Pioglitazone does not increase cerebral glucose utilisation in a murine model of Alzheimer’s disease and decreases it in wild-type mice (original) (raw)
Abstract
Aims/hypothesis
Clinical trials are in progress to test thiazolidinediones in neurodegenerative diseases such as Alzheimer’s disease that involve deficiencies in brain glucose metabolism. While thiazolidinediones enhance glucose uptake in non-cerebral tissues, their impact on brain energy metabolism has not been investigated in vivo. We thus determined whether the thiazolidinedione pioglitazone reverses the decrease in cerebral glucose utilisation (CGU) in a model of brain metabolic deficiency related to Alzheimer’s disease. Results are relevant to diabetes because millions of diabetic patients take pioglitazone as an insulin-sensitising drug, and diabetes increases the risk of developing Alzheimer’s disease.
Materials and methods
The regional pattern of CGU was measured with the 2-deoxy [14C] glucose autoradiographic technique in adult awake mice overexpressing transforming growth factor β1 (TGFβ1), and in wild-type littermates. Mice were treated with pioglitazone for 2 months.
Results
Measurement of CGU in 27 brain regions confirmed that TGFβ1 overexpression induced hypometabolism across the brain. Pioglitazone did not reverse the effect of TGFβ1 overexpression and decreased regional CGU in control animals by up to 23%. The extent of the regional CGU decrease induced by pioglitazone, but not that induced by TGFβ1, correlated strongly with basal CGU, suggesting that the higher the local metabolic rate the greater the reduction of CGU effected by pioglitazone.
Conclusions/interpretation
In contrast to its stimulatory effect in non-cerebral tissues, chronic treatment with pioglitazone decreases CGU in vivo. This evidence does not support the hypothesis that pioglitazone could act as a metabolic enhancer in Alzheimer’s disease, and raises the question of how thiazolidinediones could be beneficial in neurodegenerative diseases.
Similar content being viewed by others
Introduction
Thiazolidinediones such as pioglitazone are commonly used to treat type 2 diabetes because they exert a number of effects on glucose and lipid metabolism, resulting in reduced insulin resistance and increased glucose uptake and utilisation in non-cerebral tissues [1, 2]. The finding that thiazolidinediones are potent inhibitors of inflammation [3–5] has prompted several clinical trials testing the effects of thiazolidinediones in neurological diseases such as Alzheimer’s disease and multiple sclerosis, in which inflammation contributes to pathogenesis [6]. The anti-inflammatory properties of pioglitazone in the brain have also been shown in murine models of acute [7] and chronic [8] inflammation, when this thiazolidinedione was administered to the animals orally. Both the metabolic and inflammatory actions of thiazolidinediones have been attributed to changes in gene expression mediated by peroxisome proliferator-activated nuclear receptor (PPAR)-γ, of which thiazolidinediones are ligands [3]. However, several recent studies point to the existence of PPAR-independent effects of thiazolidinediones, such as the regulation of mitochondrial respiration [9–12]. Impaired mitochondrial activity has been related to insulin resistance [13] and correlated with the clinical state in Alzheimer’s disease [14]. These data raise the question of whether thiazolidinediones can determine the metabolic rate of the brain.
So far, the evaluation of cerebral glucose utilisation (CGU), a quantitative measure of neuronal activity that principally reflects integrated synaptic function [15], has not been used to demonstrate whether thiazolidinediones alter brain energy metabolism in vivo. In vitro studies showing that pioglitazone increases glucose uptake and consumption in muscle cells [16], adipocytes [17] and astrocytes [9] have suggested that the capacity to stimulate glucose metabolism could be a general property of thiazolidinediones that is also applicable to the brain. Considering the strict dependence that the brain has on glucose as a fuel [15], a stimulatory action of thiazolidinediones on glucose metabolism would provide an additional therapeutic benefit in pathologies such as Alzheimer’s disease. A growing body of evidence supports the idea that alterations in brain glucose metabolism and insulin sensitivity or signalling contribute to the pathogenesis of Alzheimer’s disease. Regional decreases in the activity of glycolytic enzymes and in glucose utilisation have been detected in sporadic Alzheimer’s disease [18, 19], and the administration of glucose [20] or insulin [21, 22] can facilitate memory in patients. This evidence has led to the notion of metabolic insufficiency or glucoregulatory impairment in Alzheimer’s disease [23–25] and has provided a strong rationale for the therapeutic use of drugs, such as thiazolidinediones, that increase insulin sensitivity and glucose consumption. Preliminary results consistently suggest that restoring adequate levels of insulin and glucose by using a thiazolidinedione facilitates memory in patients with Alzheimer’s disease [26]. Since type 2 diabetes increases the risk of Alzheimer’s disease and causes cognitive decline [27, 28], facilitated glucose utilisation in the brain would provide additional therapeutic benefits to patients taking thiazolidinediones regularly to treat diabetes.
