The effect of renal hyperfiltration on urinary inflammatory cytokines/chemokines in patients with uncomplicated type 1 diabetes mellitus (original) (raw)
Abstract
Aims/hypothesis
High intraglomerular pressure causes renal inflammation in experimental models of diabetes. Our objective was to determine whether renal hyperfiltration, a surrogate for intraglomerular hypertension, is associated with increased excretion of urinary cytokines/chemokines in patients with type 1 diabetes mellitus.
Methods
Blood pressure, renal haemodynamic function (inulin and _para_-aminohippurate clearances for glomerular filtration rate (GFR) and effective renal plasma flow (ERPF), respectively) and urine samples were obtained during clamped euglycaemia in individuals with type 1 diabetes with either hyperfiltration (GFR determined using inulin [GFRINULIN] ≥135 ml min−1 1.73 m−2, n = 28) or normofiltration (n = 21) and healthy control individuals (n = 18).
Results
Baseline clinical characteristics, dietary sodium and protein intake and blood pressure levels were similar in the diabetic and healthy control groups. In addition, HbA1c levels were similar in the two diabetic groups. As expected baseline GFR was higher in hyperfilterers than either normofiltering diabetic patients or healthy control patients (165 ± 9 vs 113 ± 2 and 116 ± 4 ml min−1 1.73 m−2, respectively, p < 0.01). ERPF and renal blood flow were also comparatively higher and renal vascular resistance was lower in hyperfiltering patients (p < 0.01). Hyperfiltering diabetic patients had higher excretion rates for eotaxin, IFNα2, macrophage-derived chemokine, platelet-derived growth factor (PDGF)-AA, PDGF-AB/BB and granulocyte-macrophage colony-stimulating factor (p ≤ 0.01). Urinary monocyte chemoattractant protein (MCP)-1 and RANTES (regulated on activation, normal T expressed and secreted) excretion was also higher in hyperfiltering vs normofiltering diabetic individuals (p < 0.01) and fibroblast growth factor-2, MCP-3 and CD40K excretion was elevated in hyperfiltering diabetic individuals vs healthy controls (p < 0.01).
Conclusions/interpretation
Renal hyperfiltration is associated with increased urinary excretion of inflammatory cytokines/chemokines in patients with uncomplicated type 1 diabetes.
Similar content being viewed by others
Introduction
Inflammation plays a critical role in the pathogenesis of early diabetic nephropathy. In experimental models, this is in part a result of hyperglycaemia-induced neurohormonal activation, which increases intraglomerular pressure and shear stress, leading to renal inflammation [1–3]. In in vitro studies of human mesangial cells, hyperglycaemia increases the mRNA expression of monocyte chemoattractant protein (MCP-1, also known as CCL2) and vascular endothelial growth factor, which have been linked to cell apoptosis, compensatory hypertrophy and hyperfiltration in animal models of diabetes mellitus [4, 5]. In patients with type 1 diabetes, we have focused on the effect of acute hyperglycaemia on the urinary excretion of cytokines/chemokines rather than tissue expression of these factors, hypothesising that urinary excretion of inflammatory mediators correlates with tissue expression. Similar to observations made in animal studies, we demonstrated that acute hyperglycaemia influences urinary cytokine/chemokine excretion, suggesting that the link between intraglomerular pressure, inflammation and renal injury also exists in humans [1–3, 6].
From a more clinical perspective, we have demonstrated that increased urinary cytokine/chemokine excretion precedes the onset of early clinical manifestations of nephropathy, including microalbuminuria, in young patients with type 1 diabetes [7]. In patients with type 2 diabetes and more advanced disease, urinary cytokine/chemokine excretion correlates with the severity of proteinuria and in patients with type 1 diabetes, these factors correlate with declining renal function [8]. The factors that influence urinary cytokine/chemokine excretion in patients at an even earlier stage of diabetes, before the onset of proteinuria or renal function decline, remain unclear.
