Diverse roles for protein kinase C δ and protein kinase C ε in the generation of high-fat-diet-induced glucose intolerance in mice: regulation of lipogenesis by protein kinase C δ (original) (raw)
Abstract
Aims/hypothesis
This study aimed to determine whether protein kinase C (PKC) δ plays a role in the glucose intolerance caused by a high-fat diet, and whether it could compensate for loss of PKCε in the generation of insulin resistance in skeletal muscle.
Methods
Prkcd −/−, Prkce −/− and wild-type mice were fed high-fat diets and subjected to glucose tolerance tests. Blood glucose levels and insulin responses were determined during the tests. Insulin signalling in liver and muscle was assessed after acute in vivo insulin stimulation by immunoblotting with phospho-specific antibodies. Activation of PKC isoforms in muscle from Prkce −/− mice was assessed by determining intracellular distribution. Tissues and plasma were assayed for triacylglycerol accumulation, and hepatic production of lipogenic enzymes was determined by immunoblotting.
Results
Both Prkcd −/− and Prkce −/− mice were protected against high-fat-diet-induced glucose intolerance. In Prkce −/− mice this was mediated through enhanced insulin availability, while in Prkcd −/− mice the reversal occurred in the absence of elevated insulin. Neither the high-fat diet nor Prkcd deletion affected maximal insulin signalling. The activation of PKCδ in muscle from fat-fed mice was enhanced by Prkce deletion. PKCδ-deficient mice exhibited reduced liver triacylglycerol accumulation and diminished production of lipogenic enzymes.
Conclusions/interpretation
Deletion of genes encoding isoforms of PKC can improve glucose intolerance, either by enhancing insulin availability in the case of Prkce, or by reducing lipid accumulation in the case of Prkcd. The absence of PKCε in muscle may be compensated by increased activation of PKCδ in fat-fed mice, suggesting that an additional role for PKCε in this tissue is masked.
Similar content being viewed by others
Introduction
Insulin resistance is strongly associated with increased lipid accumulation in insulin target tissues such as skeletal muscle, and several mechanisms have been proposed to account for this. Much attention has focussed on the role of isoforms of the protein kinase C (PKC) family, which are lipid-activated signal transduction enzymes capable of disrupting several steps in the insulin signalling cascade [1]. PKC activation in intact cells and tissues is associated with enzyme redistribution from cytosolic to membrane locations. Thus the translocation of PKCδ, PKCε and PKCθ and, to a lesser extent, of PKCα and PKCβ has been widely reported in muscle and liver of dietary and genetic rodent models of insulin resistance as well as in obese insulin-resistant humans [1].
More recently the causative nature of the activation of specific PKC isoforms in the generation of insulin resistance by fat oversupply has been addressed using knockout mice for genes encoding the different isoforms. The absence of PKCα or PKCβ has been reported to confer minor improvements in insulin action [1]. More strikingly, PKCθ has been shown to prevent the effects of acute lipid infusion on muscle insulin sensitivity [2]. The effect of PKCε ablation on glucose tolerance has been investigated by Prkce knockdown using specific oligonucleotides and by Prkce deletion; PKCε may play a role in hepatic insulin resistance in the short term [3], and also in insulin availability in the long term [4]. Here we examine for the first time the role of PKCδ in a long-term dietary model of glucose intolerance.
Methods
Animals and dietary treatment
Knockout mice Prkcd −/− and Prkce −/− were generated as described previously [4, 5] and maintained on either a pure C57BL/6 background (Prkce −/−) or a hybrid 129/Sv × C57BL/6 background (Prkcd −/−) using heterozygous breeding pairs. Ethical approval for mouse studies was granted by the Garvan Institute/St Vincent’s Hospital Animal Ethics Committee. Male mice at 7 weeks of age were fed one of two high-fat diets, rich in either unsaturated [6] or saturated fat [4] for 3 or 16 weeks, respectively. Intraperitoneal glucose tolerance tests (IPGTTs) were performed and blood glucose, plasma insulin and tissue triacylglycerols were assayed [4].
Immunoblotting
Tissues from chow- and fat-fed mice were extracted for the analysis of insulin signalling and metabolic enzymes [4] or fractionated to determine the distribution of PKC isoforms in cytosolic and solubilised-membrane compartments [7].
