Effects of peroxisome proliferator-activated receptor (PPAR)-α and PPAR-γ agonists on glucose and lipid metabolism in patients with type 2 diabetes mellitus (original) (raw)
Introduction
Peroxisome proliferator-activated receptor (PPAR)-α and PPAR-γ are members of the nuclear receptor superfamily which have been proposed to play key roles in lipid and carbohydrate metabolism [1]. PPAR-α agonists have been used to treat hypertriacylglycerolaemia and reduce cardiovascular risk [2, 3]. Studies in rodents clearly demonstrate that PPAR-α agonists improve hepatic and muscle insulin resistance, decrease hepatic and intramuscular fat content, reduce plasma NEFA and enhance adiponectin (AD) expression [4–8]. Some [9–11], but not all [12–14], studies in humans with hypertriacylglycerolaemia and/or type 2 diabetes suggest that PPAR-α agonists, i.e. fibrates, enhance insulin sensitivity. PPAR-γ agonists (i.e. thiazolidinediones) exert their metabolic effects by binding to the PPAR-γ receptor, which is located primarily on the adipocytes [15]. Previous studies from our laboratory [16, 17] and others [18, 19] have demonstrated that thiazolidinedione (PPAR-γ agonists) treatment in patients with type 2 diabetes mellitus is associated with a reduction in plasma NEFA levels and NEFA turnover, a shift in fat distribution from visceral and hepatic to subcutaneous depots, improved hepatic and peripheral (muscle) insulin sensitivity and enhanced insulin signalling. Of note, positive relationships between increased hepatic fat content and various measures of insulin resistance have been observed in humans, independently of BMI [16, 20].
The adipocyte not only serves as a fat depot that releases NEFA, but also secretes a number of proteins or adipocytokines, including TNF-α, IL-6, leptin, resistin, AD and others, which affect hepatic and muscle insulin sensitivity and fat distribution [21]. Plasma AD levels are reduced in obese humans [22], the offspring of diabetic parents [23] and patients with type 2 diabetes [22] and are increased two- to threefold following thiazolidinedione treatment [24]. We [25] and others [19] have previously reported that PPAR-γ (thiazolidinedione) treatment causes a threefold increase in plasma AD concentration, which is closely related to the decrease in hepatic fat content and enhanced hepatic and peripheral insulin sensitivity in type 2 diabetic patients. Injection of recombinant AD in mice enhances adenosine 5′-monophosphate-activated protein kinase (AMPK) activity, increases fatty acid oxidation and reduces triacylglycerol content in muscle, improves muscle sensitivity to insulin and decreases basal hepatic glucose output [26, 27]. Increased NEFA transport/oxidation in muscle and a reduction in plasma NEFA and muscle triacylglycerol concentration have also been reported following AD administration to rodents [26]. Although studies in rodents demonstrate that PPAR-α stimulation enhances AD expression [8], the effect of PPAR-α treatment on hepatic fat content, plasma AD concentration and muscle AMPK activity in humans with type 2 diabetes has been less well studied.
The current study was designed to examine the effect of fenofibrate (FENO), a PPAR-α agonist, and pioglitazone (PIO), a PPAR-γ agonist, as monotherapy and in combination, on glucose and lipid metabolism, insulin sensitivity, plasma AD concentrations and hepatic fat content in type 2 diabetic patients.
Subjects and methods
Subjects
Fifteen patients with type 2 diabetes participated in the study (age 52 ± 3 years, BMI 31.5 ± 1.3 kg/m2, HbA1c 9.0 ± 0.7% [SEMs]). Six subjects had been taking a stable dose of sulfonylurea drug for at least 6 months before study, and nine subjects were treated with diet alone. Sulfonylurea-treated diabetic patients were included because sulfonylurea drugs have no known direct effects on insulin sensitivity [28]. Patients who had received insulin, metformin, a thiazolidinedione or lipid-lowing agent within the previous 12 months were excluded. Entry criteria included: age 30–70 years, stable body weight (±1 kg) for at least 3 months before study and fasting plasma glucose (FPG) 7.0–14.5 mmol/l. All patients were in good general health, without evidence of cardiac, hepatic, renal or other chronic diseases as determined by history, physical examination, screening blood tests and urinalysis. No subjects participated in any heavy exercise, and no subjects were taking any medications known to affect glucose metabolism. All subjects gave signed voluntary informed consent before participation. The Institutional Review Board of the University of Texas Health Science Center at San Antonio approved the protocol.
Study design
Three weeks before study, subjects met with a dietitian and were instructed to consume a weight-maintaining diet with 50% of total energy from carbohydrate, 30% from fat and 20% from protein. During the week prior to the start of PIO or FENO, all subjects received: (1) baseline measurements of FPG, plasma AD, NEFA, insulin (mean of three values drawn at 15 min intervals), fasting plasma lipids and HbA1c; (2) measurement of liver fat content with magnetic resonance spectroscopy [16, 25]; and (3) quantification of hepatic and peripheral tissue insulin sensitivity with a hyperinsulinaemic–euglycaemic clamp [29] in combination with 3-[3H]glucose. All studies were started at 06.00 hours following a 10 h overnight fast. Sulfonylurea-treated subjects discontinued their medication 48 h before the study.