Renal hyperfiltration is one of the earliest detectable abnormalities in humans and is present is approximately 40–50% of patients with uncomplicated type 1 diabetes [9]. Due to the potential role of elevated intraglomerular pressure in the pathogenesis of early renal injury in diabetes [10], the aim of this pilot study was to determine whether renal hyperfiltration, used as a surrogate marker for intraglomerular pressure, is associated with elevated urinary cytokine/chemokine excretion in patients with uncomplicated type 1 diabetes. We hypothesised that urinary cytokines/chemokines, which either increase in response to acute hyperglycaemia or precede the onset of microalbuminuria, would be elevated in patients with renal hyperfiltration, defined as a glomerular filtration rate determined using inulin (GFRINULIN) ≥135 ml min−1 1.73 m−2 [11, 12]. If urinary cytokine/chemokine excretion is elevated in high-risk groups, such as patients with hyperfiltration and in those with high-tertile normoalbuminuria [7], then the use of additional, pre-clinical biomarkers in natural history and therapeutic trials may help to clarify the role of earlier interventions in patients with type 1 diabetes.
Methods
Participants
Patients with type 1 diabetes and hyperfiltration (DM-H, n = 28) or normofiltration (DM-N, n = 21) and a matched group of healthy individuals as controls (C, n = 18) were recruited from endocrine clinics using local advertisements. Inclusion criteria were: duration of type 1 diabetes ≥5 years, age ≥18 years, blood pressure <140/90 mmHg (systolic/diastolic), no history of renal disease or macrovascular disease and participants could not be taking any regular medications other than insulin and had to be normoalbuminuric on a 24 h urine collection. Of the 49 participants with diabetes, 27 had taken part in other physiological studies within 3 years of the present set of experiments and all of these individuals were normoalbuminuric when their albumin excretion was quantified using a 24 h urine collection. Of the remaining 22 participants with diabetes, spot albumin-to-creatinine ratios were normal at their most recent clinic visits but previous 24 h urine collections were not indicated and therefore not performed as part of routine clinical care.
We aimed to study female participants during the early follicular phase of the menstrual cycle, determined by cycle day and measurement of 17β-oestradiol levels. None were using oral contraceptive medications. The local Research Ethics Board at the University Health Network (Toronto, Canada) approved the protocol and all participants gave informed consent.
Experimental design
To suppress the activity of the endogenous renin–angiotensin system and avoid the effect of high dietary protein on renal function, participants adhered to a high-sodium (>140 mmol/day) and moderate-protein (<1.5 g kg−1 day−1) diet during the 7-day period before renal haemodynamic testing and collection of the urine sample, as described previously (Table 1) [11]. In patients with diabetes, clamped euglycaemic (4–6 mmol/l) conditions were maintained for approximately 6 h preceding and during all investigations, a period of time previously demonstrated to be sufficient to influence vascular function [13]. In all phases of the experiment, blood glucose was maintained by a modified glucose clamp technique, as described previously [13]. In brief, a 16-gauge peripheral venous cannula was inserted into the left antecubital vein for infusion of glucose and insulin and a second cannula was inserted for blood sampling more distally. Blood glucose was measured every 5–10 min and the insulin infusion was adjusted to maintain euglycaemia. In healthy control individuals, studies were performed during normoglycaemic conditions. All experiments were performed in the same warm (25°C), temperature-controlled room and in a dark, quiet environment in the supine position.
Table 1 Baseline characteristics and biochemistry
Assessment of renal variables
Following maintenance of the euglycaemic clamp, a third intravenous line was inserted into the right arm and was connected to a syringe infusion pump for administration of inulin and _para_-aminohippurate (PAH). After collecting blood for inulin and blank, a priming infusion containing 25% inulin (60 mg/kg) and 20% PAH (8 mg/kg) was administered. Thereafter, inulin and PAH were infused continuously at a rate calculated to maintain their respective plasma concentrations constant at 2.0 and 0.15 mg/l. After a 90 min equilibration period, blood was collected for measurement of inulin, PAH and haematocrit. Blood was further collected every 30 min for 60 min for inulin and PAH; GFRINULIN and effective renal plasma flow (ERPF) were estimated by steady-state infusion of inulin and PAH, respectively [13].
