Autophagy deficiency in beta cells leads to compromised unfolded protein response and progression from obesity to diabetes in mice (original) (raw)
Introduction
Macroautophagy (hereafter referred to as autophagy) is a dynamic process that involves the rearrangement of subcellular membranes to sequester cytoplasm and organelles for delivery to lysosomes, where the sequestered material is degraded and recycled [1]. Because autophagy degrades damaged or unnecessary organelles and rejuvenates their function, impairment of autophagy may lead to organelle dysfunction. In fact, autophagy deficiency has been reported to cause structural abnormalities in a variety of organelles such as mitochondria and endoplasmic reticulum (ER) [2].
ER and autophagy have an intimate relationship. ER has been reported to be a source of isolation membrane in the process of autophagosome formation [3]. Both ER and autophagy are involved in the removal of unnecessary proteins [4]. The autophagic process has been reported to be activated by ER stress and to play a cytoprotective role in such a state [5–7]. While the effects of ER stress on autophagy have been widely investigated, the role of autophagy in unfolded protein response (UPR)—the cellular response against ER stress—has not been thoroughly studied.
With respect to the ER, pancreatic beta cells are unique in that they are constantly exposed to an unusually heavy ER load due to a large insulin requirement. Thus pancreatic beta cells are particularly susceptible to ER stress, which is aggravated by obesity, insulin resistance, lipids and other physiological or pathological stimuli [8]. In pancreatic islets of human type 2 diabetes, evidence for increased ER stress has been demonstrated [9].
From experiments with mice with beta cell-specific deletion of the autophagy-related 7 (Atg7) gene, we have previously reported that autophagy is important in the maintenance of beta cell mass, structure and function [10]. The ATG7 molecule is an E1-like enzyme that is critical for expansion and completion of autophagosomes [2]. However, the role of beta cell autophagy in the pathogenesis of murine diabetes has not been addressed using mice with beta cell-specific Atg7 deletion, although signs of altered autophagy have been seen in islets of human type 2 diabetes [11]. In this study, to investigate the relationship between beta cell autophagy and diabetes, we looked at the role of autophagy in UPR of pancreatic beta cells and the susceptibility of autophagy-deficient beta cells to ER stressors. We found that autophagy-deficient beta cells are prone to ER stress inflicted as lipid injury or obesity, and autophagy deficiency in conjunction with obesity synergistically leads to overt diabetes, which was not observed in obese mice or mice with autophagy-deficient beta cells alone.
Methods
Mice
To generate obese mice with beta cell-specific autophagy deficiency, we crossed rat insulin promoter (RIP)-Cre +;Atg7 F/W mice [10] with ob/w mice (Jackson Laboratory, Bar Harbor, ME, USA). Mouse genotyping, monitoring of blood glucose levels and a glucose tolerance test were performed as described [10]. Metabolic studies were carried out using male mice of each genotype. Green fluorescent protein (GFP)-microtubule-associated protein (MAP) light chain 3 (LC3 +) mice [12] (kindly provided by N. Mizushima, Tokyo Medical and Dental University, Tokyo, Japan) were also bred with ob/w mice to generate GFP-LC3 +-ob/ob mice. All animal experiments were conducted in accordance with the institutional guidelines of Samsung Medical Center.
Cells
NIT-1 cells (SV40 T-transformed insulinoma cells) were cultured in DMEM/10% FBS.
Real-time RT-PCR
RNA was prepared from isolated islet cells using TRIzol Reagent (Invitrogen, Carlsbad, CA, USA). cDNA was synthesised using Superscript II (Invitrogen) and oligo(dT)12–18 primer. Expression of UPR-related genes, antioxidant genes and p85α and p85β subunits of phosphoinositide 3-kinase (PI3K) was examined by real-time RT-PCR analysis using specific primers (electronic supplementary material [ESM] Table 1) [13].
Beta cell mass and apoptosis
Relative beta cell mass was estimated as described [10]. Briefly, after insulin immunohistochemistry of paraffin-embedded sections, point counting was carried out using a grid. An average of 7,000 points/mouse was counted. To detect apoptotic beta cells, TUNEL staining combined with insulin immunohistochemistry was conducted as described [14]. For quantification of apoptotic beta cells, more than 50 islets from three parallel sections obtained at different cut levels were analysed per mouse.
Beta cell function
Serum insulin levels before and 15 min after glucose challenge were determined using an ELISA kit (Shibayagi Co, Gunma, Japan) to calculate the insulinogenic index (∆insulin15 min [pmol/l]/∆glucose15 min [mmol/l]) [10].
Fluorescent microscopy
Nitrated proteins were detected by fluorescent microscopy (Nikon, Melville, NY, USA) after immunostaining of paraffin-embedded sections using a nitrotyrosine-specific antibody (Chemicon, Temecula, CA, USA). Accumulation of ubiquitin and p62 was examined using specific antibodies as reported [10]. GFP puncta were identified by fluorescent microscopy as described [12].
Islet cell death
Primary pancreatic islets were isolated from fasted mice using the collagenase digestion technique [15]. Islets were placed on Millicell culture plate inserts (Millipore, Carrigtwohill, Ireland) and treated with thapsigargin (Tg; Sigma, St Louis, MO, USA), palmitic acid (PA; Sigma) or oleic acid (OA; Sigma) as described [16]. To measure cell death, we determined oligonucleosomes released into the culture supernatant fraction by ELISA using a commercial kit (Roche, Mannheim, Germany) according to the manufacturer’s instructions. _N_-Acetylcysteine (NAC; Sigma) or Ebselen (Sigma) were used as antioxidants in cell death assays.
Western blotting
Western blotting was performed using antibodies specific for GFP (Santa Cruz Biotechnology, Santa Cruz, CA, USA), LC3 (Novus, Littleton, CO, USA), p62 (Abnova, Jhongli, Taiwan), phospho-eukaryotic translation initiation factor, subunit 1 alpha (p-EIF2α; Cell Signaling, Danvers, MA, USA), C/EPB homologous protein 10 (CHOP; Santa Cruz), heat shock protein 5 (BIP; Stressgen, Antwerpen, Belgium) or β-actin (Santa Cruz) as described previously [17].