Following completion of these studies, subjects were assigned randomly (using a table of random numbers) to receive FENO, 200 mg/day (n = 8), or PIO, 45 mg/day (n = 7), for 3 months and this was followed by addition of the other agent for an additional 3 months in an open-label study. Three patients in each group were on sulfonylureas at the time of randomisation and these were continued during the entire duration of the study. All subjects underwent a repeat insulin clamp study and measurement of hepatic fat content by proton spectroscopy at the end of 3 and 6 months. During the entire 6 month treatment period, subjects returned to the Clinical Research Center every 2 weeks at 08.00 hours following an overnight fast for measurement of FPG, body weight and blood pressure. Fasting plasma lipids (total cholesterol, triacylglycerol, HDL-cholesterol and LDL-cholesterol) were measured monthly. HbA1c was measured twice during week 12 and twice during week 24. On each visit, dietary adherence was reinforced.
Hyperinsulinaemic–euglycaemic clamp
Insulin sensitivity was assessed with a hyperinsulinaemic–euglycaemic clamp, as previously described [29]. At 06.00 hours (−180 min), a prime (9.25 × 105 Bq × FPG/5.55)–continuous (9.25 × 103 Bq/min) infusion of 3-[3H]glucose was started via a catheter placed into an antecubital vein and continued throughout the study. A second catheter was inserted retrogradely into a vein on the dorsum of the hand, which was then placed in a heated box (60°C). Baseline arterialised venous blood samples for determination of plasma 3-[3H] glucose radioactivity and plasma glucose, NEFA and insulin concentrations were drawn at −30, −20, −10, −5 and 0 min. At time zero, a prime-continuous infusion of human regular insulin (Novolin; Novo Nordisk Pharmaceuticals, Princeton, NJ, USA) was started at a rate of 80 mU min−1 m−2 and continued for 240 min. After the start of insulin no glucose was infused until the plasma glucose concentration declined to 5.55 mmol/l, at which level it was maintained. During the insulin clamp, arterialised blood samples were collected every 5 min for plasma glucose determination and a 20% glucose infusion was adjusted, based upon the negative feedback principle, to maintain plasma glucose at approximately 5.55 mmol/l. Blood samples for determination of plasma insulin, NEFA and 3-[3H]glucose specific activity were collected every 10–15 min during the insulin clamp.
Vastus lateralis muscle biopsy
A needle biopsy of the vastus lateralis was obtained under local anaesthesia in the fasting condition, immediately before the hyperinsulinaemic–euglycaemic clamp in six of the eight subjects who received FENO monotherapy for 3 months followed by the addition of PIO for an additional 3 months (two subjects did not consent to the muscle biopsy procedure). The sample (~100 mg) was frozen in liquid nitrogen for subsequent analyses.
Liver fat content (proton magnetic resonance spectroscopy)
Liver fat content was assessed by magnetic resonance spectroscopy as previously described [16, 25].
AMPK phosphorylation and protein production
For western blotting, muscle lysates were prepared as previously described [30]. Proteins (50 μg) from the muscle lysates were separated by 10% SDS-PAGE and transferred to nitrocellulose membranes. After blocking the membranes with 5% BSA/Tris-buffered saline with Tween 20, they were probed overnight at 4°C with antibodies against anti-phosphorylated AMPK (Thr172) (1:1,000) (Cell Signaling, Beverly, MA, USA). AMPK phosphorylation was normalised for total AMPK α protein content using a pan-α antibody (1:1,000) (Upstate Biotechnology, Lake Placid, NY, USA). Bound antibodies were detected using anti-rabbit immunoglobulin–horseradish peroxidase-linked antibody and ECL reagents (Perkin Elmer, Boston, USA), and the bands were quantified using ImageQuant software (Molecular Dynamics, Sunnyvale, CA, USA).
Analytical determinations
Plasma glucose was measured by the glucose oxidase method (Beckman Instruments, Fullerton, CA, USA). Plasma insulin was measured by RIA (Diagnostic Products Corporation, Los Angeles, CA, USA). 3-[3H]glucose specific activity was determined on deproteinised barium/zinc plasma samples. Plasma NEFA concentration was determined by an enzymatic calorimetric quantification method (Wako Chemicals, Nuess, Germany). Plasma AD concentration was measured by RIA (Linco Research, St Charles, MO, USA).
Calculations
Under steady-state post-absorptive conditions the rate of endogenous glucose appearance (_R_a) was calculated as the 3-[3H]glucose infusion rate (dpm/min) divided by the steady-state plasma 3-[3H]glucose specific activity (dpm/mg). In the post-absorptive state, the hepatic insulin resistance index was calculated as the product of endogenous glucose production (EGP) and fasting plasma insulin concentration. During the hyperinsulinaemic–euglycaemic clamp the rate of total-body glucose appearance (_R_a) was calculated using Steele’s equation [31], using a distribution volume of 250 ml/kg. EGP was calculated by subtracting the exogenous glucose infusion rate from _R_a. The rate of insulin-stimulated total-body glucose disposal (R d) was determined by adding the rate of residual EGP to the exogenous glucose infusion rate.
Statistical analysis
Statistical calculations were performed with StatView for Windows, version 5.0 (SAS Institute, Cary, NC, USA). Values before and after treatment were compared using paired t tests. Data are presented as means ± SEM. p < 0.05 was considered to be statistically significant. Linear regression analysis was used to examine the relationship between plasma AD and hepatic fat content.
Results
Metabolic parameters are shown in Tables 1 and 2. After 3 months of FENO monotherapy, FPG, NEFA, insulin concentrations and HbA1c did not change, while fasting plasma triacylglycerol declined (p < 0.01). Total cholesterol and LDL-cholesterol (both p < 0.05) declined significantly, while HDL-cholesterol (p < 0.05) increased after 3 months of FENO.