We thus sought to determine whether thiazolidinediones affect brain glucose metabolism in vivo. To this end we tested the effect of pioglitazone treatment on regional CGU using the 2-deoxyglucose autoradiographic technique [15]. We compared the cerebral pattern of CGU in normal mice with that of mice overexpressing transforming growth factor β1 (TGFβ1). When produced in excess, TGFβ1 causes robust astrocytosis and microglia activation, vascular deposition of fibrillar material, decreased CGU and impaired cerebral blood flow [29–31]. This recently corroborated murine model [32] presents a condition of chronic inflammation [8] and oxidative stress [32] associated with metabolic deficiency which makes them suitable for testing in vivo the combined anti-inflammatory and metabolic actions of thiazolidinediones. Our findings reveal unexpected actions of thiazolidinediones on brain glucose metabolism, which raise the question of whether thiazolidinediones are appropriate for the treatment of neurodegenerative diseases in which deficiencies in glucose metabolism occur.
Materials and methods
Animal groups and treatment
The animals used in this study were heterozygous male and female mice genetically modified to produce a constitutively active form of TGFβ1 under the control of the GFAP promoter on a C57BL/6 background [29], crossed with wild-type C57BL/6 mice (CERJ, Le Genest-St-Isle, France). Control animals were wild-type littermates. Animals were treated with pioglitazone (Takeda Pharmaceuticals, Lincolnshire, IL, USA) provided in the food at 120 μg/g. The treatment started at 2 months of age, soon after weaning, and lasted 2 months. The daily intake of pioglitazone was approximately 18 mg/kg, which yields low micromolar serum levels of pioglitazone. We had previously shown that orally administered pioglitazone modified TGFβ1-induced pathology [8]: it clearly reduced astrogliosis and microglia activation while increasing hydrocephalus. In addition, pioglitazone caused a small (20–30%) but statistically significant reduction in the level of amyloid β42 but not β40 peptide, in both control and TGFβ mice [8]. These data demonstrate that thiazolidinedione does cross the blood–brain barrier at a functionally effective concentration. The physiological variables of the four experimental groups investigated, including body weights before and after the treatment, are given in Table 1. Mice were used in accordance with the European Community’s regulations on the care and use of laboratory animals (Préfecture de Paris, authorisations 75-071 to P. Lacombe).
Table 1 Physiological parameters in the four experimental groups
Measurement of glucose utilisation
Glucose utilisation was measured by the 2-deoxy-d-[14C]glucose ([14C]DG) technique [15], adapted to the awake mouse [31]. A dose of 5.5 MBq/kg of [14C]DG (2,035 MBq/mmol; NEN/Perkin Elmer Life Sciences: Courtaboeuf-Les Ulis, Essonne, France) dissolved in 45 μl of saline was infused through the femoral vein. The concentration of [14C]DG in the brain was assessed by densitometric analysis of autoradiograms of 20 μm-thick brain sections [31]. For methodological reasons, animals were fasted overnight before CGU measurement. Data are expressed as regional CGU with respect to the value found in the white matter (genu of the corpus callosum) of the same animal, and calculated as a ratio of tracer concentration in Bq/g. Absolute CGU values (mean±SEM) for the genu of the corpus callosum (μmol 100 g−1 min−1) were 34.7±5.0 and 38.7±4.4 for wild-type mice on the control and pioglitazone diets, respectively, and 39.7±5.1 and 33.2±3.3 for TGFβ1-overexpressing mice on the control and pioglitazone diets, respectively. Groups did not differ significantly in this CGU value.
Statistical analysis
Data were analysed by one- or two-way ANOVA, as appropriate, using StatView software (SAS Institute, Cary, NC, USA); p values are given for the ANOVA. Linear regression analysis was used to evaluate the relationship between the changes in CGU induced by either TGFβ1 overexpression or pioglitazone and regional CGU values. The p value of the correlation coefficient is given.
Results
Neither TGFβ1 overexpression nor pioglitazone significantly altered the physiological parameters at the time of measurement (Table 1). The effects of TGFβ1 overexpression and pioglitazone on regional CGU levels are shown in Figs. 1, 2 and 3. The representative autoradiograms displayed in Fig. 1 provide a first indication that both TGFβ1 and pioglitazone reduced CGU across the brain. CGU values obtained in three distinct functional systems displayed significant decreases in several brain regions of TGFβ1-overexpressing mice (Fig. 2a), in accordance with previous studies [30, 31]. Figure 2b shows that pioglitazone did not significantly reverse the decreases caused by TGFβ1, nor did it increase CGU in wild-type mice (Fig. 2c). Rather, the thiazolidinedione reduced CGU in wild-type mice. As the differences in several regions were close to significance, we grouped together the regions within functional systems. The results displayed in Fig. 3 show that both TGFβ1 overexpression and pioglitazone alone significantly reduced the functional activity in the three systems, whereas pioglitazone in TGFβ1 mice did not. Whether the effects of pioglitazone were affected by TGFβ1 overexpression was determined by evaluating the interaction between TGFβ1 and pioglitazone. When whole functional systems were considered, interaction was significant in the sensory and motor systems (Fig. 3), validating the abatement of pioglitazone responses in TGFβ1 mice.