Electron microscopy
Ultrastructural changes in cells at the organelle level were examined using electron microscopy (EM; JEM-1010; Jeol, Peabody, MA, USA) as described [10]. Briefly, the pancreas was fixed in 2.5% glutaraldehyde/0.1 mol/l NaH2PO4/Na2HPO4 buffer (pH 7.2). Tissue was then post-fixed with 1% OsO4 and embedded in Epon812. Ultrathin sections were mounted and stained with uranyl acetate/lead citrate. The number of autophagosomes was counted by scanning randomly at ×10,000–50,000 magnification [10].
Proteolysis
Degradation of long-lived proteins was determined as described [2] with modifications. Briefly, cells were plated on collagen-coated plates for 24 h. Cells were then labelled with 18.5 kBq/ml [14C]leucine (Perkin Elmer, Waltham, MA, USA) for 24 h. After culture for 2 h in a chase medium to allow degradation of short-lived proteins, cells were incubated with serum-free medium containing test reagents and 2 mmol/l unlabelled leucine at 37°C for 3–6 h in the presence or absence of 10 μg/ml each of E64d/pepstatin A and 20 mmol/l NH4Cl. Aliquots of the medium were precipitated with trichloroacetic acid, and proteolysis was measured as the percentage of released radioactivity relative to the initial radioactivity.
Statistical analysis
All values were expressed as means±SD from three to four independent experiments performed in triplicate. Two-tailed Student’s t test was used to compare the values between two groups. One-way ANOVA with Tukey’s test was used to compare values between multiple groups. Repeated-measures ANOVA with Bonferroni correction was used to compare multiple inter-related measurements between groups. A p value of less than 0.05 was considered significant.
Results
Compromised UPR machinery in autophagy-deficient beta cells
Because we have observed abnormal ER distension in autophagy-deficient beta cells of RIP_-Cre_ +;Atg7 F/F mice (Atg7 ∆β-cell mice) [10], we first studied whether basal constitutive expression of genes related to UPR is altered in Atg7_-deficient beta cells. Contrary to our expectation that UPR gene expression would be upregulated as a manifestation of ER stress, the expression of diverse UPR-associated genes, such as Eif2α (also known as Eif2s1), Atf4, Chop (also known as Ddit3), Gadd34 (also known as Ppp1r15a), Ero1α (also known as Ero1l), Ero1β (also known as Ero1lb), Bip (also known as Hspa5), Grp94, Erp72 (also known as Pdia4), Xbp1, Erdj4 (also known as Dnajb9), Edem (also known as Edem1), Hrd1, Ubc7, Herpud1, Herpud2 and p58 IPK, was significantly downregulated in pancreatic beta cells from Atg7 ∆β-cell mice compared with littermate RIP_-Cre −;Atg7 F/F mice (Atg7 F/F mice; p < 0.05–0.001; Fig. 1a, b), suggesting that basal UPR is compromised in autophagy-deficient beta cells. Expression of genes related to protection against ER stress, such as Sod1, Sod2, Ho1, Gpx1, Gpx2, Ucp2, Ppargc1b and Pparg [13], was also significantly downregulated in autophagy-deficient beta cells (p < 0.05–0.001; Fig. 1c). Basal production of representative UPR proteins such as BIP or CHOP and phosphorylation of eIF2α also tended to be diminished in islets from Atg7 ∆β-cell mice compared with littermate Atg7 F/F mice (ESM Fig. 1), supporting the hypothesis that UPR machinery is compromised.
Fig. 1
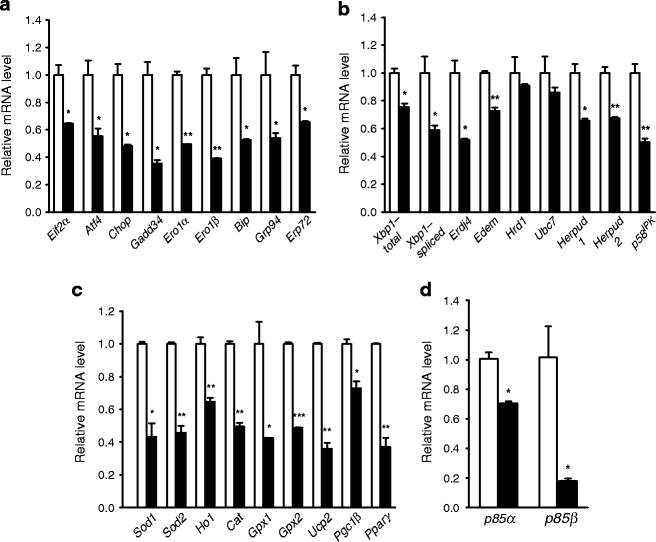
Expression of UPR-related genes in autophagy-deficient islets. Pancreatic islets were isolated from Atg7 ∆β-cell (black bar) and Atg7 F/F (white bar) mice as previously described [10]. Real-time RT-PCR was performed using primer sets specific for diverse genes of UPR (a,b), antioxidant responses (c) and p85α or p85β subunit of PI3K (d). Data are representative of four independent experiments. *p < 0.05, **p < 0.01, ***p < 0.001
Next, we studied the potential mechanism of the compromised expression of UPR machinery. We studied the production of non-catalytic regulatory subunits of PI3K because p85α and p85β regulatory subunits of PI3K have been reported to bind to X-box binding protein 1 (XBP-1) in an insulin-dependent manner and to be important in the basal and stimulated expression of UPR genes [18, 19]. In addition, production of p85α and p85β subunits of PI3K was decreased in the insulin-resistance state, and restoration of their production improved the deficient UPR in obese mice [18]. As hypothesised, mRNA expression of both p85α and p85β subunits was significantly reduced in islets of Atg7 ∆β-cell mice compared with Atg7 F/F mice (Fig. 1d; p < 0.05 for both subunits), suggesting that deficient production of regulatory subunits of PI3K may be responsible for the compromised UPR machinery in autophagy-deficient beta cells.