Sample collection and analytical methods
Blood samples collected for inulin and PAH determinations were immediately centrifuged at 3,000 rpm for 10 min at 4°C. Plasma was separated, placed on ice and then stored at −70°C before the assay. Inulin and PAH were measured in serum by colorimetric assays using anthrone and _N_-(1-naphthyl)ethylenediamine, respectively [14–[16](/article/10.1007/s00125-013-2857-5#ref-CR16 "Florijn KW, Barendregt JN, Lentjes EG et al (1994) Glomerular filtration rate measurement by "single-shot" injection of inulin. Kidney Int 46:252–259")]. The mean of two baseline clearance periods represents GFRINULIN and ERPF, expressed per 1.73 m2. Renal blood flow (RBF) was derived using ERPF/(1 − haematocrit) and renal vascular resistance (RVR) was derived by dividing the mean arterial pressure by the RBF. All renal haemodynamic measurements were adjusted for body surface area [14, [16](/article/10.1007/s00125-013-2857-5#ref-CR16 "Florijn KW, Barendregt JN, Lentjes EG et al (1994) Glomerular filtration rate measurement by "single-shot" injection of inulin. Kidney Int 46:252–259")].
Upon arrival at the renal physiology laboratory, participants were asked to void and this final volume completed the 24 h urine collection. Participants then voided again mid-morning at approximately 10:00 hours to clear the bladder of urine that was produced while the euglycaemic clamp was being stabilised. The subsequent spot urine sample that was used for the analysis was collected before the first set of renal haemodynamic variables were measured. This 50 ml mid-stream sterile urine specimen was used to measure levels of cytokines/chemokines (see electronic supplementary materials [ESM] Table 1) using an established Cytokine/Chemokine Panel Luminex Assay (Eve Technologies, Calgary, AB, Canada) corrected for urine creatinine concentration [6, 7]. Immediately after collection, urine was centrifuged at 96 g for 15 min to remove cells, then separated into 1 ml portions and frozen at −80°C. One day before use, urine was thawed at 4°C. Due to the protocol for handling urine specimens, this analysis only included spot urine samples and not the timed collections that were obtained before the study visit. Furthermore, we only included analytes that either increase in response to hyperglycaemia or that are associated with the highest tertile of normoalbuminuria in our previous work [6, 7]. Our analysis therefore included eotaxin, fibroblast growth factor-2 (FGF-2), granulocyte–macrophage colony-stimulating factor (GM-CSF), IFNα2, IL-6, IL-12, monocyte chemoattractant protein-3 (MCP-3), macrophage-derived chemokine (MDC), macrophage inflammatory protein-1α (MIP-1α), TNFβ, sCD40 ligand (sCD40K), regulated on activation normal T cell expressed and secreted (RANTES), platelet-derived growth factor (PDGF)-AA, PDGF-AB/BB and monocyte chemoattractant protein-1 (MCP-1). We limited our analysis to these factors to maintain statistical power, to minimise false-positive results and to further elucidate mechanisms that may link high intraglomerular pressure with factors that may promote initiation of renal disease. The investigator performing data analysis was blinded to all study variables.
The accuracy and precision of the urinary cytokine/chemokine assay is available through the vendor at www.millipore.com/userguides/tech1/proto_mpxhcyto-60k (28 January 2013). The detection limits of our assays have been published previously [7].
Urinary albumin excretion rate was determined from a 24 h urine collection by immunoturbidimetry. HbA1c was measured by high-performance liquid chromatography [17]. Plasma cystatin C was also measured using previously described standard methods, and GFRCYSTATIN C was then calculated using the MacIssac formula [18, 19].