Fig. 1
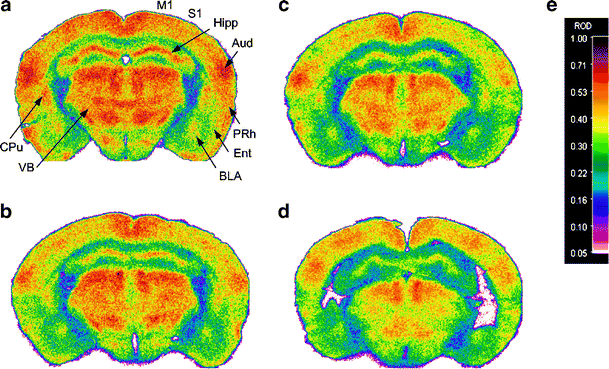
Representative autoradiograms of CGU in wild-type littermates (a, b) and mice overexpressing TGFβ1 (c, d). Mice were fed with control pellets (a, c) or pioglitazone (b, d). Brain sections were taken at the level of the anterior thalamus. Both TGFβ1 overexpression and pioglitazone appeared to reduce CGU compared with wild-type mice fed control food. Mice overexpressing TGFβ1 showed more pronounced decreases in CGU in structures of the hippocampal formation (Hipp) and in the ventral thalamus (VB). Treatment with pioglitazone reduced glucose utilisation mostly in cortical areas (S1), the striatum (CPu, here the tail of the caudate nucleus) and the ventral thalamus. The same colour scale and corresponding relative optical density (ROD) (e) were used for the four autoradiograms. Regional changes in CGU are detailed in Fig. 2. For abbreviations, see legend Fig. 2
Fig. 2
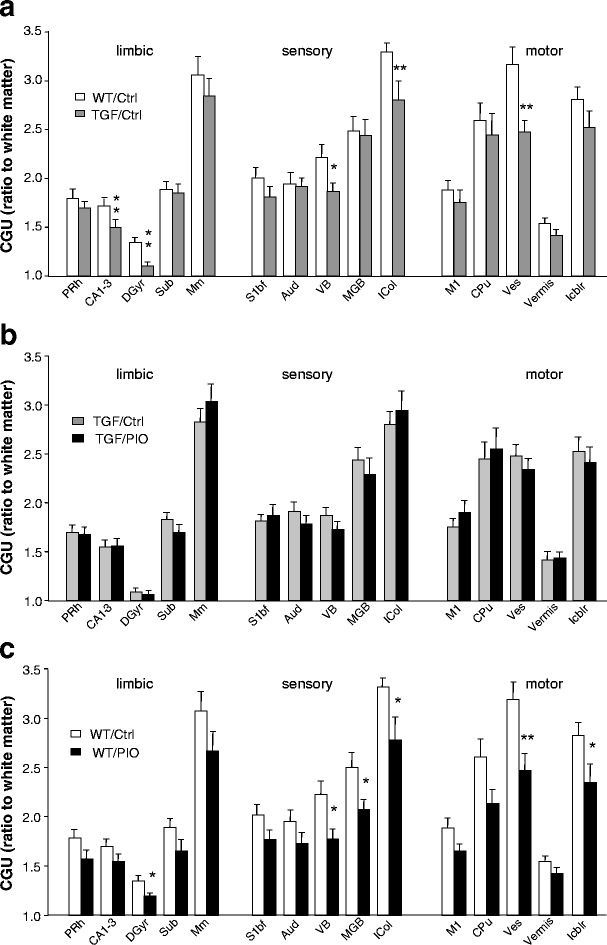
Regional distribution of the effects of TGFβ1 overexpression and/or pioglitazone on CGU. a Effect of TGFβ1 overexpression. A significant decrease in CGU was observed in five individual regions. b Effect of pioglitazone in mice overexpressing TGFβ1. In general, pioglitazone did not reverse the effect of TGFβ1. c Effect of pioglitazone in wild-type mice. Pioglitazone appeared to reduce CGU on its own, significantly so in six regions. Values are mean ± SEM of the ratio of regional CGU:white matter (genu of the corpus callosum) in the same animal (_n_=8 or 9 animals per group). * p<0.05, ** p<0.01 (one-way ANOVA). ADAV Anterodorsal thalamic nucleus, Aud primary auditory cortex, BLA basolateral amygdala, CA1-3 fields of the dorsal hippocampus, CA-pyr layer of hippocampal pyramidal cells seen on Nissl-stained sections, CPu caudate putamen, Ent entorhinal cortex, GP globus pallidus, DGyr dentate gyrus (layer of hippocampal molecular cells), Icol inferior colliculi (dark appearance on autoradiograms, i.e. activated layer), Icbl intracerebellar nuclei, LGB lateral geniculate body, MGB medial geniculate body, Mm hypothalamic mamillary nuclei, Pnc pontine reticular nucleus, caudal, Prh perirhinal cortical area, S1bf primary somatosensory barrel field cortex, S1J primary somatosensory jaw region cortex, Scol superior colliculi, SN substantia nigra, Sub subiculum, Tu olfactory tubercles, VB ventrobasal thalamic nuclei, Ves vestibular nuclei, Vermis cerebellar vermis, Vis primary visual cortex
Fig. 3
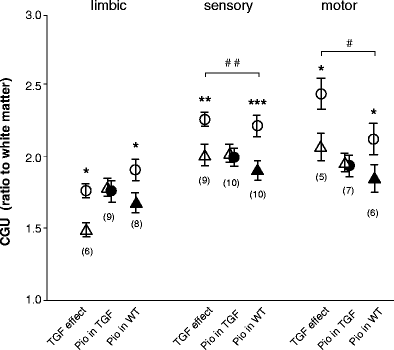
Comparative effects of TGFβ1 overexpression and/or pioglitazone (Pio) on the CGU of three functionally distinct neural systems: limbic (nine regions), sensory (ten regions) and motor (seven regions). System CGU values are compared using three evaluations: (1) effects of TGFβ1 (TGF/Ctrl vs WT/Ctrl); (2) effects of pioglitazone in mice overexpressing TGFβ1 (TGF/pioglitazone vs TGF/Ctrl); and (3) effects of pioglitazone in WT mice (WT/pioglitazone vs WT/Ctrl). Open circles, WT/Ctrl; open triangles, TGF/Ctrl; filled circles, TGF/Pio; filled triangles, WT/Pio. Significance was attained using all three evaluation systems for the effects of TGFβ1 overexpression and of pioglitazone in wild-type mice, but not for the effects of pioglitazone in mice overexpressing TGFβ1. Interaction between the effects of TGFβ1 and pioglitazone was significant in the sensory and motor systems, indicating that pioglitazone responses were reduced by TGFβ1 overexpression. Values are mean ± SEM of the average CGU in each evaluation system. The numbers in parentheses indicate the numbers of regions that provided the best significance. * p<0.05, ** p<0.01, *** p<0.001 (one-way ANOVA); # p<0.05, ## p<0.01 (two-way ANOVA)