Susceptibility of autophagy-deficient beta cells to lipid injury in vitro
Considering a previous paper showing that appropriate UPR is important for survival in the presence of ER stress [20], compromised UPR expression in _Atg7_-deficient beta cells may impair their adaptation to ER stress. To address this issue, we treated primary islet cells from Atg7 ∆β-cell mice with Tg, a classic ER stressor, and assessed cell death by measuring oligonucleosome release. Death of primary pancreatic islet cells from Atg7 ∆β-cell mice caused by Tg was more pronounced than in control Atg7 F/F mice, probably because of insufficient UPR in response to ER stress (p < 0.05–0.001; Fig. 2a). Because Tg is a strong and unphysiological stressor that acts by chemically inhibiting sarcoplasmic/endoplasmic reticulum Ca2+-ATPase (SERCA) and dissipating ER Ca2+ [21], we next used lipid as a more physiological ER stressor. Lipids are relevant to the pathogenesis of diabetes because obesity and lipid injury induce ER stress by sustained regulation of SERCA activity through metabolism [22]. First, we studied whether UPR is activated by NEFAs in vitro. For this purpose, we treated primary islet cells with 600 μmol/l PA or 1,200 μmol/l OA for 24 h, which did not affect viability of islet cells from Atg7 F/F or Atg7 ∆β-cell mice (ESM Fig. 2). Treatment of primary islet cells from Atg7 F/F mice with PA or OA induced enhanced expression of the majority of UPR genes, suggesting increased demand for UPR, although the detailed expression patterns of individual genes were not identical between PA and OA (p < 0.05; Fig. 2c–f). We next examined whether UPR machinery in autophagy-deficient beta cells is able to meet the increased demand for UPR caused by NEFAs. Expression of UPR genes after treatment with PA or OA was lower in primary islet cells from Atg7 ∆β-cell mice compared with Atg7 F/F mice (p < 0.05–0.01; Fig. 2c–f), suggesting an unmet need for UPR in autophagy-deficient beta cells. Because these results indicated that lipid overload is a type of ER stress and that demand for UPR is increased by lipid, we studied the susceptibility of _Atg7_-deficient beta cells to lipid injury. Pancreatic islet cells from Atg7 ∆β-cell mice were more susceptible to treatment with PA for 48 h than wild-type beta cells at a high concentration of PA (p < 0.01; Fig. 2b), suggesting that autophagy deficiency renders beta cells more vulnerable to lipid injury causing ER stress, probably because of insufficient UPR.
Fig. 2
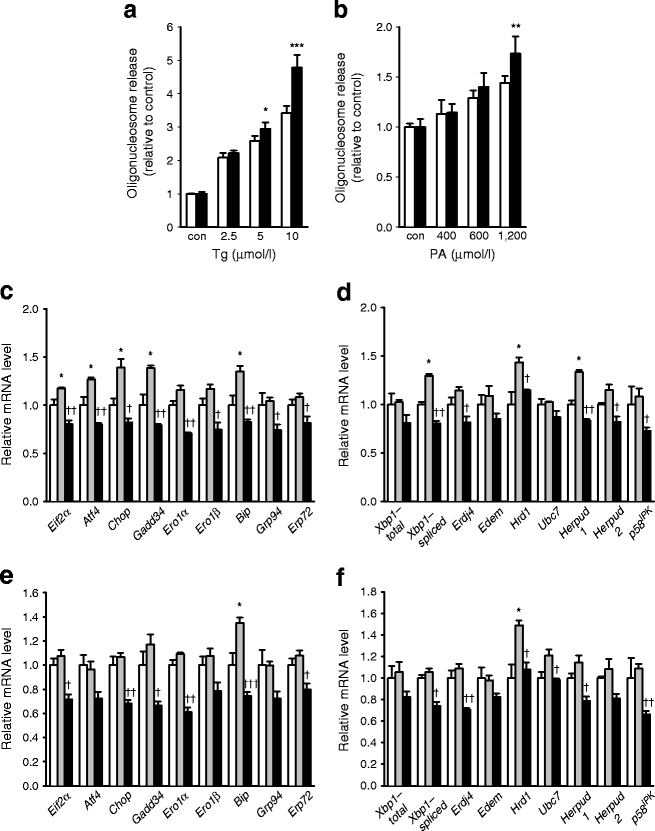
Susceptibility of autophagy-deficient islet cells to ER stressors. Primary pancreatic islets were treated with increasing concentrations of Tg for 24 h (a) or PA for 48 h (b), and cell death was determined as described in the Methods, and analysed by repeated-measures ANOVA with Bonferroni correction (con, control). Black bar, Atg7 ∆β-cell; white bar, Atg7 F/F. c–f Primary islet cells from Atg7 ∆β-cell and Atg7 F/F mice were treated with 600 μmol/l PA (c,d) or 1,200 μmol/l OA (e,f) for 24 h, and UPR gene induction was determined by real-time RT-PCR analysis as in Fig. 1a. c,d White bar, Atg7 F/F; grey bar, Atg7 F/F, PA; black bar, Atg7 ∆β-cell, PA. e,f White bar, Atg7 F/F; grey bar, Atg7 F/F, OA; black bar, Atg7 ∆β-cell, OA. Data are representative of four independent experiments. *p < 0.05, **p < 0.01, ***p < 0.001 vs Atg7 F/F; † p < 0.05, †† p < 0.01, ††† p < 0.001 vs Atg7 F/F, PA (c,d) or OA (e,f)
Susceptibility of autophagy-deficient beta cells to lipid injury in vivo
As these results suggested that autophagy deficiency of beta cells may lead to impaired adaptive response and increased susceptibility to ER stressors such as lipid, we next studied whether autophagy-deficient beta cells are vulnerable to ER stress in vivo. For this purpose, we bred RIP-Cre +;Atg7 F/W mice with ob/w mice to derive RIP-Cre +;Atg7 F/F-ob/ob (Atg7 ∆β-cell -ob/ob) mice. Pancreatic islets of Atg7 ∆β-cell-ob/ob mice had characteristic features of both mice with defective beta cell autophagy (frequent vacuolated cells sometimes containing insulin+ materials or cell debris in Atg7 ∆β-cell -ob/w mice) [10] and obese mice (hypertrophy, dilated capillaries and displacement of exocrine cells into islets in Atg7 F/F-ob/ob mice; Fig. 3a). Inclusion bodies containing p62 and ubiquitin were also observed in pancreatic islets from Atg7 ∆β-cell-ob/ob mice, similar to Atg7 ∆β-cell mice [10] (ESM Fig. 3). EM showed severe ER distension in beta cells from Atg7 ∆β-cell-ob/ob mice, while only minimal and moderate ER distension were observed in those from Atg7 F/F -ob/ob and Atg7 ∆β-cell-ob/w mice, respectively (Fig. 3b). Using these mice, we examined whether the relationship between lipid injury and UPR observed in vitro holds true in vivo. Expression of UPR-related genes was increased in pancreatic beta cells from Atg7 F/F -ob/ob mice compared with Atg7 F/F -ob/w mice (p < 0.05–0.001; Fig. 4a,b), which is in line with in vitro results (Fig. 2c–f) and suggests increased demand for UPR in obesity. Induction of UPR genes in beta cells from Atg7 ∆β-cell-ob/ob mice was significantly lower than in beta cells from Atg7 F/F-ob/ob mice for most UPR genes (p < 0.05–0.001; Fig. 4a,b), suggesting that UPR as an adaptive response to obesity is insufficient in autophagy-deficient beta cells.
