovery of type 1 diabetes. Diabetes 50:217–226
Article PubMed CAS Google Scholar - Gepts W, De Mey J (1978) Islet cell survival determined by morphology. An immunocytochemical study of the islets of Langerhans in juvenile diabetes mellitus. Diabetes 27(Suppl 1):251–261
PubMed Google Scholar - Lohr M, Kloppel G (1987) Residual insulin positivity and pancreatic atrophy in relation to duration of chronic type 1 (insulin-dependent) diabetes mellitus and microangiopathy. Diabetologia 30:757–762
Article PubMed CAS Google Scholar - Pipeleers D, Ling Z (1992) Pancreatic beta cells in insulin-dependent diabetes. Diabetes Metab Rev 8:209–227
Article PubMed CAS Google Scholar - Meier JJ, Bhushan A, Butler AE, Rizza RA, Butler PC (2005) Sustained beta-cell apoptosis in patients with long-standing type 1 diabetes: indirect evidence for islet regeneration? Diabetologia 48:2221–2228
Article PubMed CAS Google Scholar - Kurrer MO, Pakala SV, Hanson HL, Katz JD (1997) Beta cell apoptosis in T cell-mediated autoimmune diabetes. Proc Natl Acad Sci USA 94:213–218
Article PubMed CAS Google Scholar - Chatenoud L, Thervet E, Primo J, Bach JF (1994) Anti-CD3 antibody induces long-term remission of overt autoimmunity in nonobese diabetic mice. Proc Natl Acad Sci USA 91:123–127
Article PubMed CAS Google Scholar - Butler AE, Janson J, Bonner-Weir S et al (2003) Beta-cell deficit and increased beta-cell apoptosis in humans with type 2 diabetes. Diabetes 52:102–210
Article PubMed CAS Google Scholar - Yoon KH, Ko SH, Cho JH et al (2003) Selective beta-cell loss and alpha-cell expansion in patients with type 2 diabetes mellitus in Korea. J Clin Endocrinol Metab 88:2300–2308
Article PubMed CAS Google Scholar - Melton LJ 3rd, Palumbo PJ, Chu CP (1983) Incidence of diabetes mellitus by clinical type. Diabetes Care 6:75–86
Article PubMed Google Scholar - Kassem SA, Ariel I, Thornton PS, Scheimberg I, Glaser B (2000) Beta-cell proliferation and apoptosis in the developing normal human pancreas and in hyperinsulinism of infancy. Diabetes 49:1233–1325
Article Google Scholar - Bouwens L, Pipeleers DG (1998) Extra-insular beta cells associated with ductules are frequent in adult human pancreas. Diabetologia 41:629–633
Article PubMed CAS Google Scholar - Bouwens L, Rooman I (2005) Regulation of pancreatic beta-cell mass. Physiol Rev 85:1255–1270
Article PubMed CAS Google Scholar - Warren S, Root HF (1925) The pathology of diabetes, with special reference to pancreatic regeneration. Am J Pathol 1:415–430
Google Scholar - Dor Y, Brown J, Martinez OI, Melton DA (2004) Adult pancreatic beta-cells are formed by self-duplication rather than stem-cell differentiation. Nature 429:41–46
Article PubMed CAS Google Scholar - Georgia S, Bhushan A (2004) Beta cell replication is the primary mechanism for maintaining postnatal beta cell mass. J Clin Invest 114:963–968
PubMed CAS Google Scholar - Tsai EB, Sherry NA, Palmer JP, Herold KC (2006) The rise and fall of insulin secretion in type 1 diabetes mellitus. Diabetologia 49:261–270
Article PubMed CAS Google Scholar - Meier JJ, Ritzel RA, Maedler K, Gurlo T, Butler PC (2005) Increased vulnerability of newly forming beta-cells to cytokine-induced cell death. Diabetologia 49:83–89
Article PubMed CAS Google Scholar - Maedler K, Schumann DM, Iwakura Y, Donath MY (2005) The physiological role of interleukin-1beta is mediated by IL-1Ra secretion of human pancreatic islets. Diabetes 54(Suppl 1):A387 (Abstract)
Google Scholar - Sreenan S, Pick AJ, Levisetti M et al (1999) Increased beta-cell proliferation and reduced mass before diabetes onset in the nonobese diabetic mouse. Diabetes 48:989–996