To better characterise the respective effects of TGFβ1 overexpression and pioglitazone, we then analysed the distribution of the changes in CGU that these factors induced in the brain. Figure 4 shows the percentage changes in the group overexpressing TGFβ1 and the pioglitazone-treated group with regard to control animals. What differentiated the distribution of their effects in the brain is that TGFβ1 was more effective in structures of the hippocampal formation (dentate gyrus, −16%; CA1-3, −14%), where pioglitazone displayed only a minor effect. Conversely, pioglitazone reduced CGU more prominently; for example, in the striatum (−18%) and the medial geniculate body (−17%). Furthermore, a region-to-region analysis of the relationship between CGU changes and basal CGU (i.e. the CGU of wild-type mice on the control diet) confirmed a difference in the actions of TGFβ1 and pioglitazone (Fig. 5). Regional effects of TGFβ1 overexpression appeared unrelated to the basal glucose metabolic rate of the region (Fig. 5a), whereas decreases in CGU caused by pioglitazone correlated significantly (p<0.0001) with basal CGU (Fig. 5b). This indicates that the higher the basal metabolic rate the more responsive was the region to the action of pioglitazone.
Fig. 4
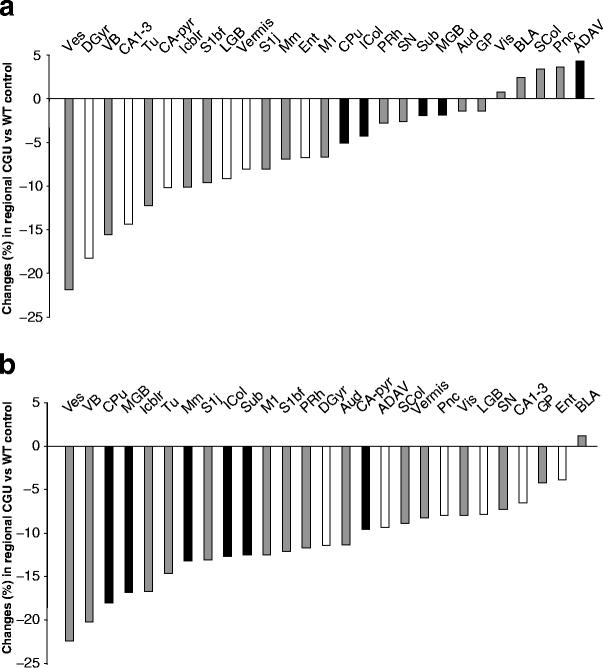
Differential distribution of the regional percentage changes in CGU induced by TGFβ1 overexpression (a) or pioglitazone (b). Changes are CGUTGF or CGUPIO minus GCUWT divided by CGUWT×100, where PIO is pioglitazone and WT is wild-type. Regions are ranked by the extent of CGU change. Both TGFβ1 overexpression and treatment with pioglitazone induced decreases in CGU exceeding 20%. However, many of the regions most sensitive to TGFβ1 differed from those most sensitive to pioglitazone. White bars, regions with greater sensitivity to overexpression of TGFβ1 than to pioglitazone; black bars, regions with greater sensitivity to pioglitazone than to overexpression of TGFβ1. For abbreviations see legend of Fig. 2
Fig. 5
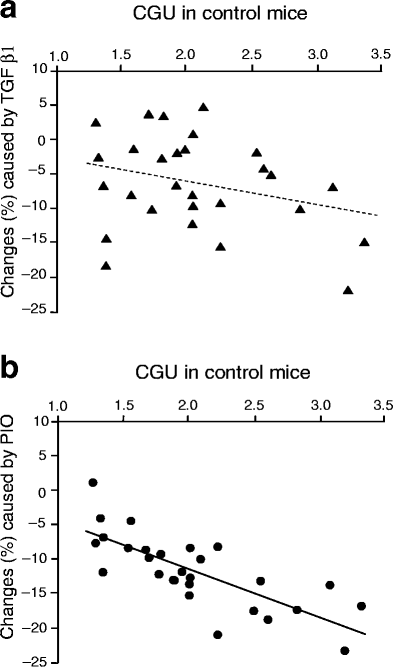
Region-to-region correlation between the decrease in CGU caused by TGFβ1 overexpression (a) or pioglitazone (PIO) (b) in CGU control mice and CGU levels in wild-type mice on the control diet. A clear positive correlation was found for pioglitazone but not for TGFβ1, indicating that regional responsiveness to pioglitazone increased with increasing regional basal CGU. For abbreviations see legend of Fig. 2. _p_>0.05 (a); p<0.0001 (b)
Discussion
The main finding of this study is that pioglitazone decreased CGU in multiple brain regions, and that an essential feature of this action is its proportionality to the basal CGU. This is the only case in which pioglitazone has been found to reduce glucose metabolism, since all previous reports showed that this and other thiazolidinediones increase glucose uptake in cultured cells [9, 16, 17] and in vivo, as recently shown in the myocardium of diabetic patients [33]. In addition, pioglitazone did not reverse the decreases in CGU associated with overexpression of TGFβ1, despite the fact that this thiazolidinedione efficiently inhibits the inflammatory reaction in this model [8]. Together, these data support a dual but distinctive action of pioglitazone on glucose metabolism and inflammatory processes in the brain. As glucose utilisation reflects neuronal activity in normal, resting physiological conditions [15, 34], this finding prompts the question of the effects of thiazolidinediones on brain function. This issue is of current interest since (1) several million diabetic patients take pioglitazone, (2) these patients suffer from a greater rate of cognitive decline [27, 28], and (3) pioglitazone is being tested for the treatment of neurological diseases (for information about clinical trials in progress see [6]). Below we discuss the possible mechanisms underlying the changes in brain glucose utilisation induced by pioglitazone in vivo, compared with the effects in cultured cells, and the therapeutic implications.