Fig. 3
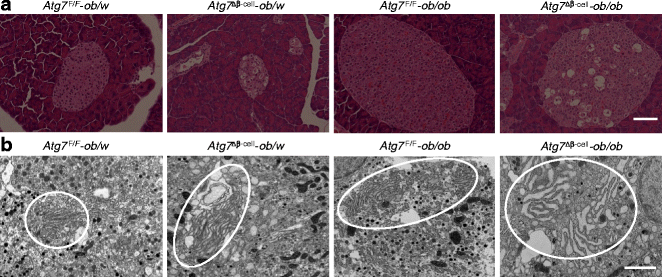
Islet morphology of obese mice with beta cell-specific autophagy deficiency. a Atg7 ∆β-cell-ob/ob mice were produced by breeding, and paraffin-embedded pancreatic sections from these mice and control mice were subjected to light microscopy after haematoxylin and eosin staining (scale bar, 100 μm). b EM was performed as described in the Methods. ER in the ellipses shows minimal, moderate and severe distension in beta cells from Atg7 F/F-ob/ob, Atg7 ∆β-cell-ob/w and Atg7 ∆β-cell-ob/ob mice, respectively, compared with normal ER from Atg7 F/F-ob/w mice (scale bar, 1 μm)
Fig. 4
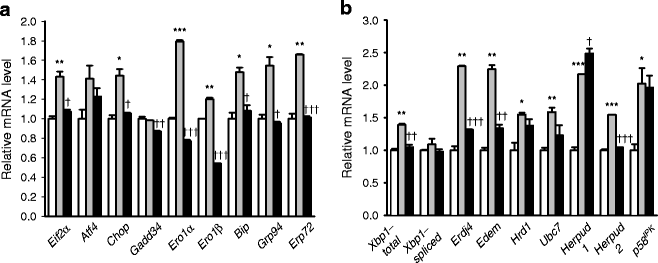
a, b Unmet need for UPR in autophagy-deficient beta cells of Atg7 ∆β-cell-ob/ob mice. Pancreatic islets were isolated from Atg7 F/F-ob/w (white bar), Atg7 F/F-ob/ob (grey bar) and Atg7 ∆β-cell-ob/ob (black bar) mice, and real-time RT-PCR was conducted as in Fig. 1a,b. Data are representative of four independent experiments. *p < 0.05, **p < 0.01, ***p < 0.001 vs Atg7 F/F-ob/w mice; † p < 0.05, †† p < 0.01, ††† p < 0.001 vs Atg7 F/F-ob/ob mice
Next, we studied whether the unmet need for UPR in Atg7 ∆β-cell-ob/ob mice leads to increased beta cell damage in vivo and aggravation of the glucose profile. Mice with obesity alone (Atg7 F/F-ob/ob) or with defective beta cell autophagy alone (Atg7 ∆β-cell-ob/w) developed hyperglycaemia but not diabetes, consistent with previous papers [10] (Fig. 5a). Strikingly, mice with both obesity and defective beta cell autophagy (Atg7 ∆β-cell-ob/ob mice) developed severe diabetes characterised by markedly elevated non-fasting blood glucose levels (p < 0.001 compared with the other three groups of mice; Fig. 5a). The fasting blood glucose level of _Atg7_ ∆β-cell-_ob/ob_ mice was also significantly higher than that of the other types of mice (_p_ < 0.001 compared with other groups; Fig. 5b). An intraperitoneal glucose tolerance test showed that _Atg7_ ∆β-cell-_ob/ob_ mice had markedly aggravated glucose intolerance compared with the other groups of mice (_p_ < 0.05–0.001; Fig. 5c). To examine the possible deleterious effect of RIP-_Cre_ transgene expression itself [23] on diabetes in _Atg7_ ∆β-cell-_ob/ob_ mice, we studied the glucose profile of our Rip-_Cre_ + mice. Fasting blood glucose level and glucose tolerance after i.p. glucose injection in Rip-_Cre_ + mice did not differ from RIP-_Cre_ − control littermates (_p_ > 0.1, p > 0.1; ESM Fig. 4), eliminating a possible contribution of RIP-Cre transgene expression to diabetes in Atg7 ∆β-cell-ob/ob mice.