Statistical methods
Between-group differences were determined by repeated measures ANOVA, corrected for multiple comparisons, with p = 0.01 (SPSS v.14, Armonk, NY, USA). Our sample size calculation was based on differences in urinary MCP-1 excretion due to the consistent relationship between this cytokine and renal disease [20]. Our previous data have shown that the SD for MCP-1 is 17 units [7]. To have an 80% power to detect a significant 25-unit between-group difference in MCP-1 [21, 22], for a two-sided test with p = 0.01 and Z α = 2.58, the sample size should be ≥16 per group. Participants with diabetes were analysed on the basis of filtration status determined using inulin clearance as described above. Filtration status was determined at the end of the study once inulin assays were complete for the entire cohort. Between-group differences using GFRCYSTATIN C were not assessed because of the expected underestimation of GFRINULIN within the hyperfiltration range [18]. As a consequence, only 7/28 hyperfilterers by GFRINULIN could still be classified as hyperfilterers using GFRCYSTATIN C. Urinary cytokine/chemokine excretion was also correlated with GFRINULIN and GFRCYSTATIN C in the entire 49-member group of participants with diabetes using the Spearman correlation coefficient. The University Health Network (Toronto, Canada) Ethics Board approved the protocol and patients gave informed consent.
Results
Baseline characteristics
DM-H and DM-N participants were young, normotensive, normoalbuminuric men and women with suboptimal glycaemic control levels commonly observed in this age group [23] (Table 1). Participants with diabetes were otherwise similar to controls in terms of age, BMI, sex distribution, oestrogen levels and sodium and protein intake.
For baseline haemodynamic variables, as expected, baseline GFR was higher in DM-H than in DM-N and healthy control participants (p < 0.01). ERPF and RBF were also higher and RVR was lower in DM-H patients (p < 0.01). Renal haemodynamic variables were similar in the control and DM-N groups. Differences in blood pressure between the three groups were not significant.
Filtration status and urinary excretion of cytokines/chemokines
The urinary excretion of each cytokine/chemokine in each group is represented in Fig. 1,a–k. Compared with the DM-N and C groups, the DM-H group exhibited higher urinary eotaxin, IFNα2, MDC, PDGF-AA, PDGF-AB/BB and GM-CSF (Fig. 1, p < 0.01). Urinary MCP-1 and RANTES levels were higher in the DM-H group than in the DM-N group (_p_ < 0.01); between-group interactions for DM-H vs C were not significant. For MCP-3 and FGF-2, urinary excretion was elevated in the DM-H group compared with C (Fig. 1, _p_ < 0.01), and sCD40K excretion was higher in both diabetic groups compared with C (Fig. 1). Between-group differences in IL-6 excretion for control (0.18 ± 0.05 pmol/l) vs DM-N (0.28 ± 0.05) vs DM-H (0.38 ± 0.05) showed the same trend as the other factors but did not reach significance (_p_ = 0.09). Urinary IL-2, IL-12 and MIP1α excretion was not detectable in >50% of samples, and these factors were therefore excluded from the analysis.