An increase in glucose uptake is suggestive of a neural activation [34]. The increases in glucose uptake in cultured astrocytes or in brain slices were not restricted to pioglitazone, as they were mimicked by other thiazolidinediones, although with different potencies of action [9]. A clue to the underlying mechanism is provided by the findings that pioglitazone reduces mitochondrial respiration and that the lactate content increases in parallel [9]. The triple event of increased glucose uptake and lactate release concomitant with decreased respiration has also been described in skeletal muscle [10]. Such a combination suggests a cause–effect relationship in which the increased glucose uptake may represent an adaptive response, producing ATP via a highly glucose-consuming pathway, which generates lactate. This response would compensate for the decreased oxygen-dependent production of ATP resulting from impaired respiration. If so, the increased CGU could be considered an acute reaction that protects against alterations in mitochondrial function. Interestingly, the effect of thiazolidinediones on mitochondrial respiration appeared to be independent of PPAR [9, 11].
A major difference between in vitro and our in vivo studies is that the former provide information about effects occurring as early as 2 h after the addition of pioglitazone, whereas the latter reflect the effects of 2 months of treatment aimed at modelling the chronic actions of the thiazolidinedione. In brain slices the stimulatory effect of pioglitazone on glucose uptake dissipates several days after initial exposure to the thiazolidinedione [9]. This reveals the transient nature of the rapid increase in glucose uptake. Another difference is that the metabolically available glucose, which is in large excess in vitro, may be limited in vivo. Hence, increased glucose utilisation in vivo, if any, may be short-lasting and limited. Conceivably, reduction in glucose utilisation in pioglitazone-treated mice could represent chronic down regulation of aerobic glucose metabolism in the brain resulting from a reduction in mitochondrial respiration, as shown in cultures. This process would be initiated to maintain the balance between aerobic and anaerobic utilisation of glucose so as to avoid excessive glucose consumption. It presumably consists of multiple adaptive changes, which would account for the regionally differentiated sensitivity to pioglitazone, including local re-equilibration of the glucose metabolic pathways and improvement of the insulin signalling pathway [35, 36]. As a result, the highest, potentially ‘excessive’ metabolic rate in vivo would be preferentially depressed. Why this action of pioglitazone is specific to the brain remains unclear. The differing effects of TGFβ1 overexpression on CGU throughout the brain indicate the involvement of other mechanisms of action. The decrease in CGU induced by TGFβ1 is probably due to glia inflammation and fibrosis-derived damage. Indeed, the hippocampal formation, in which several regions showed large decreases in CGU, also displayed the strongest inflammation [8]. In regions where a significant interaction was found between pioglitazone and TGFβ1, no further hypometabolism occurred with pioglitazone treatment, as if a minimal metabolic rate had already been attained owing to TGFβ1.