Fig. 5
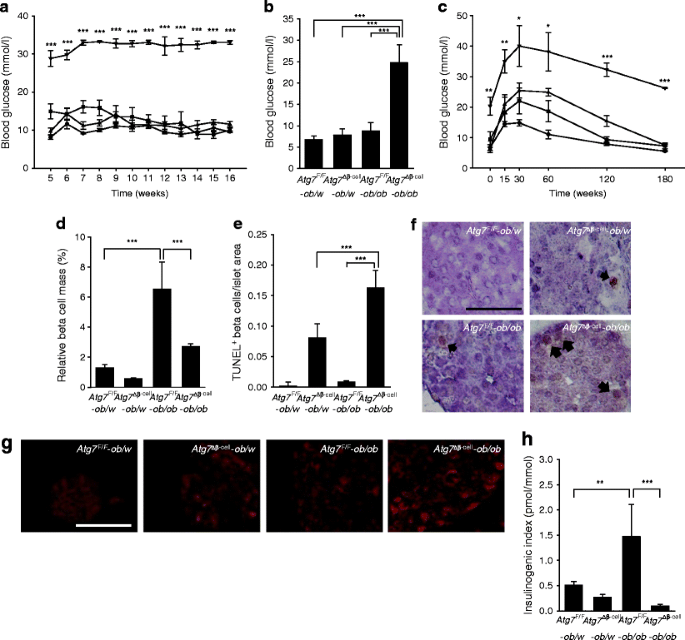
Blood glucose level and beta cells from Atg7 ∆β-cell-ob/ob mice. a Non-fasting blood glucose levels were monitored in Atg7 F/F-ob/w (n = 7; circle), Atg7 ∆β-cell-ob/w (n = 6; triangle), Atg7 F/F-ob/ob (n = 5; square) and Atg7 ∆β-cell-ob/ob mice (n = 4; upside down triangle). b Fasting blood glucose levels were determined in 14-week-old Atg7 F/F-ob/w (n = 3), Atg7 ∆β-cell-ob/w (n = 7), Atg7 F/F-ob/ob (n = 7) and Atg7 ∆β-cell-ob/ob mice (n = 9), and analysed by ANOVA with Tukey’s test. c An i.p. glucose tolerance test was performed as described in the Methods, and the results for Atg7 ∆β-cell-ob/ob mice were compared with those for the other types of mice (n = 4, each group). Beta cell mass (d) and apoptotic beta cell number (e) were determined as described in the Methods. f Representative TUNEL+ beta cells in each type of mouse (arrows). g Pancreatic sections were subjected to immunofluorescent staining using antibody to nitrotyrosine. h Serum insulin levels before (0 min) and 15 min after i.p. glucose challenge were determined by ELISA, and the insulinogenic index was calculated (n = 4, each group). Scale bars, 50 μm. *p < 0.05, **p < 0.01, ***p < 0.001
To investigate the mechanism of severe diabetes in Atg7 ∆β-cell-ob/ob mice, we measured beta cell mass. The beta cell mass of Atg7 F/F-ob/ob mice was increased compared with that of Atg7 F/F -ob/w mice (p < 0.001), as expected (Fig. 5d), and the beta cell mass of _Atg7_ ∆β-cell-_ob/ob_ mice was significantly lower than that of _Atg7_ F/F-_ob/ob_ mice (_p_ < 0.001), indicating that decreased beta cell mass is responsible for the severe diabetes seen in _Atg7_ ∆β-cell-_ob/ob_ mice (Fig. 5d). To determine the cause of the decreased beta cell mass in _Atg7_ ∆β-cell-_ob/ob_ mice, we conducted TUNEL staining combined with insulin immunohistochemistry. The number of apoptotic beta cells was significantly increased in islets from _Atg7_ ∆β-cell-_ob/ob_ mice compared with _Atg7_ ∆β-cell-_ob/w_ or _Atg7_ F/F-_ob/ob_ mice (_p_ < 0.001 for both comparisons; Fig. 5e,f), showing increased susceptibility of autophagy-deficient beta cells to ER stress in vivo. To investigate further the mechanism of increased death of autophagy-deficient beta cells in obese mice, we studied possible changes in reactive oxygen species (ROS) level. Dysfunctional mitochondria or ER in autophagy-deficient cells may produce more ROS and, further, may not be able to cope with increased stress such as metabolic load [24]. Nitrotyrosine staining showed increased accumulation of peroxynitrite-modified tyrosine residues, reflecting ROS damage to proteins [13] in beta cells from _Atg7_ ∆β-cell-_ob/ob_ mice compared with _Atg7_ F/F-_ob/ob_ or _Atg7_ ∆β-cell-_ob/w_ mice (Fig. 5g). However, the increase in death of primary islet cells from _Atg7_ ∆β-cell mice induced by PA was not significantly inhibited by pretreatment with antioxidants such as NAC or Ebselen (_p_ > 0.1, p > 0.1; ESM Fig. 5), suggesting that factors other than ROS may also contribute to the increased susceptibility of autophagy-deficient beta cells to lipid injury. We next studied the functional significance of the decreased beta cell mass in Atg7 ∆β-cell-ob/ob mice. The insulinogenic index, representing the functional beta cell response to change in blood glucose level, was increased in Atg7 F/F-ob/ob mice compared with Atg7 F/F-ob/w mice (p < 0.01). However, it was remarkably suppressed in Atg7 ∆β-cell-ob/ob mice compared with Atg7 F/F-ob/ob mice (p < 0.001; Fig. 5h), showing a severe functional defect in the beta cells causing diabetes in Atg7 ∆β-cell-ob/ob mice.