Statistics
All results are expressed as means ± SEM. Statistical calculations were performed using Statview SE + Graphics for Macintosh (Abacus Concepts, Berkeley, CA, USA).
Results
The effect of Prkcd deletion on glucose tolerance in fat-fed mice
To investigate the role of PKCδ in the dysregulation of glucose homeostasis by lipid oversupply, we examined glucose tolerance in Prkcd −/− mice and wild-type litter mates fed either chow or a well-characterised high-fat diet rich in safflower oil [6]. The high-fat diet did not cause a significant increase in body weight but tended to increase muscle triacylglycerol content (Electronic supplementary material [ESM] Fig. 1) and, most importantly, caused significant glucose intolerance in wild-type mice, whereas Prkcd deletion protected against this impairment (Fig. 1a). Prkcd deletion also improved glucose tolerance in chow-fed mice (Fig. 1a), most probably through enhanced insulin sensitivity, as circulating insulin was diminished (Fig. 1b).
Fig. 1
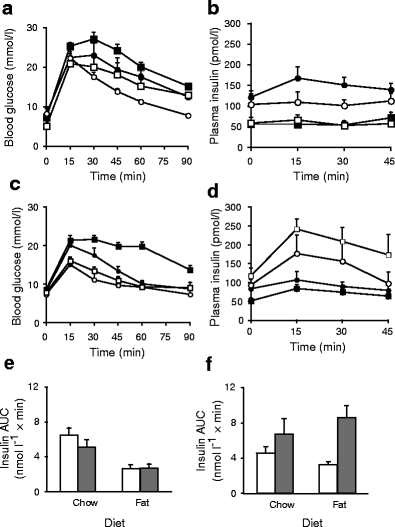
Effect of high-fat diet and Prkcd or Prkce deletion on glucose tolerance in mice. a Blood glucose levels during IPGTT of wild-type mice (black squares, n = 5) and Prkcd −/− litter mates (white squares, n = 5) fed a diet rich in unsaturated fat for 3 weeks, and wild-type mice (black circles, n = 11) and Prkcd −/− litter mates (white circles, n = 6) fed a standard chow diet as controls. ANOVA: p < 0.05 for effect of diet; p < 0.001 for effect of Prkcd deletion. b Insulin levels during the IPGTT of wild-type and Prkcd −/− mice (symbols as in a). ANOVA: p < 0.001 for effect of diet; p < 0.02 for effect of Prkcd deletion in chow-fed mice. c Blood glucose levels during IPGTT of wild-type (black squares, n = 17) and Prkce −/− (white squares, n = 15) mice fed an unsaturated-fat diet for 3 weeks, and wild-type (black circles, n = 12) and Prkce −/− (white circles, n = 9) mice fed a standard chow diet as controls. ANOVA: p < 0.001 for effect of fat diet in wild-type mice; p < 0.001 for effect of Prkce deletion in fat-fed mice. d Insulin levels during the IPGTT of wild-type and Prkce −/− mice (symbols as in c). ANOVA: p < 0.01 for effect of fat diet in wild-type mice; p < 0.001 for effect of Prkce deletion; p < 0.05 for effect of fat diet in Prkce −/− mice. The areas under the curve for insulin were calculated for the IPGTTs in (e) Prkcd −/− mice (grey bars) and (f) Pkcε −/− mice (grey bars) and wild-type litter mates (white bars). ANOVA: p < 0.005 for effect of diet in Prkcd −/− mice; p < 0.001 for effect of genotype in Prkce −/− mice
Distinct roles for PKCδ and PKCε in glucose homeostasis
These results contrast with those we have previously reported for Prkce deletion [4], where there was no obvious improvement in insulin sensitivity but a protection against glucose intolerance caused by a high-saturated-fat diet through enhanced insulin availability. For direct comparison, Prkce −/− mice were fed the same fat diet as above, which is rich in unsaturated fat, and exhibited similar increases in muscle triacylglycerol levels but no gross changes in body weight (ESM Fig. 1). Although these animals were also more glucose tolerant than the corresponding chow- or fat-fed wild-type mice (Fig. 1c), in this case we observed greater insulin responses to the glucose challenge in Prkce −/− mice (Fig. 1d). This again suggests that the improved glucose tolerance observed in Prkce −/− mice is best explained by enhanced insulin availability rather than an improvement in insulin sensitivity. This difference between PKCε- and PKCδ-deficient animals is clearly seen by comparing the respective areas under the curves of insulin during IPGTT (Fig. 1e, f).