Table 1 Anthropometric and metabolic parameters in eight type 2 diabetic patients at baseline, after 3 months of FENO monotherapy, and after 3 months of FENO plus PIO combination therapy (FENO+PIO)
Table 2 Anthropometric and metabolic parameters in seven type 2 diabetic patients at baseline, after 3 months of PIO monotherapy, and after 3 months of FENO plus PIO combination therapy (FENO+PIO)
Following PIO monotherapy for 3 months, FPG, NEFA, insulin concentrations (all p < 0.05), HbA1c (p < 0.01) and fasting plasma triacylglycerol (p < 0.05) decreased significantly, while HDL-cholesterol increased (p < 0.05). Total cholesterol and LDL-cholesterol did not change significantly following PIO monotherapy.
Addition of FENO to PIO did not significantly change FPG, NEFA, insulin or HbA1c; total cholesterol, LDL-cholesterol and plasma triacylglycerol concentrations decreased (p < 0.05) and HDL-cholesterol increased (p < 0.05). Addition of PIO to FENO reduced FPG (p < 0.05), HbA1c (p < 0.05), fasting plasma NEFA (p < 0.01) and plasma triacylglycerol (p < 0.01) concentrations and increased HDL-cholesterol (p < 0.05); total cholesterol and LDL-cholesterol did not change significantly.
Euglycaemic–hyperinsulinaemic clamp
The steady-state plasma glucose concentrations during the 240 min hyperinsulinaemic–euglycaemic clamp were similar before and after 3 months of FENO monotherapy (5.8 ± 0.2 vs 5.8 ± 0.2 mmol/l) and PIO monotherapy (5.8 ± 0.2 vs 5.7 ± 0.2 mmol/l). During the insulin clamp studies performed after the addition of PIO to FENO and FENO to PIO, the steady-state plasma glucose levels were similar (5.7 ± 0.2 mmol/l). The steady-state plasma insulin concentrations during the insulin clamp studies were similar before and after FENO therapy (848 ± 44 vs 825 ± 34 pmol/l) and following the addition of PIO (798 ± 30 pmol/l). Similarly, the steady-state plasma insulin concentrations during the insulin clamp studies were similar before and after PIO therapy (791 ± 50 vs 753 ± 39 pmol/l) and following the addition of FENO (779 ± 48 pmol/l).
R d (Fig. 1) did not change significantly following FENO monotherapy (22.7 ± 4.4 to 24.9 ± 4.4 μmol kg−1 min−1). Following the addition of PIO to FENO monotherapy , R d increased significantly (24.9 ± 4.4 to 36.1 ± 2.2 μmol kg−1 min−1, p < 0.005). Following 3 months of PIO monotherapy, R d increased significantly (23.8 ± 3.8 to 40.5 ± 4.4 μmol kg−1 min−1, p < 0.005). The addition of FENO to PIO treatment did not significantly change R d (40.5 ± 4.4 to 38.8 ± 4.9 μmol kg−1 min−1).
Fig. 1
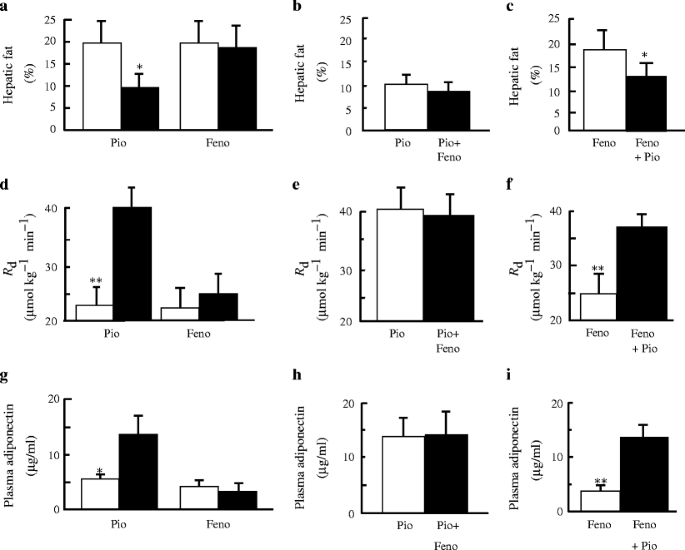
a Effect of FENO (Feno) monotherapy and PIO (Pio) monotherapy for 3 months on hepatic fat content (a), R d (d) and plasma adiponectin levels (g). Effect of FENO added for 3 months to PIO therapy on hepatic fat content (b), R d (e) and plasma adiponectin levels (h). Effect of PIO for 3 months added to FENO therapy on hepatic fat content (c), R d (f) and plasma adiponectin levels (i). *p < 0.05; **p < 0.005. Means ± SEM
Suppression of plasma NEFA during the insulin clamp was similar before and after FENO treatment (145 ± 28 to 120 ± 18 μmol/l). Following the addition of PIO, suppression of plasma NEFA during the insulin clamp was significantly increased (120 ± 18 to 40 ± 8 μmol/l, p < 0.005). Suppression of plasma NEFA was enhanced after 3 months of PIO monotherapy (143 ± 13 to 84 ± 27 μmol/l, p < 0.05) and did not change significantly following the addition of FENO (84 ± 27 to 57 ± 14 μmol/l). The decrease in fasting plasma NEFA concentration in PIO-treated type 2 diabetic patients was strongly correlated with the improvement in R d (r = −0.51, p < 0.05).