Effect of lipid on autophagy
Because these results suggested that autophagy is a protective mechanism against the ER stress induced by obesity, and its deficiency leads to compromised UPR in response to ER stress, we wondered whether autophagy level or activity is changed by lipid injury. To determine changes in autophagy level with obesity in vivo, we bred ob/w mice with GFP-LC3 + mice, which express GFP fused to LC3 globally. In these mice, GFP puncta represent autophagosomes, and thus direct visualisation of autophagy level is possible in vivo [12]. The number of GFP puncta, reflecting autophagy level, appeared to be increased in pancreatic islets from GFP-LC3 +-ob/ob mice compared with GFP-LC3 +-ob/w mice (Fig. 6a), suggesting that autophagy level is increased by obesity in vivo. Autophagosome number determined by EM was also significantly increased in pancreatic islet cells from ob/ob mice compared with ob/w mice (p < 0.05; Fig. 6b,c). As the number of LC3 puncta and autophagosomes was increased by blockade of the autophagic process at the lysosomal step rather than increasing autophagic activity, we studied cleavage of GFP in GFP-LC3 + mice, which reflects autophagic degradation or cleavage of substrates in lysosomes and thus autophagic activity [17]. Indeed, cleaved GFP was observed in islets of GFP-LC3 +-ob/ob mice and not in those of GFP-LC3 +-ob/w mice (Fig. 6d), suggesting that the increased number of autophagosomes in the islets of GFP-LC3 +-ob/ob mice is due to increased autophagic activity. An increase in LC3-II level reflected by increases in both GFP-LC3-II and endogenous LC3-II was also observed in islets from GFP-LC3 +-ob/ob mice (Fig. 6d), supporting the finding of increased autophagic activity in islets of ob/ob mice. However, the p62 level in islets from GFP-LC3 +-ob/ob mice was elevated compared with GFP-LC3 +-ob/w mice, which appears to be inconsistent with increased autophagic activity [25]. To resolve this inconsistency, we performed a proteolysis assay which represents the eventual degradation of autophagic protein substrates [25]. Lysosomal degradation of long-lived proteins determined by measuring release of incorporated [14C]leucine was significantly inhibited by treatment of NIT-1 insulinoma cells with PA or OA (p < 0.01–0.001; Fig. 6e–g). These results suggest that, while lipids appear to increase autophagic activity to eliminate lipids through ‘lipophagy’ [26], lipid overload decreases proteolysis, probably due to shunting or sequestration of the autophagic process and machinery toward lipophagy.
Fig. 6
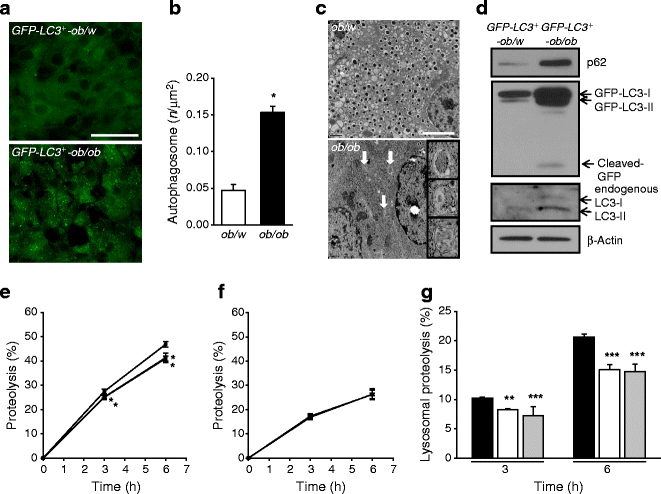
Increased autophagy level and autophagic activity in islets of ob/ob mice. a GFP-LC3 +-ob/ob mice were bred as described in the Methods, and GFP puncta in pancreatic islets of fed GFP-LC3 +-ob/ob and GFP-LC3 +-ob/w mice were visualised by fluorescent microscopy (scale bar, 50 μm). b The number of autophagosomes in islets of ob/ob and control ob/w mice was counted by EM (c). Arrows indicate representative autophagosomes (scale bar, 1 μm). d Pancreatic islets were isolated from GFP-LC3 +-ob/ob and GFP-LC3 +-ob/w mice, and western blotting was conducted to examine conversion of LC3-I into LC3-II, GFP cleavage and p62 accumulation using specific antibodies. e–f After labelling of NIT-1 cells with [14C]leucine and 2 h incubation to allow degradation of short-lived proteins, cells were treated with 300 μmol/l PA or OA for 3–6 h. The percentage release of radioactivity was measured with (f) or without (e) E64d/pepstatin A/NH4Cl. Lysosomal proteolysis (g) was calculated by subtracting values in f from those in e, and analysed by repeated-measures ANOVA with Bonferroni correction. KRB, Krebs–Ringer bicarbonate buffer. Black bar, control; white bar, PA; grey bar, OA. Data are representative of three independent experiments. *p < 0.05, **p < 0.01, ***p < 0.001
Discussion
In view of the close relationship between the ER and autophagic machinery [3, 4], it is not surprising that UPR and autophagy are inter-related. Our data show that autophagy is necessary for UPR constitutively and also in response to ER stress in pancreatic beta cells. Probably because of insufficient UPR, autophagy-deficient beta cells were found to be susceptible to ER stressors in vitro and in vivo, which is consistent with previous reports suggesting that UPR is not only a marker of, but also an adaptation to, ER stress, and that insufficient UPR in the presence of ER stress renders cells prone to cell death [20]. Our data from Atg7 ∆β-cell mice are different from previous studies showing increased UPR in autophagy-deficient tissue [27, 28], which may be attributable to peculiarities of pancreatic beta cells with respect to ER stress [29].
In our experiment to elucidate the mechanism of compromised expression of UPR genes, we observed reduced production of p85α and p85β non-catalytic subunits of PI3K, which would lead to deficient accumulation of nuclear XBP-1 and reduced expression of UPR genes [18, 19]. While direct targets of p85α and p85β subunits are XBP-1 downstream genes, previous papers have reported that the expression of other UPR genes such as Chop or genes of the ATF4 and ATF6 pathways is also affected by p85α or p85β deficiency [19]. Such results suggest that different pathways of UPR influence and interact with each other through negative and positive feedback regulation, and explain our finding that diverse UPR genes not restricted to XBP-1 pathways were downregulated in autophagy-deficient beta cells. The mechanism of reduced production of PI3K p85α and β subunits is not known. In ob/ob mice, the production of PI3K p85α and -β subunits is reduced [18], which may be due to insulin resistance and chronic insufficiency of insulin action on PI3K. Because insulin release from autophagy-deficient beta cells is lower than from wild-type beta cells [10], chronic insufficiency of ambient insulin around islet cells from Atg7 Δβ-cell mice may lead to reduced production of p85α and β subunits.