PKCδ activation is enhanced in skeletal muscle of Prkce−/− mice
PKCα, PKCθ and PKCε were found to translocate from cytosolic to membrane fractions of muscle from wild-type fat-fed mice resulting in an increased membrane/cytosol ratio, indicating activation (ESM Figs 2 and 3). Fat feeding also induced diminished cytosolic levels of PKCδ (ESM Fig. 2d), most likely resulting from translocation to, and subsequent downregulation in, membrane fractions. This PKCδ response was enhanced in fat-fed Prkce −/− mice (ESM Fig. 2d), suggesting that PKCδ could compensate for the absence of PKCε in the generation of muscle insulin resistance.
Prkcd deletion reduces lipid accumulation in fat-fed mice
However, although the diet high in unsaturated fat causes muscle insulin resistance in rats within 3 weeks [6, 7], the improvement in glucose homeostasis observed in Prkcd −/− mice was not associated with the reversal of any defect in proximal insulin signal transduction in skeletal muscle (ESM Fig. 4) or in whole body insulin tolerance (ESM Fig. 5). This suggested that this diet may cause a more subtle defect at the level of the liver, rather than in peripheral tissues, which is PKCδ dependent. Maximal insulin-stimulated phosphorylation of protein kinase B (Akt) and insulin receptor substrate 1 (IRS-1) in liver was also not affected by the diet (ESM Fig. 6). However, liver and plasma triacylglycerol levels and epididymal fat mass were all reduced in Prkcd −/− mice in comparison to fat-fed wild-type litter mates (Fig. 2a–f). This reduction in lipid deposition was associated with reduced production of sterol regulatory element binding transcription factor 1 (SREBF1), fatty acid synthase and acetyl-CoA carboxylase in liver (Fig. 2i–l) indicating that PKCδ plays a role in the regulation of hepatic lipogenesis. Most likely as a consequence, fasting plasma glucose was also reduced in Prkcd −/− mice (Fig. 2g, h). In contrast, there was no difference in these variables between fat-fed Prkce −/− and control mice.
Fig. 2
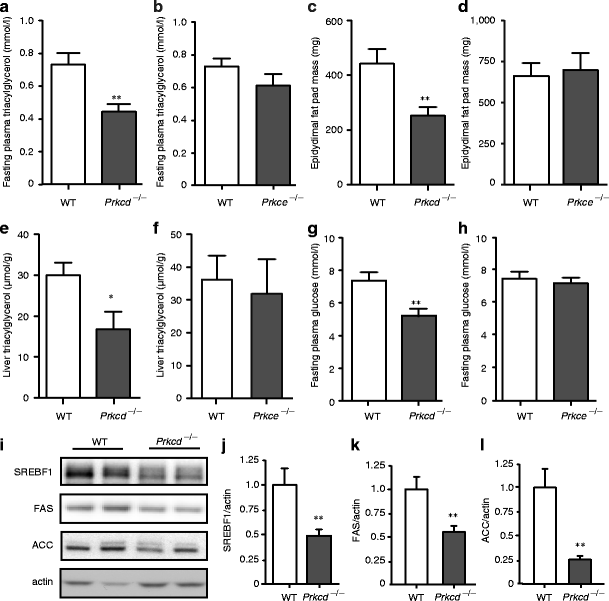
Prkcd deletion improves lipid profiles and reduces lipogenic protein production in fat-fed mice. Fat-fed Prkcd −/− but not Prkce −/− mice exhibit reduced (a, b) plasma triacylglycerol, (c, d) epididymal fat pad mass, (e, f) liver triacylglycerol and (g, h) fasting plasma glucose. Student’s t test, Prkcd −/− vs wild-type (WT) mice: *p < 0.05, **p < 0.01. i Production of SREBF1 (j), fatty acid synthase (FAS) (k) and acetyl-CoA carboxylase (l) (ACC) protein was determined in liver from fat-fed mice by immunoblotting and densitometry. Student’s t test, Prkcd −/− vs wild-type mice: **p < 0.01
Discussion
We have demonstrated an inhibitory role for PKCδ in glucose homeostasis, which appears to contribute significantly to the whole body glucose intolerance caused by a high-fat diet. In fact, this is the first demonstration of a role for any PKC isoform consistent with modulating insulin action in a long-term model of lipid oversupply. Roles for PKCθ (in muscle [2]) and PKCε (in liver [3]), proposed on the basis of short-term lipid infusion or 3 day diets, have not been confirmed when high-fat diets have been extended for several weeks [4, 8]. Such a role for PKCδ is in contrast to our earlier findings concerning PKCε, where deletion of Prkce has a greater effect on insulin secretion. While deletion of either of the genes encoding the isoforms improves glucose tolerance, a difference in action is supported by the comparison of the effects of the fat diet on insulin responses during IPGTT. Loss of PKCε gives rise to an improved insulin response which most likely compensates for insulin resistance. On the other hand, the lower insulin levels observed during IPGTT in both chow- and fat-fed