Article PubMed CAS Google Scholar - Pour PM, Schmied BM, Ulrich AB et al (2001) Abnormal differentiation of islet cells in pancreatic cancer. Pancreatology 1:110–116
Article PubMed CAS Google Scholar - Chari ST, Zapiach M, Yadav D, Rizza RA (2005) Beta-cell function and insulin resistance evaluated by HOMA in pancreatic cancer subjects with varying degrees of glucose intolerance. Pancreatology 5:229–233
Article PubMed CAS Google Scholar - Zhang ZL, Constantinou D, Mandel TE, Georgiou HM (1994) Lymphocyte subsets in thymus and peripheral lymphoid tissues of aging and diabetic NOD mice. Autoimmunity 17:41–48
Article PubMed CAS Google Scholar - Foulis AK, Stewart JA (1984) The pancreas in recent-onset type 1 (insulin-dependent) diabetes mellitus: insulin content of islets, insulitis and associated changes in the exocrine acinar tissue. Diabetologia 26:456–461
Article PubMed CAS Google Scholar - Meier JJ, Bhushan A, Butler PC (2006) The potential for stem cell therapy in diabetes. Pediatr Res 59:65R–73R
Article PubMed Google Scholar - Chong AS, Shen J, Tao J et al (2006) Reversal of diabetes in non-obese diabetic mice without spleen cell-derived beta cell regeneration. Science 311:1774–1775
Article PubMed CAS Google Scholar - Nishio J, Gaglia JL, Turvey SE et al. (2006) Islet recovery and reversal of murine type 1 diabetes in the absence of any infused spleen cell contribution. Science 311:1775–1778
Article PubMed CAS Google Scholar - Suri A, Calderon B, Esparza TJ et al (2006) Immunological reversal of autoimmune diabetes without hematopoietic replacement of beta cells. Science 311:1778–1780
Article PubMed CAS Google Scholar - Keymeulen B, Vandemeulebroucke E, Ziegler AG et al (2005) Insulin needs after CD3-antibody therapy in new-onset type 1 diabetes. N Engl J Med 352:2598–2608
Article PubMed CAS Google Scholar
Acknowledgements
These studies were supported by funding from the Juvenile Diabetes Research Foundation, the Larry L. Hillblom Foundation and the German Research Foundation (Deutsche Forschungsgemeinschaft, Me 2096/2-1). We are grateful to our colleagues in the Larry Hillblom Islet Research Center, A. Bhushan, K. Maedler, L. Haataja and T. Gurlo, for their excellent suggestions.
Author information
Authors and Affiliations
- Larry Hillblom Islet Research Center, UCLA David Geffen School of Medicine, 24–130 Warren Hall, 900 Veteran Avenue, Los Angeles, CA, 90095-7073, USA
J. J. Meier, A. E. Butler, R. Galasso & P. C. Butler - Division of Endocrinology, Diabetes and Hypertension, UCLA David Geffen School of Medicine, Los Angeles, CA, USA
J. C. Lin & D. S. Martinez
Authors
- J. J. Meier
You can also search for this author inPubMed Google Scholar - J. C. Lin
You can also search for this author inPubMed Google Scholar - A. E. Butler
You can also search for this author inPubMed Google Scholar - R. Galasso
You can also search for this author inPubMed Google Scholar - D. S. Martinez
You can also search for this author inPubMed Google Scholar - P. C. Butler
You can also search for this author inPubMed Google Scholar
Corresponding author
Correspondence toP. C. Butler.
Additional information
J. J. Meier and J. C. Lin contributed equally to this work.
Rights and permissions
About this article
Cite this article
Meier, J.J., Lin, J.C., Butler, A.E. et al. Direct evidence of attempted beta cell regeneration in an 89-year-old patient with recent-onset type 1 diabetes.Diabetologia 49, 1838–1844 (2006). https://doi.org/10.1007/s00125-006-0308-2
- Received: 20 March 2006
- Accepted: 08 April 2006
- Published: 27 June 2006
- Issue Date: August 2006
- DOI: https://doi.org/10.1007/s00125-006-0308-2