Because of insufficient UPR machinery, autophagy-deficient beta cells could not adapt to an environment in which increased demand for UPR exists such as obesity, which led to increased beta cell apoptosis, decreased beta cell mass, decreased insulin secretion, and diabetes in Atg7 ∆β-cell-ob/ob mice. Diabetes in Atg7 ∆β-cell-ob/ob mice is not related to a previously reported disturbance in glucose profile by RIP-Cre expression itself [23] because the glucose profile and glucose tolerance were normal in our RIP-Cre + colony, as previously reported [30]. Our data are consistent with a previous paper showing a paucity of compensatory beta cell hyperplasia in beta cell-specific autophagy-deficient mice fed a high-fat diet due to increased death and impaired proliferation [31], while diabetes was not observed in these mice. Consistent with our in vivo data showing an increase in death of beta cells in Atg7 ∆β-cell-ob/ob mice, autophagy-deficient beta cells were also more susceptible to PA in vitro. These data are in line with a recent report showing increased susceptibility of insulinoma cells to ER stressors after knockdown of ubiquitin fold modifier 1 (Ufm1), the expression of which is induced by ER stress [32]. However, an increased death rate was observed only at high concentrations of PA. The long-term effect of increased NEFA concentration in vivo may be difficult to recapitulate in vitro in a relatively short time unless high concentrations of NEFAs are used.
Increased death of autophagy-deficient beta cells in Atg7 ∆β-cell-ob/ob mice may be partly due to increased ROS production as shown by increased nitrotyrosine staining. Our results are consistent with recent reports showing increased ROS damage in beta cells with genetic deficiency of UPR [13] or autophagy-deficient kidney cells [33]. However, NAC and Ebselen both failed to suppress the increased death rate of autophagy-deficient beta islet cells after PA treatment, suggesting a possible contribution from other pathways.
Previous papers have reported a role for autophagy in the regulation of intracellular lipids in a process called ‘lipophagy’ [26]. However, no unanimous consensus has been reached about the effect of lipid on autophagy level or autophagic activity [26, 28, 31, 34, 35]. Such inconsistencies may be due to dissimilar assay methods or difference in the cell or tissue types used. Differentiation of autophagy level and autophagic activity is technically difficult, particularly in vivo, which may lead to invalid conclusions [25]. Here, we used obese GFP-LC3 + mice, which should provide the most reliable results regarding in vivo autophagic activity [17, 25]. In pancreatic islets of GFP-LC3 +-ob/ob mice, we observed not only increased LC3 puncta but also cleavage of GFP. These results demonstrate that the lysosomal steps of the autophagic process had occurred and autophagic activity was indeed increased in pancreatic islets of ob/ob mice. We have also observed increased GFP cleavage in the liver and muscle of GFP-LC3 +-ob/ob mice (Y. Lim, K.Y. Hur, W. Quan and M.-S. Lee, unpublished results). Therefore our data show that both autophagy level and autophagic activity are increased by lipid or obesity in multiple tissues in vivo. While GFP cleavage liberating free GFP molecules has been regarded as some of the most convincing evidence of autophagic activity, a recent paper reported that the amount of free GFP fragments can be affected by lysosomal acidity and may not reflect autophagic activity in certain conditions [36].
While we showed increased autophagic activity caused by lipid injury, autophagic protein degradation or proteolysis of long-lived proteins was reduced by lipid, consistent with a previous paper [34]. p62 level may be increased by lipid injury despite increased autophagic flux because the level of p62, a substrate of selective autophagy, may reflect proteolysis rather than apparent autophagic activity. In contrast, LC3 binding to lipid droplets and conversion may occur without p62 [26, 35]. Further studies will be necessary to elucidate the mechanism and significance of the changes in autophagy associated with obesity and lipid injury.
In summary, we found that beta cell autophagy is necessary for appropriate UPR machinery constitutively and in response to ER stress such as lipid injury. Our finding that overt diabetes developed in Atg7 ∆β-cell-ob/ob mice, which was not observed in Atg7 ∆β-cell mice or ob/ob mice, suggests the possibility that autophagy deficiency in pancreatic beta cells due to genetic predilection or other causes such as aging may be a factor in the progression from obesity to diabetes.
References
- Klionsky DJ, Emr SD (2000) Autophagy as a regulated pathway of cellular degradation. Science 290:1717–1721
Article PubMed CAS Google Scholar - Komatsu M, Waguri S, Ueno T et al (2005) Impairment of starvation-induced and constitutive autophagy in Atg7-deficient mice. J Cell Biol 169:425–434
Article PubMed CAS Google Scholar - Matsunaga K, Morita E, Saitoh T et al (2009) Autophagy requires endoplasmic reticulum targeting of the PI3-kinase complex via Atg14L. J Cell Biol 190:511–521
Article Google Scholar - Rubinsztein DC (2006) The roles of intracellular protein-degradation pathways in neurodegeneration. Nature 443:780–786
Article PubMed CAS Google Scholar - Kouroku Y, Fujita E, Tanida I et al (2007) ER stress (PERK/eIF2alpha phosphorylation) mediates the polyglutamine-induced LC3 conversion, an essential step for autophagy formation. Cell Death Differ 14:230–239
Article PubMed CAS Google Scholar - Ogata M, S-i H, Saito A et al (2006) Autophagy is activated for cell survival after endoplasmic reticulum stress. Mol Cell Biol 26:9220–9231
Article PubMed CAS Google Scholar - Yorimitsu T, Nair U, Yang Z, Klionsky DJ (2006) Endoplasmic reticulum stress triggers autophagy. J Biol Chem 281:30299–30304