Basal rates of EGP were similar before and after FENO treatment (11.6 ± 1.0 vs 11.1 ± 0.5 μmol kg−1 min−1) and did not change significantly following the addition of PIO (11.1 ± 1.0 μmol kg−1 min−1). Basal rates of EGP also were similar before and after PIO treatment (11.1 ± 0.5 vs 10.5 ± 0.5 μmol kg−1 min−1) and did not change after the addition of FENO (11.1 ± 0.5 μmol kg−1 min−1). Insulin-mediated suppression of EGP, determined during the 210–240 min period of the hyperinsulinaemic–euglycaemic clamp, was similar before and after FENO treatment (0.9 ± 0.4 vs 0.8 ± 0.2 μmol kg−1 min−1, respectively). Following the addition of PIO, EGP during the insulin clamp was significantly decreased (to 0.1 ± 0.1 μmol kg−1 min−1, p < 0.05). Insulin-mediated suppression of EGP during the hyperinsulinaemic–euglycaemic clamp, was enhanced after PIO treatment (0.8 ± 0.2 vs 0.1 ± 0.1 μmol kg−1 min−1, p < 0.05). Following the addition of FENO, EGP during the insulin clamp did not change significantly (0.1 ± 0.1 μmol kg−1 min−1).
Plasma AD concentration
Plasma AD (4.3 ± 1.0 to 4.1 ± 0.8 μg/ml) did not change following FENO treatment (Fig. 1). After the addition of PIO, plasma AD levels increased almost threefold (4.1 ± 0.8 to 13.1 ± 2.5 μg/ml, p < 0.005). Following 3 months of PIO monotherapy, plasma AD increased significantly (5.5 ± 0.9 to 13.8 ± 3.5 μg/ml, p < 0.03). Addition of FENO to PIO did not change plasma AD levels (13.8 ± 3.5 to 14.1 ± 4.5 μg/ml). Taken collectively, plasma AD levels before and after PIO treatment correlated with R d (r = 0.51, p < 0.05).
Hepatic fat content
FENO monotherapy (Fig. 1) did not significantly change hepatic fat content (20.5 ± 3.3 to 18.3 ± 3.1%, p = NS). Following the addition of PIO to FENO, hepatic fat content declined significantly (18.3 ± 3.1 to 13.5 ± 2.1%, p < 0.03). After PIO monotherapy, hepatic fat content decreased significantly (20.4 ± 4.8 to 10.2 ± 2.5%, p < 0.02), and was not changed further by the addition of FENO (9.0 ± 2.1%). Taken collectively, plasma AD levels before and after PIO treatment correlated negatively with hepatic fat content (r = −0.52, p < 0.05).
AMPK phosphorylation and protein production
Muscle AMPK activity, as determined by the phosphorylated AMPK:total AMPK ratio, did not change significantly following FENO treatment (1.2 ± 0.2 to 1.2 ± 0.2) (Fig. 2). Following the addition of PIO, muscle AMPK activity increased almost twofold (1.2 ± 0.2 to 2.2 ± 0.3, p < 0.01). Taken collectively, before and after the addition of PIO, muscle AMPK activity was positively correlated with plasma AD concentration (r = 0.72, p < 0.01) and R d (r = 0.49, p < 0.10) but not with fasting plasma NEFA levels (r = 0.02, p = NS). The increase in AMPK phosphorylation was not explained by changes in AMPK α protein content (data not shown).
Fig. 2
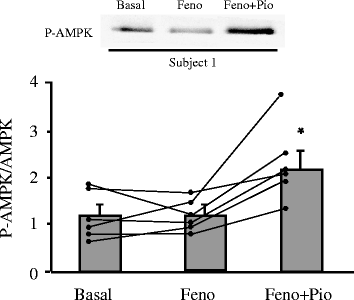
Effect of FENO (Feno) alone for 3 months and addition of PIO (Pio) to FENO for 3 months on skeletal muscle AMPK activity expressed as the phosphorylated AMPK (P-AMPK):total AMPK ratio in six type 2 diabetic patients. *p < 0.01. Means ± SEM
Discussion
In the present study we examined the effect of PIO, a PPAR-γ agonist, and FENO, a PPAR-α agonist, on glucose and lipid metabolism, insulin sensitivity, plasma AD concentration and hepatic fat content in type 2 diabetic patients. Within the therapeutic and supra-therapeutic range of plasma PIO concentrations, this thiazolidinedione does not manifest any PPAR-α activity [32], whereas FENO is a pure PPAR-α agonist [33, 34]. While the PPAR-α effect of FENO is considered to be moderate, it is the only fibrate (along with gemfibrozil) approved for use in humans. Consistent with previous studies in man [16–19, 35], PIO in the present study enhanced adipocyte sensitivity to the antilipolytic effect of insulin, leading to a decrease in fasting plasma NEFA concentration and improved insulin-mediated suppression of plasma NEFA concentration. These results are consistent with the concept that the insulin-sensitising effect of PPAR-γ agonists, at least in part, are indirect and mediated via a reduction in plasma NEFA and intramyocellular concentration of toxic lipid metabolites [1, 35].