Although the rationale for the therapeutic use of thiazolidinediones to compensate for hypometabolism in Alzheimer’s disease has received strong support [20–22], the data presented here suggest that long-term administration of pioglitazone may not enhance the capacity of the brain to metabolise glucose in Alzheimer’s disease. However, this may not necessarily mean that pioglitazone is deleterious to the brain. First, the possibility cannot be ruled out that the brain utilises more of a fuel other than glucose, such as lactate of peripheral origin [37]. Second, pioglitazone treatment may be protective by limiting the highest rates of regional glucose oxidative metabolism and the concomitant oxidative stress [14, 32, 38, 39], in association with the benefits derived from the attenuation of inflammation [5]. Finally, pioglitazone can induce the expression of proteins related to the stress response [40] and mitochondrial membrane hyperpolarisation that can safeguard the cells against an excessive metabolic rate [9]. This scenario would be akin to a ‘preconditioning’ effect in which a drug is beneficial by mildly impairing mitochondrial respiration. In support of these ideas, pioglitazone has been shown to be protective in situations of 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine administration in the mouse model of Parkinson’s disease [7] and in acute metabolic stress caused by ischaemia [41]. Extrapolating these findings to diabetic patients should nevertheless be done with caution. Indeed, no cognitive impairment has been reported in patients treated with pioglitazone, and first results from a clinical trial testing the thiazolidinedione rosiglitazone in Alzheimer’s disease point to preserved cognition [26]. A very recent study in mice modelling Alzheimer’s disease suggests that rosiglitazone attenuates learning and memory deficits by lowering glucocorticoid actions [[42](/article/10.1007/s00125-006-0326-0#ref-CR42 "Pedersen WA, McMillan PJ, Kulstad JJ, Leverenz JB, Craft S, Haynatzki GR (2006) Rosiglitazone attenuates learning and memory deficits in Tg2576 Alzheimer mice. Exp Neurol (Epub ahead of print) DOI 10.1016/j.expneurol.2006.01.018
")\]. However, unlike pioglitazone, rosiglitazone does not cross the blood–brain barrier. Therefore, any effect on cognition by rosiglitazone ought to be initiated outside the brain, while pioglitazone would act both centrally and peripherally. Hence, it is premature to conclude that pioglitazone would mimic the beneficial effects of rosiglitazone. Forthcoming studies should address more adequately the repercussions on brain function of the use of different thiazolidinediones, and define the mechanisms by which thiazolidinediones could provide therapeutic benefits in neurological disorders.Abbreviations
CGU:
cerebral glucose utilisation
PPAR:
peroxisome-proliferator activated receptor
TGFβ1:
transforming growth factor β1
WT:
wild-type
References
- Hammarstedt A, Sopasakis VR, Gogg S, Jansson PA, Smith U (2005) Improved insulin sensitivity and adipose tissue dysregulation after short-term treatment with pioglitazone in non-diabetic, insulin-resistant subjects. Diabetologia 48:96–104
Article PubMed CAS Google Scholar - Kubota N, Terauchi Y, Kubota T et al (2006) Pioglitazone ameliorates insulin resistance and diabetes by both adiponectin dependent and independent pathway. J Biol Chem 281:8748–8755
Article PubMed CAS Google Scholar - Heneka MT, Klockgether T, Feinstein DL (2000) Peroxisome proliferator-activated receptor-γ ligands reduce neuronal inducible nitric oxide synthase expression and cell death in vivo. J Neurosci 20:6862–6867
PubMed CAS Google Scholar - Cunard R, Eto Y, Muljadi JT, Glass CK, Kelly CJ, Ricote M (2004) Repression of IFN-gamma expression by peroxisome proliferator-activated receptor gamma. J Immunol 172:7530–7536
PubMed CAS Google Scholar - Schutz B, Reimann J, Dumitrescu-Ozimek L et al (2005) The oral antidiabetic pioglitazone protects from neurodegeneration and amyotrophic lateral sclerosis-like symptoms in superoxide dismutase-G93A transgenic mice. J Neurosci 25:7805–7812
Article PubMed CAS Google Scholar - Feinstein DL (2004) Contrasting the neuroprotective and gliotoxic effects of PPARγ agonists. Drug Discov Today Ther Strategy 1:29–34
Article CAS Google Scholar - Breidert T, Callebert J, Heneka MT, Landreth G, Launay JM, Hirsch EC (2002) Protective action of the peroxisome proliferator activated receptor-gamma agonist pioglitazone in a mouse model of Parkinson’s disease. Neurochemistry 82:615–624
Article CAS Google Scholar - Lacombe P, Mathews PM, Schmidt SD et al (2004) Effect of anti-inflammatory agents on transforming growth factor beta overexpressing mouse brains: a model revised. J Neuroinflammation 1:1–17