Article PubMed CAS Google Scholar - Scheuner D, Vander Mierde D, Song B et al (2005) Control of mRNA translation preserves endoplasmic reticulum function in beta cells and maintains glucose homeostasis. Nat Med 11:757–764
Article PubMed CAS Google Scholar - Laybutt DR, Preston AM, Akerfeldt MC et al (2007) Endoplasmic reticulum stress contributes to beta cell apoptosis in type 2 diabetes. Diabetologia 50:752–763
Article PubMed CAS Google Scholar - Jung HS, Chung KW, Kim JW et al (2008) Loss of autophagy diminishes pancreatic β-cell mass and function with resultant hyperglycemia. Cell Metab 8:318–324
Article PubMed CAS Google Scholar - Masini M, Bugliani M, Lupi R et al (2009) Autophagy in human type 2 diabetes pancreatic beta cells. Diabetologia 52:1083–1086
Article PubMed CAS Google Scholar - Mizushima N, Yamamoto A, Matsui M, Yoshimori T, Ohsumi Y (2004) In vivo analysis of autophagy in response to nutrient starvation using transgenic mice expressing a fluorescent autophagosome marker. Mol Biol Cell 15:1101–1111
Article PubMed CAS Google Scholar - Back SH, Scheuner D, Han J et al (2009) Translation attenuation through eIF2alpha phosphorylation prevents oxidative stress and maintains the differentiated state in beta cells. Cell Metab 10:13–26
Article PubMed CAS Google Scholar - Kim YH, Kim S, Kim KA et al (1999) Apoptosis of pancreatic beta-cells detected in accelerated diabetes of NOD mice: no role of Fas-Fas ligand interaction in autoimmune diabetes. Eur J Immunol 29:455–465
Article PubMed CAS Google Scholar - Chang I, Cho N, Kim S et al (2004) Role of calcium in pancreatic islet cell death by IFN-gamma/TNF-alpha. J Immunol 172:7008–7014
PubMed CAS Google Scholar - Han MS, Park SY, Shinzawa K et al (2008) Lysophosphatidylcholine as a death effector in lipoapoptosis of hepatocytes. J Lipid Res 49:84–97
Article PubMed CAS Google Scholar - Hosokawa N, Hara Y, Mizushima N (2006) Generation of cell lines with tetracycline-regulated autophagy and a role for autophagy in controlling cell size. FEBS Lett 580:2623–2629
Article PubMed CAS Google Scholar - Park SW, Zhou Y, Lee J et al (2010) The regulatory subunits of PI3K, p85α and p85β, interact with XBP-1 and increase its nuclear translocation. Nat Med 16:429–437
Article PubMed CAS Google Scholar - Winnay JN, Boucher J, Mori MA, Ueki K, Kahn CR (2010) A regulatory subunit of phosphoinositide 3-kinase increases the nuclear accumulation of X-box-binding protein-1 to modulate the unfolded protein response. Nat Med 16:438–445
Article PubMed CAS Google Scholar - Merksamer PI, Trusina A, Papa FR (2008) Real-time redox measurements during endoplasmic reticulum stress reveal interlinked protein folding functions. Cell 135:933–947
Article PubMed CAS Google Scholar - Li WW, Alexander SA, Cao X, Lee AS (1993) Transactivation of the grp78 promoter by Ca2+ depletion. J Biol Chem 268:12003–12009
PubMed CAS Google Scholar - Cunha DA, Hekerman P, Ladrière L et al (2008) Initiation and execution of lipotoxic ER stress in pancreatic beta-cells. J Cell Sci 121:2308–2318
Article PubMed CAS Google Scholar - Lee JY, Ristow M, Lin X, White MF, Magnuson MA, Hennighausen L (2006) RIP-Cre revisited, evidence for impairments of pancreatic beta-cell function. J Biol Chem 281:2649–2653
Article PubMed CAS Google Scholar - Nakahira K, Haspel JA, Rathinam VAK et al (2011) Autophagy proteins regulate innate immune responses by inhibiting the release of mitochondrial DNA mediated by the NALP3 inflammasome. Nat Immunol 12:222–231
Article PubMed CAS Google Scholar - Klionsky DJ, Abeliovich H, Agostinis P et al (2008) Guidelines for the use and interpretation of assays for monitoring autophagy in higher eukaryotes. Autophagy 4:151–175
PubMed CAS Google Scholar - Singh R, Kaushik S, Wang Y et al (2009) Autophagy regulates lipid metabolism. Nature 458:1131–1135
Article PubMed CAS Google Scholar - Nakai A, Yamaguchi O, Takeda T et al (2007) The role of autophagy in cardiomyocytes in the basal state and in response to hemodynamic stress. Nat Med 13:619–624
Article PubMed CAS Google Scholar - Yang L, Li P, Fu S, Calay ES, Hotamisligil GS (2010) Defective hepatic autophagy in obesity promotes ER stress and causes insulin resistance. Cell Metab 11:467–478
Article PubMed CAS Google Scholar - Gomez E, Powell ML, Bevington A, Herbert TP (2008) A decrease in cellular energy status stimulates PERK-dependent eIF2alpha phosphorylation and regulates protein synthesis in pancreatic beta-cells. Biochem J 410:485–493
Article PubMed CAS Google Scholar - Kim S, Millet I, Kim HS et al (2007) NF-kappa B prevents beta cell death and autoimmune diabetes in NOD mice. Proc Natl Acad Sci USA 104:1913–1918
Article PubMed CAS Google Scholar - Ebato C, Uchida T, Arakawa M et al (2008) Autophagy is important in islet homeostasis and compensatory increase of beta cell mass in response to high-fat diet. Cell Metab 8:325–332
Article PubMed CAS Google Scholar - Lemaire K, Moura RF, Granvik M et al (2011) Ubiquitin fold modifier 1 (UFM1) and its target UFBP1 protect pancreatic beta cells from ER stress-induced apoptosis. PLoS One 6:e18517
Article PubMed CAS Google Scholar - Mathew R, Karp CM, Beaudoin B et al (2009) Autophagy suppresses tumorigenesis through elimination of p62. Cell 137:1062–1075
Article PubMed CAS Google Scholar - Koga H, Kaushik S, Cuervo AM (2010) Altered lipid content inhibits autophagic vesicular fusion. FASEB J 24:3052–3065
Article PubMed CAS Google Scholar - Shibata M, Yoshimura K, Tamura H et al (2010) LC3, a microtubule-associated protein1A/B light chain3, is involved in cytoplasmic lipid droplet formation. Biochem Biophys Res Commun 393:274–279
Article PubMed CAS Google Scholar - Ni HM, Bockus A, Wozniak AL et al (2011) Dissecting the dynamic turnover of GFP-LC3 in the autolysosome. Autophagy 7:1–17
Article Google Scholar