In a recent study [36], PIO was shown to enhance the clearance of very-low-density-lipoprotein (VLDL) triacylglycerol-rich particles from the circulation. However, since substrate (plasma NEFA and glucose) delivery to the liver plays an important role in hepatic triacylglycerol synthesis [37], one could hypothesise that part of PIO’s triacylglycerol-lowering effect could be related to the reduction in plasma NEFA and/or glucose concentrations. FENO did not significantly reduce fasting plasma NEFA or glucose concentrations and did not alter adipocyte sensitivity to insulin, yet it reduced the plasma triacylglycerol concentration by a similar amount to that observed with PIO. These findings indicate that FENO exerts a direct inhibitory effect on hepatic VLDL synthesis/secretion or enhances triacylglycerol clearance from the circulation [38, 39]. Fibrates, which possess modest PPAR-α activity, stimulate intravascular lipoprotein lipolysis, enhancing lipoprotein lipase (LPL) activity and reducing the expression of apoCIII, a natural LPL inhibitor. They also inhibit triacylglycerol synthesis and VLDL production by favouring fatty acid uptake and retention, by enhancing fatty acid catabolism and by reducing fatty acid synthesis in hepatocytes [40]. As hypotriacylglycerolaemic drugs, fibrates also induce the expression of apolipoprotein A5 (APOA5), an important regulator of plasma triacylglycerol levels [41]. The results of the present study suggest that FENO directly inhibit hepatic VLDL synthesis, possibly by upregulating APOA5. In contrast, PIO does not alter APOA5 expression in patients with type 2 diabetes [36].
Despite the widespread use of PPAR-α agonists (i.e. FENO and gemfibrozil) for the treatment of hypertriacylglycerolaemia in type 2 diabetic patients, their effect on insulin sensitivity in humans has not been rigorously examined and conflicting results have been reported [9–14]. Studies in rodents conclusively demonstrate that PPAR-α activation with FENO and other more potent PPAR-α agents enhances peripheral (muscle) and hepatic insulin sensitivity [4–8]. Although PPAR-α-null mice do not manifest any obvious alteration in insulin sensitivity [42], PPAR-α activation in nutritional (high-fat diet), genetic (Zucker obese fa/fa rat), and lipoatrophic (A-ZIP/F-1) models of insulin resistance markedly improves insulin sensitivity [4, 7, 43] and reduces visceral fat in the two former models. Intracellular fatty acids and their derivatives interfere with insulin-stimulated glucose metabolism, through an effect on the insulin-signalling pathway, possibly by activating protein kinase C [1, 44]. In rodents it has been suggested that PPAR-α activation increases fatty acid oxidation, thus decreasing intramyocellular lipid content and improving insulin sensitivity [4–7]. In contrast, PPAR-α-null mice are protected from high-fat diet-induced insulin resistance [42]. Furthermore, while in both man and rodents fibrates decrease plasma triacylglycerol, their effects on HDL occur in an opposite manner in these species. Whereas in humans plasma HDL-cholesterol consistently increases with fibrate treatment, a pronounced decrease is observed in rodents. In rats, fibrate treatment lowers hepatic, but not intestinal, apoliprotein (apo) A-I levels, due to a decreased transcription rate of the apoA-I gene in liver. In contrast, the transcription rate of the human apoA-I gene is induced by PPAR-α via its interaction with a positive PPAR response element located in the human apoA-I gene promoter liver-specific enhancer [45]. At present, there is no clear explanation for the markedly discrepant results obtained in rodents vs humans. Nonetheless, the present results clearly demonstrate that in man the PPAR-α agonist, FENO, does not enhance insulin sensitivity in either peripheral tissues (muscle), liver or adipocytes and has no effect on circulating NEFA levels in type 2 diabetic patients. These results may provide a potential explanation for the lack of an effect of FENO on the primary cardiovascular outcome in the recently published FIELD study [46]. However, it should be noted that gemfibrozil treatment in type 2 diabetic patients has previously been shown to reduce the risk of macrovascular complications in the VA-HIT study [47], suggesting that the two fibrates may have differing effects on macrovascular complications in type 2 diabetic patients.
Studies in animals indicate that PPAR-α agonists increase hepatic fat oxidation and reduce hepatic fat content in association with enhanced insulin-mediated suppression of EGP [3, 7, 43, 48]. Although hepatic fat oxidation was not measured in the present study, we did not observe a significant reduction in hepatic fat content or an increase in hepatic insulin sensitivity (in contrast to PIO therapy) following FENO therapy. The lack of effect of FENO on hepatic fat content in the present study serves to emphasise further the differences between the actions of PPAR-α agonist in humans vs rodents.
Of note, we observed an additive effect of combined PIO plus FENO therapy on the reduction in plasma triacylglycerol concentration in the present study, suggesting that the mechanisms responsible for triacylglycerol lowering may be complementary [49, 50]. Further, the results of the present study suggest that a combination of PPAR-γ and PPAR-α therapy (dual PPAR therapy) may be an effective means to lower elevated plasma triacylglycerol concentrations in type 2 diabetic patients [51]. The results of the present study also demonstrate that the decrease in plasma triacylglycerol levels following combined PPAR-α and PPAR-γ therapy is associated with an additive effect to increase plasma HDL-cholesterol levels.