Prkcd −/− mice, when compared with wild-type mice, suggest that Prkcd −/− mice are more insulin sensitive. While PKCδ has also been implicated in the inhibition of insulin signal transduction through serine phosphorylation of IRS-1 [9], our findings suggest an effect of the kinase on lipogenesis, especially in the liver, which may indirectly influence insulin sensitivity. This could be through subtle effects on insulin signalling which we were unable to detect in tissues from mice injected with a maximal bolus of insulin. Previously, only atypical PKC activity has been linked to SREBF1 production and hepatic lipid synthesis [10], making this a novel PKCδ-dependent regulatory mechanism.
We also present evidence that PKCδ has the potential to compensate for Prkce deletion, at least in skeletal muscle. Fat feeding resulted in a decrease in the cytosolic component of PKCδ, a pattern which has previously been described for both PKCθ and PKCδ under similar conditions, suggestive of increased chronic activation [1], and this redistribution was increased in Prkce −/− mice. It is possible that an enhanced PKCδ response prevented an improvement in insulin action, as opposed to insulin secretion, in Prkce −/− mice. Compensation by PKCδ has previously been described for specific PKCε-dependent effects in heart muscle [11].
The diet high in unsaturated fat had inhibitory effects on insulin levels during IPGTT. This is in agreement with previous work comparing the effects of lard-based (i.e. containing saturated NEFA) and soy-based (containing polyunsaturated NEFA) high-fat diets on insulin secretion [12]. Importantly, however, we show that Prkce deletion is able to overcome the suppression of insulin secretion caused by unsaturated NEFA oversupply.
In conclusion, we have highlighted an inhibitory role for PKCδ which appears to be mediated by effects on lipogenesis and insulin sensitivity rather than insulin production, and which could compensate for Prkce deletion in knockout mice. Our findings underscore the utility of targeting PKCε to overcome beta cell dysfunction as a treatment of type 2 diabetes, and further suggest that combined inhibition of PKCε and PKCδ might be beneficial in overcoming insulin resistance.
Abbreviations
Akt:
Protein kinase B
IPGTT:
Intraperitoneal glucose tolerance test
IRS-1:
Insulin receptor substrate 1
PKC:
Protein kinase C
SREBF1:
Sterol regulatory element binding transcription factor 1
References
- Schmitz-Peiffer C, Biden TJ (2008) Protein kinase C function in muscle, liver, and beta-cells and its therapeutic implications for type 2 diabetes. Diabetes 57:1774–1783
Article CAS PubMed Google Scholar - Kim JK, Fillmore JJ, Sunshine MJ et al (2004) PKC-θ knockout mice are protected from fat-induced insulin resistance. J Clin Invest 114:823–827
CAS PubMed Google Scholar - Samuel VT, Liu ZX, Wang A et al (2007) Inhibition of protein kinase Cε prevents hepatic insulin resistance in nonalcoholic fatty liver disease. J Clin Invest 117:739–745
Article CAS PubMed Google Scholar - Schmitz-Peiffer C, Laybutt DR, Burchfield JG et al (2007) Inhibition of PKCε improves glucose-stimulated insulin secretion and reduces insulin clearance. Cell Metab 6:320–328
Article CAS PubMed Google Scholar - Leitges M, Mayr M, Braun U et al (2001) Exacerbated vein graft arteriosclerosis in protein kinase C delta-null mice. J Clin Invest 108:1505–1512
CAS PubMed Google Scholar - Kraegen EW, Clark PW, Jenkins AB, Daley EA, Chisholm DJ, Storlien LH (1991) Development of muscle insulin resistance after liver insulin resistance in high-fat fed rats. Diabetes 40:1397–1403
Article CAS PubMed Google Scholar - Schmitz-Peiffer C, Browne CL, Oakes ND et al (1997) Alterations in the expression and cellular localization of protein kinase C isozymes ε and θ are associated with insulin resistance in skeletal muscle of the high-fat-fed rat. Diabetes 46:169–178