Fig. 1
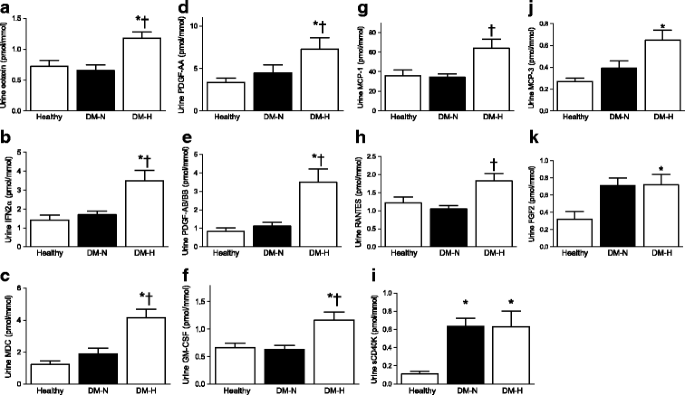
Urinary excretion of cytokines/chemokines in patients with type 1 diabetes mellitus. *p values for DM-H vs healthy controls: eotaxin p = 0.003; IFN p = 0.006; MDC p < 0.0001; FGF2 p = 0.007; PDGF-AA p = 0.007; PDGF-AB/BB p = 0.001; sCD40K p < 0.001; GM-CSF p < 0.0001; MCP-3 p = 0.001. †p values for DM-H vs DM-N: eotaxin p = 0.003; IFN2α p = 0.001; MDC p = 0.0001; PDGF-AA p = 0.01; PDGF-AB/BB p = 0.003; RANTES p = 0.001; GM-CSF p = 0.007 MCP-1 p = 0.007
In the group of 49 participants with diabetes, GFRINULIN correlated with urinary excretion levels of eotaxin (r = 0.467, p = 0.001), GM-CSF (r = 0.355, p = 0.013), MCP-1 (r = 0.327, p = 0.023), MCP-3 (r = 0.298, p = 0.04), MDC (r = 0.345, p = 0.016), PDGF-AB/BB (r = 0.296, p = 0.046), PDGF-AA (r = 0.369, p = 0.010), RANTES (r = 0.381, p = 0.008) and sCD40K (r = 0.372, p = 0.009). When urinary excretion levels of cytokines/chemokines were correlated with GFRCYSYATIN C in the diabetic cohort, similar interactions were observed between GFRCYSYATIN C and eotaxin, GM-CSF, MCP-1, MCP-3, MDC, PDGF-AA and sCD40K (ESM Table 2). In addition, GFRCYSYATIN C correlated with FGF2 and TNF (ESM Table 2). In contrast with GFRINULIN, the interactions between GFRCYSYATIN C and RANTES and PDGF-AB/BB were not significant and IL-6 did not correlate with GFRINULIN or GFRCYSYATIN C.
Discussion
Experimental data have suggested the association of high intraglomerular pressure with increased shear stress in diabetes, leading to renal injury [1–3]. The pathogenesis of hyperfiltration remains controversial; both haemodynamic (the ‘haemodynamic hypothesis’) and tubuloglomerular feedback mechanisms (the ‘tubular hypothesis’) have been implicated [24]. Animal studies of type 1 diabetes and translational physiology experiments in humans have associated hyperfiltration with lower levels of RVR [11–13, 25–27]. This is likely to be based on: (1) hyperglycaemia-induced increases in nitric oxide and vasodilatory prostanoid bioactivity in the pre-glomerular, afferent renal circulation [11, 13, 24]; (2) changes in tubuloglomerular feedback, due to increased sodium–glucose co-transport at the proximal tubule. Increased proximal tubular sodium reabsorption decreases distal delivery to the macula densa, resulting in afferent vasodilatation [28]. Since hyperfiltration, used as a surrogate for intraglomerular pressure in humans, also contributes to the initiation of diabetic nephropathy in some but not all studies [10, 29], we hypothesised that DM-H patients would also exhibit increased urinary excretion of inflammatory mediators that have been linked with kidney injury. We included a panel of analytes that either increase acutely in response to clamped hyperglycaemia or that are associated with the highest tertile of normoalbuminuria in young type 1 diabetes patients [6, 7].