FENO monotherapy did not alter the plasma AD concentration. Previous studies from our laboratory have shown a strong relationship between reduced plasma AD levels and both hepatic and peripheral (muscle) tissue insulin resistance in type 2 diabetic patients [25]. One of the primary effects of AD is to enhance intracellular AMPK activity and increase fatty acid oxidation in muscle, leading to a decrease in intracellular fatty acid metabolites (i.e. FACoAs diacylglycerol, ceramides) and enhanced insulin signal transduction [26, 52]. In contrast to the lack of an effect of FENO therapy on insulin sensitivity, plasma AD levels and muscle AMPK phosphorylation, PIO therapy was associated with a threefold increase in plasma AD concentrations and a twofold increase in skeletal muscle AMPK activity. Although we did not determine muscle AMPK activity in the diabetic patients who received PIO monotherapy in the present study, we have demonstrated previously that PIO monotherapy significantly increases AMPK activity [53]. To the best of our knowledge, this is the first study to examine the effect of combined PPAR-α and PPAR-γ agonists on plasma AD levels and skeletal muscle AMPK activity in type 2 diabetic subjects. Our results demonstrate that FENO, a PPAR-α agonist, has no stimulatory effect to increase plasma AD levels, when given as monotherapy or when added to PIO monotherapy. In contrast, PIO, when given as monotherapy or added to FENO-treated patients, is associated with a marked increase in plasma AD concentration. FENO monotherapy has no effect on skeletal muscle AMPK activity, whereas addition of PIO to FENO-treated diabetic patients stimulates AMPK phosphorylation. These results suggest that enhanced peripheral (muscle) insulin sensitivity following PIO treatment may be associated with at least two distinct mechanisms: (1) AD-mediated enhanced intracellular AMPK activity, leading to increased muscle fatty acid oxidation, reduction in intramyocellular fatty acid metabolites and enhanced insulin signal transduction [52]; and (2) a reduction in plasma NEFA concentration following enhanced adipocyte insulin sensitivity and decreased lipolysis [35], leading to a decrease in intramyocellular lipid metabolites.
In summary, treatment of type 2 diabetic patients with a PPAR-α agonist (FENO) has no effect on plasma glucose/NEFA/AD concentrations, hepatic fat content, muscle AMPK activity, or hepatic, adipocyte and peripheral (muscle) tissue insulin sensitivity despite a reduction in plasma triacylglycerol levels and an increase in HDL-cholesterol. Addition of FENO to PIO-treated patients causes a further reduction in plasma triacylglycerol levels and a further increase in HDL-cholesterol, but has no effect on glycaemic control, peripheral tissue (muscle), hepatic or adipocyte insulin sensitivity, hepatic fat content or plasma AD levels beyond that observed with PIO monotherapy.
References
- Bays H, Mandarino L, DeFronzo RA (2004) Role of the adipocyte, free fatty acids, and ectopic fat in pathogenesis of type 2 diabetes mellitus: peroxisomal proliferator-activated receptor agonists provide a rational therapeutic approach. J Clin Endocrinol Metab 89:463–478
Article PubMed CAS Google Scholar - Kendall DM (2005) The dyslipidaemia of diabetes mellitus: giving triglycerides and high-density lipoprotein cholesterol a higher priority? Endocrinol Metab Clin North Am 34:27–48
Article PubMed CAS Google Scholar - Rubins HB, Robins SJ, Collins D et al (1999) Gemfibrozil for the secondary prevention of coronary heart disease in men with low levels of high-density lipoprotein cholesterol. N Engl J Med 341:410–418
Article PubMed CAS Google Scholar - Ye JM, Iglesias MA, Watson DG et al (2003) PPARalpha/gamma ragaglitazar eliminates fatty liver and enhances insulin action in fat-fed rats in the absence of hepatomegaly. Am J Physiol Endocrinol Metab 284:E531–E540
PubMed CAS Google Scholar - Ye JM, Doyle PJ, Iglesias MA et al (2001) Peroxisome proliferator-activated receptor (PPAR)-alpha activation lowers muscle lipids and improves insulin sensitivity in high fat-fed rats: comparison with PPAR-gamma activation. Diabetes 50:411–417
Article PubMed CAS Google Scholar - Kim H, Haluzik M, Asghar Z et al (2003) Peroxisome proliferator-activated receptor-alpha agonist treatment in a transgenic model of type 2 diabetes reverses the lipotoxic state and improves glucose homeostasis. Diabetes 52:1770–1778
Article PubMed CAS Google Scholar - Guerre-Millo M, Gervois P, Raspe E et al (2000) Peroxisome proliferator-activated receptor alpha activators improve insulin sensitivity and reduce adiposity. J Biol Chem 275:16638–16642
Article PubMed CAS Google Scholar - Choi KC, Ryu OH, Lee KW et al (2005) Effect of PPAR-alpha and -gamma agonist on the expression of visfatin, adiponectin, and TNF-alpha in visceral fat of OLETF rats. Biochem Biophys Res Commun 336:747–753
Article PubMed CAS Google Scholar - Idzior-Walus B, Sieradzki J, Rostworowski W et al (2000) Effects of comicronised fenofibrate on lipid and insulin sensitivity in patients with polymetabolic syndrome X. Eur J Clin Invest 30:871–878