Article CAS Google Scholar - Dello-Russo C, Gavrilyuk V, Weinberg G et al (2003) Peroxisome proliferator-activated receptor gamma thiazolidinedione agonists increase glucose metabolism in astrocytes. J Biol Chem 278:5828–5836
Article PubMed CAS Google Scholar - Brunmair B, Staniek K, Gras F et al (2004) Thiazolidinediones, like metformin, inhibit respiratory complex I. A common mechanism contributing to their anti-diabetic actions? Diabetes 53:1052–1059
Article PubMed CAS Google Scholar - Feinstein DL, Spagnolo A, Akar C et al (2005) Receptor-independent actions of PPAR thiazolidinedione agonists: is mitochondrial function the key? Biochem Pharmacol 70:177–188
Article PubMed CAS Google Scholar - Konrad D, Rudich A, Bilan PJ et al (2005) Troglitazone causes acute mitochondrial membrane depolarisation and an AMPK-mediated increase in glucose phosphorylation in muscle cells. Diabetologia 48:954–966
Article PubMed CAS Google Scholar - Petersen KF, Dufour S, Shulman GI (2005) Decreased insulin-stimulated ATP synthesis and phosphate transport in muscle of insulin-resistant offspring of type 2 diabetic parents. PLoS Med 2:e233
Article PubMed CAS Google Scholar - Bubber P, Haroutunian V, Fisch G, Blass JP, Gibson GE (2005) Mitochondrial abnormalities in Alzheimer brain: mechanistic implications. Ann Neurol 57:695–703
Article PubMed CAS Google Scholar - Sokoloff L (1981) Localization of functional activity in the central nervous system by measurement of glucose utilization with radioactive deoxyglucose. J Cereb Blood Flow Metab 1:7–36
PubMed CAS Google Scholar - El-Kebbi IM, Roser S, Pollet RJ (2004) Regulation of glucose transport by pioglitazone in cultured muscle cells. Metabolism 43:953–958
Article Google Scholar - Shimaya A, Kurosaki E, Shioduka K, Nakano R, Shibasaki M, Shika H (1998) YM268 increases the glucose uptake, cell differentiation and mRNA expression of glucose transporter 3T3-L1 adipocytes. Horm Metab Res 30:543–548
Article PubMed CAS Google Scholar - Sorbi S, Bird ED, Blass JP (1983) Decreased pyruvate dehydrogenase complex activity in Huntington and Alzheimer brain. Ann Neurol 13:72–78
Article PubMed CAS Google Scholar - de Leon MJ, Convit A, Wolf OT et al (2001) Prediction of cognitive decline in normal elderly subjects with 2-[18F]fluoro-2-deoxyglucose/positron-emission tomography (FDC/PET). Proc Natl Acad Sci USA 98:10966–10971
Article PubMed Google Scholar - Craft S, Zallen G, Baker LD (1992) Glucose and memory in mild senile dementia of the Alzheimer type. J Clin Exp Neuropsychol 14:253–267
Article PubMed CAS Google Scholar - Craft S, Asthana S, Cook DG et al (2003) Insulin dose-response effects on memory and plasmid amyloid precursor protein in Alzheimer’s disease: interactions with apolipoprotein E genotype. Psychoneuroendocrinology 28:809–822
Article PubMed CAS Google Scholar - Watson GS, Craft S (2004) Modulation of memory by insulin and glucose: neuropsychological observations in Alzheimer’s disease. Eur J Pharmacol 490:97–113
Article PubMed CAS Google Scholar - Gibson GE, Sheu KF, Blass JP (1998) Abnormalities of mitochondrial enzymes in Alzheimer disease. J Neural Trans 105:855–870
Article CAS Google Scholar - Convit A, Wolf OT, Tarshish C, de Leon MJ (2003) Reduced glucose tolerance is associated with poor memory performance and hippocampal atrophy among normal elderly. Proc Natl Acad Sci USA 100:2019–2022
Article PubMed CAS Google Scholar - Hoyer S (2004) Glucose metabolism and insulin receptor signal transduction in Alzheimer’s disease. Eur J Pharmacol 490:115–125
Article PubMed CAS Google Scholar - Watson GS, Cholerton BA, Reger MA et al (2005) Preserved cognition in patients with early Alzheimer disease and amnestic mild cognitive impairment during treatment with rosiglitazone: a preliminary study. Am J Geriatr Psychiatry 13:950–958
Article PubMed Google Scholar - Arvanitakis Z, Wilson RS, Bienias JL, Evans DA, Bennett DA (2004) Diabetes mellitus and risk of Alzheimer’s disease and decline in cognitive function. Arch Neurol 61:661–666
Article PubMed Google Scholar - Cukierman T, Gerstein HC, Williamson JD (2005) Cognitive decline and dementia in diabetes – systematic overview of prospective observational studies. Diabetologia 48:2460–2469
Article PubMed CAS Google Scholar - Wyss-Coray T, Lin C, Sanan DA, Mucke L, Masliah E (2000) Chronic overproduction of transforming growth factor-β1 by astrocytes promotes Alzheimer’s disease-like microvascular degeneration in transgenic mice. Am J Pathol 156:139–150
PubMed CAS Google Scholar - Wyss-Coray T, Lin C, Von Euw D, Masliah E, Mucke L, Lacombe P (2000) Alzheimer’s disease-like cerebrovascular pathology in TGFβ1 transgenic mice and functional metabolic correlates. Ann NY Acad Sci 903:317–323