Article CAS PubMed Google Scholar - Gao Z, Wang Z, Zhang X et al (2007) Inactivation of PKCθ leads to increased susceptibility to obesity and dietary insulin resistance in mice. Am J Physiol 292:E84–E91
CAS Google Scholar - Waraich RS, Weigert C, Kalbacher H et al (2008) Phosphorylation of Ser357 of rat insulin receptor substrate-1 mediates adverse effects of protein kinase C-delta on insulin action in skeletal muscle cells. J Biol Chem 283:11226–11233
Article CAS PubMed Google Scholar - Matsumoto M, Ogawa W, Akimoto K et al (2003) PKC lambda in liver mediates insulin-induced SREBP-1c expression and determines both hepatic lipid content and overall insulin sensitivity. J Clin Invest 112:935–944
CAS PubMed Google Scholar - Gray MO, Zhou HZ, Schafhalter-Zoppoth I, Zhu PL, Mochly-Rosen D, Messing RO (2004) Preservation of base-line hemodynamic function and loss of inducible cardioprotection in adult mice lacking protein kinase C-ε. J Biol Chem 279:3596–3604
Article CAS PubMed Google Scholar - Dobbins RL, Szczepaniak LS, Myhill J et al (2002) The composition of dietary fat directly influences glucose-stimulated insulin secretion in rats. Diabetes 51:1825–1833
Article CAS PubMed Google Scholar
Acknowledgements
This work was supported by grants from the National Health and Medical Research Council of Australia and from the Diabetes Australia Research Trust. The authors wish to acknowledge the expert technical assistance of the Garvan Biological Testing Facility and M. Liao, Diabetes and Obesity Program, Garvan Institute of Medical Research, New South Wales, Australia. We would also like to thank R. Laybutt, Diabetes and Obesity Program, Garvan Institute, for helpful discussions.
Duality of interest
The authors declare that there is no duality of interest associated with this manuscript.
Author information
Authors and Affiliations
- Diabetes and Obesity Program, Garvan Institute of Medical Research, 384 Victoria Street, Darlinghurst, New South Wales, 2010, Australia
G. Frangioudakis, J. G. Burchfield, S. Narasimhan, G. J. Cooney, T. J. Biden & C. Schmitz-Peiffer - St Vincent’s Clinical School, University of New South Wales, Sydney, New South Wales, Australia
G. J. Cooney, T. J. Biden & C. Schmitz-Peiffer - Biotechnology Centre of Oslo, University of Oslo, Oslo, Norway
M. Leitges
Authors
- G. Frangioudakis
You can also search for this author inPubMed Google Scholar - J. G. Burchfield
You can also search for this author inPubMed Google Scholar - S. Narasimhan
You can also search for this author inPubMed Google Scholar - G. J. Cooney
You can also search for this author inPubMed Google Scholar - M. Leitges
You can also search for this author inPubMed Google Scholar - T. J. Biden
You can also search for this author inPubMed Google Scholar - C. Schmitz-Peiffer
You can also search for this author inPubMed Google Scholar
Corresponding author
Correspondence toC. Schmitz-Peiffer.
Electronic supplementary material
Below is the link to the electronic supplementary material.
ESM Fig. 1
Effects of high-fat diet and Prkcd and Prkce deletion on body weight and muscle triacylglycerol content. Body weight (a) and triacylglycerol content of quadriceps muscle (b) of wild-type and Prkcd −/− mice fed an unsaturated-fat diet for 3 weeks, and wild-type and Prkcd −/− mice fed a standard chow diet as controls. (c) Body weight and (d) muscle triacylglycerol content of wild-type and Prkce −/− mice fed the unsaturated fat diet for 3 weeks, and wild-type and Prkce −/− mice fed a standard chow diet as controls. ANOVA: p < 0.09 and p < 0.01, effect of diet on muscle triacylglycerol in (b) and (d), respectively (PDF 240 kb).