Article PubMed CAS Google Scholar - Avogaro A, Beltramello P, Marin R et al (1995) Insulin action and glucose metabolism are improved by gemfibrozil treatment in hypertriglyceridaemic patients. Atherosclerosis 113:117–124
Article PubMed CAS Google Scholar - Shen DC, Fuh MM, Shieh SM et al (1991) Effect of gemfibrozil treatment in sulfonylurea-treated patients with noninsulin-dependent diabetes mellitus. J Clin Endocrinol Metab 73:503–510
Article PubMed CAS Google Scholar - Vega GL, Cater NB, Hadizadeh DR 3rd et al (2003) Free fatty acid metabolism during fenofibrate treatment of the metabolic syndrome. Clin Pharmacol Ther 74:236–244
Article PubMed CAS Google Scholar - Vuorinen-Markkola H, Yki-Järvinen H, Taskinen M-R (1993) Lowering of triglycerides by gemfibrozil affects neither the glucoregulatory nor antilipolytic effect of insulin in type 2 (non-insulin-dependent) diabetic patients. Diabetologia 36:161–169
Article PubMed CAS Google Scholar - Sane T, Vuorinen-Markkola H, Yki-Järvinen H, Taskinen M-R (1995) Decreasing triglyceride by gemfibrozil therapy does not affect the glucoregulatory or antilipolytic effect of insulin in nondiabetic subjects with mild hypertriglyceridaemia. Metabolism 44:589–596
Article PubMed CAS Google Scholar - Mukherjee R, Jow L, Croston GE, Paterniti JR Jr (1997) Identification, characterization, and tissue distribution of human peroxisome proliferator-activated receptor (PPAR) isoforms PPARγ2 vs PPARγ1 and activation with retinoid X receptor agonists and antagonists. J Biol Chem 272:8071–8076
Article PubMed CAS Google Scholar - Bajaj M, Suraamornkul S, Pratipanawatr T et al (2003) Pioglitazone reduces hepatic fat content and augments splanchnic glucose uptake in patients with type 2 diabetes. Diabetes 52:1364–1370
Article PubMed CAS Google Scholar - Miyazaki Y, Mahankali A, Matsuda M et al (2002) Effect of pioglitazone on abdominal fat distribution and insulin sensitivity in type 2 diabetic patients. J Clin Endocrinol Metab 87:2784–2791
Article PubMed CAS Google Scholar - Mayerson AB, Hundal RS, Dufour S et al (2002) The effects of rosiglitazone on insulin sensitivity, lipolysis, and hepatic and skeletal muscle triglyceride content in patients with type 2 diabetes. Diabetes 51:797–802
Article PubMed CAS Google Scholar - Tiikkainen M, Hakkinen AM, Korsheninnikova E, Nyman T, Makimattila S, Yki-Jarvinen H (2004) Effects of rosiglitazone and metformin on liver fat content, hepatic insulin resistance, insulin clearance, and gene expression in adipose tissue in patients with type 2 diabetes. Diabetes 53:2169–2176
Article PubMed CAS Google Scholar - Seppala-Lindroos A, Vehkavaara S, Hakkinen AM et al (2002) Fat accumulation in the liver is associated with defects in insulin suppression of glucose production and serum free fatty acids independent of obesity in normal men. J Clin Endocrinol Metab 87:3023–3028
Article PubMed CAS Google Scholar - Roden M (2006) Mechanisms of disease: hepatic steatosis in type 2 diabetes-pathogenesis and clinical relevance. Nat Clin Pract Endocrinol Metab 2:335–348
Article PubMed CAS Google Scholar - Weyer C, Funahashi T, Tanaka S et al (2001) Hypoadiponectinaemia in obesity and type 2 diabetes: close association with insulin resistance and hyperinsulinaemia. J Clin Endocrinol Metab 86:1930–1935
Article PubMed CAS Google Scholar - Civitarese AE, Jenkinson CP, Richardson D et al (2004) Adiponectin receptors gene expression and insulin sensitivity in non-diabetic Mexican Americans with or without a family history of type 2 diabetes. Diabetologia 47:816–820
Article PubMed CAS Google Scholar - Maeda N, Takahashi M, Funahashi T et al (2001) PPARgamma ligands increase expression and plasma concentrations of adiponectin, an adipose-derived protein. Diabetes 50:2094–2099
Article PubMed CAS Google Scholar - Bajaj M, Suraamornkul S, Piper P et al (2004) Decreased plasma adiponectin concentrations are closely related to hepatic fat content and hepatic insulin resistance in pioglitazone-treated type 2 diabetic patients. J Clin Endocrinol Metab 89:200–206
Article PubMed CAS Google Scholar - Yamauchi T, Kamon J, Waki H et al (2001) The fat-derived hormone adiponectin reverses insulin resistance associated with both lipoatrophy and obesity. Nat Med 7:941–946
Article PubMed CAS Google Scholar - Combs TP, Berg AH, Obici S, Scherer PE, Rossetti L (2001) Endogenous glucose production is inhibited by the adipose-derived protein Acrp30. J Clin Invest 108:1875–1881
Article PubMed CAS Google Scholar - DeFronzo RA (1999) Pharmacologic therapy for type 2 diabetes mellitus. Ann Intern Med 131:281–303
PubMed CAS Google Scholar - DeFronzo R, Tobin J, Andres R (1979) Glucose clamp technique: a method for quantifying insulin secretion and resistance. Am J Physiol 232:E214–E223
Google Scholar - Musi N, Fujii N, Hirshman MF et al (2001) AMP-activated protein kinase (AMPK) is activated in muscle of subjects with type 2 diabetes during exercise. Diabetes 50:921–927
Article PubMed CAS Google Scholar - Steele R (1959) Influence of glucose loading and of injected insulin on hepatic glucose output. Ann N Y Acad Sci 82:420–430