Article PubMed CAS Google Scholar - Gaertner RF, Wyss-Coray T, Von Euw D, Lesné S, Vivien D, Lacombe P (2005) Reduced brain tissue perfusion in TGFβ1 transgenic mice showing Alzheimer’s disease-like cerebrovascular abnormalities. Neurobiol Dis 19:38–46
Article PubMed CAS Google Scholar - Tong XK, Nicolakakis N, Kocharyan A, Hamel E (2005) Vascular remodeling versus amyloid beta-induced oxidative stress in the cerebrovascular dysfunctions associated with Alzheimer’s disease. J Neurosci 25:11165–1174
Article PubMed CAS Google Scholar - Lautermäki R, Airaksinen KE, Seppanen M et al (2005) Rosiglitazone improves myocardial glucose uptake in patients with type 2 diabetes and coronary artery disease: a 16-week randomized, double-blind, placebo-controlled study. Diabetes 54:2787–2794
Article Google Scholar - Magistretti PJ, Pellerin L (1996) Cellular bases of brain energy metabolism and their relevance to functional brain imaging: evidence for a prominent role of astrocytes. Cerebral Cortex 6:50–61
Article PubMed CAS Google Scholar - Carro E, Torres-Aleman I (2004) The role of insulin and insulin-like growth factor-I in the molecular and cellular mechanisms underlying the pathology of Alzheimer’s disease. Eur J Pharmacol 490:127–133
Article PubMed CAS Google Scholar - Steen E, Terry BM, Rivera EJ et al (2005) Impaired insulin and insulin-like growth factor expression and signaling mechanisms in Alzheimer’s disease – is this type 3 diabetes? J Alzheimers Dis 7:63–80
CAS Google Scholar - Smith D, Pernet A, Hallett WA, Bingham E, Marsden PK, Amiel SA (2003) Lactate: a preferred fuel for human brain metabolism in vivo. J Cereb Blood Flow Metab 23:658–664
Article PubMed CAS Google Scholar - Collino M, Aragno M, Mastrocola R et al (2006) Modulation of the oxidative stress and inflammatory response by PPAR-gamma agonists in the hippocampus of rats exposed to cerebral ischemia/reperfusion. Eur J Pharmacol 530:70–80
Article PubMed CAS Google Scholar - Lortz S, Gurgul-Convey E, Lenzen S, Tiedge M (2005) Importance of mitochondrial superoxide dismutase expression in insulin-producing cells for the toxicity of reactive oxygen species and proinflammatory cytokines. Diabetologia 48:1541–1548
Article PubMed CAS Google Scholar - Dehmer T, Heneka MT, Sastre M, Dichgans J, Schulz JB (2003) Protection by pioglitazone in the MPTP model of Parkinson’s disease correlates with IkappaB alpha induction and block of NFkappaB and iNOS activation. J Neurochem 88:494–501
Article CAS Google Scholar - Sundararajan S, Gamboa JL, Victor NA, Wanderi EW, Lust WD, Landreth GE (2005) Peroxisome proliferator-activated receptor-gamma ligands reduce inflammation and infarction size in transient focal ischemia. Neuroscience 130:685–696
Article PubMed CAS Google Scholar - Pedersen WA, McMillan PJ, Kulstad JJ, Leverenz JB, Craft S, Haynatzki GR (2006) Rosiglitazone attenuates learning and memory deficits in Tg2576 Alzheimer mice. Exp Neurol (Epub ahead of print) DOI 10.1016/j.expneurol.2006.01.018
Acknowledgements
The authors thank G. Landreth and M. Heneka for providing the animal food containing pioglitazone and R. Sercombe for critical reading of the manuscript. E. Galea was the recipient of an award from Association France Alzheimer.
Duality of interest
The authors declare that they have no duality of interest.
Author information
Authors and Affiliations
- Institute of Neurosciences, Autonoma University, and Catalonian Institute for Advanced Studies (ICREA), Barcelona, Spain
E. Galea - Neuroanesthesiology, University of Illinois at Chicago, Chicago, IL, USA
D. L. Feinstein - Genetics of Vascular Disease, INSERM U740, University of Paris 7, Paris, France
P. Lacombe - Génétique des Maladies Vasculaires, INSERM U740, Faculté de Médecine, Lariboisière-St Louis, 10 avenue de Verdun, 75010, Paris, France
P. Lacombe
Authors
- E. Galea
You can also search for this author inPubMed Google Scholar - D. L. Feinstein
You can also search for this author inPubMed Google Scholar - P. Lacombe
You can also search for this author inPubMed Google Scholar
Corresponding author
Correspondence toP. Lacombe.
Rights and permissions
About this article
Cite this article
Galea, E., Feinstein, D.L. & Lacombe, P. Pioglitazone does not increase cerebral glucose utilisation in a murine model of Alzheimer’s disease and decreases it in wild-type mice.Diabetologia 49, 2153–2161 (2006). https://doi.org/10.1007/s00125-006-0326-0
- Received: 02 March 2006
- Accepted: 25 April 2006
- Published: 08 July 2006
- Issue Date: September 2006
- DOI: https://doi.org/10.1007/s00125-006-0326-0