ESM Fig. 2
a Total PKCε content in muscle from chow- and fat-fed wild-type and Prkcd −/− mice (n = 5 per group). b Total PKCδ levels in muscle from chow- and fat-fed wild-type and Prkce −/− mice (n = 3 per group). PKC protein in total quadriceps muscle extracts was assessed by immunoblotting and corrected for total tubulin expression. (c–f) Effects of high-fat diets and Prkce deletion on PKC translocation in skeletal muscle. Representative immunoblots of PKC isoform distribution in cytosolic (C) and solubilised membrane (M) fractions of quadriceps muscle from chow- and fat-fed mice are shown, together with the results of densitometry (n = 5–6 mice per group). Histograms above immunoblots indicate the relative amounts of each isoform in specific fractions, while panels below indicate the ratio of membrane-associated enzyme to cytosolic enzyme, to provide an indication of activation. (c) PKCε and (d) PKCδ in muscle from mice fed a diet rich in unsaturated fat. (e) PKCε and (f) PKCδ in muscle from mice receiving a diet rich in saturated fat. ANOVA: for effect of diet, *p < 0.05, **p < 0.02; for effect of genotype on cytosolic PKC level (upper histograms) or membrane:cytosol ratio (lower histograms),† p < 0.05, †† p < 0.01 (PDF 722 kb).
ESM Fig. 3
Effects of diets high in unsaturated or saturated fat on PKCα and PKCθ distribution in skeletal muscle. Data are presented as described in the legend to ESM Fig. 2. (a) PKCα and (b) PKCθ in muscle from mice fed a diet rich in unsaturated fat, (c) PKCα and (d) PKCθ in muscle from mice receiving a diet rich in saturated fat. ANOVA for effect of unsaturated fat diet, ***p < 0.0075 (PDF 372 kb).
ESM Fig. 4
Effects of a diet high in unsaturated fat and Prkcd deletion on proximal insulin signal transduction in skeletal muscle. Acute insulin stimulation of mice was carried out as previously reported [1]. Signal transduction in quadriceps muscle extracts was assessed by immunoblotting with phospho-specific antibodies, and the results of densitometry normalised to total signalling protein in each case [1, 2]. (a) phosphotyrosine-1162/1163 insulin receptor (pY 1162/1163 IR); (b) phosphotyrosine-612 IRS-1(pY 612 IRS-1); (c) phosphoserine-473 Akt (pS 473 Akt). References 1. Cooney GJ, Lyons RJ, Crew AJ et al (2004) Improved glucose homeostasis and enhanced insulin signalling in Grb14-deficient mice. EMBO J 23:582–593. 2. Schmitz-Peiffer C, Laybutt DR, Burchfield JG et al (2007) Inhibition of PKCε improves glucose-stimulated insulin secretion and reduces insulin clearance. Cell Metab 6:320–328 (PDF 352 kb).
ESM Fig. 5
Effects of high-fat diet and Prkcd deletion on insulin tolerance. Wild-type (black squares) and Prkcd −/− mice (white squares) fed an unsaturated fat diet for 3 weeks, and wild-type mice fed a standard chow diet as controls (black circles), were subjected to insulin tolerance tests (0.75 U/kg) [1]. Blood glucose levels over 30 min are shown (n = 5 mice per group). Reference 1. Cooney GJ, Lyons RJ, Crew AJ et al (2004) Improved glucose homeostasis and enhanced insulin signalling in Grb14-deficient mice. EMBO J 23:582–593. (PDF 22 kb).
ESM Fig. 6
Effects of a diet high in unsaturated fat and Prkcd deletion on proximal insulin signal transduction in liver. Acute insulin stimulation of mice and immunoblotting was carried out as for ESM Fig. 4. (a) phosphotyrosine-612 IRS-1(pY 612 IRS-1); (b) phosphoserine-473 Akt (pS 473 Akt) (PDF 160 kb).
Rights and permissions
About this article
Cite this article
Frangioudakis, G., Burchfield, J.G., Narasimhan, S. et al. Diverse roles for protein kinase C δ and protein kinase C ε in the generation of high-fat-diet-induced glucose intolerance in mice: regulation of lipogenesis by protein kinase C δ.Diabetologia 52, 2616–2620 (2009). https://doi.org/10.1007/s00125-009-1543-0
- Received: 08 May 2009
- Accepted: 02 September 2009
- Published: 07 October 2009
- Issue Date: December 2009
- DOI: https://doi.org/10.1007/s00125-009-1543-0