Article PubMed CAS Google Scholar - Lehmann JM, Moore LB, Smith-Oliver TA et al (1995) An antidiabetic thiazolidinedione is a high affinity ligand for peroxisome proliferator-activated receptor gamma (PPAR gamma). J Biol Chem 270:12953–12956
Article PubMed CAS Google Scholar - Schoonjans K, Staels B, Auwerx J (1996) Role of the peroxisome proliferator activated receptor (PPAR) in mediating effects of fibrates and fatty acids on gene expression. J Lipid Res 37:907–925
PubMed CAS Google Scholar - Staels B, Fruchart JC (2005) Therapeutic roles of peroxisome proliferator-activated receptor agonists. Diabetes 54:2460–2470
Article PubMed CAS Google Scholar - Miyazaki Y, Glass L, Triplitt C et al (2001) Effect of rosiglitazone on glucose and free fatty acid metabolism in type 2 diabetic patients. Diabetologia 44:2210–2219
Article PubMed CAS Google Scholar - Nagashima K, Lopez C, Donovan D et al (2005) Effects of the PPARgamma agonist pioglitazone on lipoprotein metabolism in patients with type 2 diabetes mellitus. J Clin Invest 115:1323–1332
Article PubMed CAS Google Scholar - Greenfield M, Kolterman O, Olefsky J, Reaven GM (1980) Mechanism of hypertriglyceridaemia in diabetic patients with fasting hyperglycaemia. Diabetologia 18:441–446
Article PubMed CAS Google Scholar - Auboeuf D, Rieusset J, Fajas L et al (1997) Tissue distribution and quantification of the expression of the peroxisome proliferator activated receptors and of LXR mRNAs in human: effect of obesity and NIDDM in adipose tissue. Diabetes 46:1319–1327
Article PubMed CAS Google Scholar - D’Costa MA, Angel A (1975) Inhibition of hormone-stimulated lipolysis by clofibrate: a possible mechanism for its hypolipidaemic action. J Clin Invest 55:138–148
PubMed CAS Google Scholar - Staels B, Dallongeville J, Auwerx J et al (1998) Mechanism of action of fibrates on lipid and lipoprotein metabolism. Circulation 98:2088–2093
PubMed CAS Google Scholar - Vu-Dac N, Gervois P, Jakel H et al (2003) Apolipoprotein A5, a crucial determinant of plasma triglyceride levels, is highly responsive to peroxisome proliferator-activated receptor alpha activators. J Biol Chem 278:17982–17985
Article PubMed CAS Google Scholar - Guerre-Millo M, Rouault C, Poulain P et al (2001) PPAR-alpha-null mice are protected from high-fat diet-induced insulin resistance. Diabetes 50:2809–2814
Article PubMed CAS Google Scholar - Chou CJ, Haluzik M, Gregory C et al (2002) WY14643, a peroxisome proliferator-activated receptor alpha (PPARalpha) agonist, improves hepatic and muscle steatosis and reverses insulin resistance in lipoatrophic A-ZIP/F-1 mice. J Biol Chem 277:24484–24489
Article PubMed CAS Google Scholar - Itani SI, Ruderman NB, Schmieder F, Boden G (2002) Lipid-induced insulin resistance in human muscle is associated with changes in diacylglycerol, protein kinase C, and IkB-α. Diabetes 51:2005–2011
Article PubMed CAS Google Scholar - Vu-Dac N, Chopin-Delannoy S, Gervois P et al (1998) The nuclear receptors peroxisome proliferator-activated receptor alpha and Rev-erbalpha mediate the species-specific regulation of apolipoprotein A-I expression by fibrates. J Biol Chem 273:25713–25720
Article PubMed CAS Google Scholar - Keech A, Simes RJ, Barter P et al (2005) Effects of long-term fenofibrate therapy on cardiovascular events in 9795 people with type 2 diabetes mellitus (the FIELD study): randomised controlled trial. Lancet 366:1849–1861
Article PubMed CAS Google Scholar - Rubins HB, Robins SJ, Collins D et al (2002) Diabetes, plasma insulin, and cardiovascular disease: subgroup analysis from the Department of Veterans Affairs high-density lipoprotein intervention trial (VA-HIT). Arch Intern Med 162:2597–2604
Article PubMed CAS Google Scholar - Staels B, Vu-Dac N, Kosykh VA et al (1995) Fibrates downregulate apolipoprotein C-III expression independent of induction of peroxisomal acyl coenzyme A oxidase: a potential mechanism for the hypolipidaemic action of fibrates. J Clin Invest 95:705–712
Article PubMed CAS Google Scholar - Auwerx J, Schoonjans K, Fruchart JC, Staels B (1996) Regulation of triglyceride metabolism by PPARs: fibrates and thiazolidinediones have distinct effects. J Atheroscler Thromb 3:81–89
PubMed CAS Google Scholar - Lefebvre AM, Peinado-Onsurbe J, Leitersdorf I et al (1997) Regulation of lipoprotein metabolism by thiazolidinediones occurs through a distinct but complementary mechanism relative to fibrates. Arterioscler Thromb Vasc Biol 17:1756–1764
PubMed CAS Google Scholar - Buse JB, Rubin CJ, Frederich R et al (2005) Muraglitazar, a dual (alpha/gamma) PPAR activator: A randomized, double-blind, placebo-controlled, 24-week monotherapy trial in adult patients with type 2 diabetes. Clin Ther 27:1181–1195
Article PubMed CAS Google Scholar - Yamauchi T, Kamon J, Minokoshi Y et al (2002) Adiponectin stimulates glucose utilization and fatty-acid oxidation by activating AMP-activated protein kinase. Nat Med 8:1288–1295
Article PubMed CAS Google Scholar - Sriwijitkamol A, Wajcberg E, DeFronzo RA, Cersosimo E, Musi N (2006) Role of AMPK in thiazolidinediones mechanism of action. Diabetes 55(Suppl 1):A337
Google Scholar