In experimental models of diabetes, hyperfiltration is associated with increased renal tissue cytokine/chemokine mRNA expression (IFN, MCP-1) [4, 30, 31]. In animal models in which diabetes is induced by streptozotocin, factors that reduce glomerular hypertrophy and hyperfiltration, such as anti-angiogenic factors, also reduce renal mRNA expression of Tnf, Il6 and Mcp-1 [32]. Our major finding in this pilot study was that renal hyperfiltration is also associated increased urinary cytokine/chemokine excretion in humans with uncomplicated type 1 diabetes. To our knowledge, this is the first time that hyperfiltration, measured using gold-standard inulin clearance techniques, has been linked with renal inflammation in humans. Although we could not perform between-group comparisons based on filtration status using GFRCYSTATIN C, due to the small numbers of hyperfiltering patients defined by cystatin C criteria, GFRCYSTATIN C also correlated with urinary cytokine/chemokine excretion in participants with diabetes. Previous work in patients with type 1 diabetes has demonstrated that urinary inflammatory mediators correlate with GFR loss and with increased urinary albumin excretion, even when the albumin-to-creatinine ratio values are within the normal range [7, 33]. Since hyperfiltration is associated with diabetic nephropathy and occurs early in the natural history of diabetes before the onset of microalbuminuria, our results may identify a potentially high-risk group that can benefit from earlier renoprotective therapies. Our findings are important from a therapeutic perspective because urine cytokines/chemokines can be influenced by renoprotective medications such as ACE inhibitors in humans [33–38]. Urine cytokines/chemokines may therefore act as future therapeutic targets for novel or existing medications and can be used by clinicians to monitor treatment failure or success. The identification of new biomarkers of renal disease initiation and progression is critical due to the widely recognised limitations of albuminuria as a clinical outcome measure [39].
Previous work has linked diabetes with renal inflammation, possibly through effects of chronic hyperglycaemia on intraglomerular pressure [1–3]. We studied patients under clamped euglycaemic conditions and using a controlled dietary preparation to isolate the effect of filtration status without the influence of acute hyperglycaemia, renal dysfunction or albuminuria [6]. Under these conditions, urinary factors that are associated with either chemotaxis or inflammation/fibrosis were elevated in DM-H [40–42]. Our results suggest that high intraglomerular pressure may provide a mechanistic link between factors that cause hyperfiltration, such as hyperglycaemia-induced renin angiotensin system activation [43], and renal inflammation, leading to the onset of clinical disease.
Although we cannot determine from the present study whether urinary excretion of cytokines/chemokines was the result of renal tubular secretion or spill-over of systemic proteins, these preliminary observations may give mechanistic insights into disease pathogenesis in hyperfiltering humans at a pre-clinical stage of the disease. In light of the emerging role for cystatin C as a clinical indicator of hyperfiltration and the association between GFRCYSYATIN C and urinary cytokines/chemokines, our results suggest that these factors may be used to monitor the biological effects of therapeutic interventions in high-risk patients with type 1 diabetes in future clinical trials [19]. Unfortunately, cystatin C and other indirect GFR measurements, such as creatinine-based estimates, are not yet accurate or precise enough to study hyperfiltration in large cohorts. As described elsewhere, the large degree of bias associated with indirect GFR measurements, such as cystatin C, may account in part for inconsistencies in the literature surrounding the possible role of hyperfiltration in the progression of renal disease [19, 29]. Consistent with our previous observations suggesting that GFRCYSYATIN C underestimates GFRINULIN, only 7/28 DM-H by GFRINULIN were still classified as hyperfilterers using GFRCYSYATIN C, making between-group comparisons unreliable. Nevertheless, the identification of early ‘pre-clinical’ risk biomarkers, including urinary cytokines/chemokines, in a larger cohort is now more vital than ever given the failure of primary renal prevention strategies using conventional agents, such as renin angiotensin system inhibitors, in an important subset of patients with diabetes [44]. Furthermore, recent natural history studies of human diabetic nephropathy have demonstrated that older paradigms involving progression from normoalbuminuria to microalbuminuria to macroalbuminuria to impaired renal function and end-stage renal disease are not accurate in many patients [45]. For example, some patients have significant initial deterioration of renal function without proteinuria whereas, in others, microalbuminuria regresses spontaneously [46]. The elucidation of novel, non-albuminuria biomarkers is therefore important for the early identification and potential risk-stratification of patients with diabetes.
We attempted to minimise the effect of the small sample size in our study by using homogeneous study groups and by using a careful pre-study preparation phase with a focus on known factors that influence neurohormonal activation, including dietary sodium intake [47]. Nevertheless, the small sample size may have limited our ability to detect between-group differences in urinary IL-6 excretion. Furthermore, because we did not study participants under conditions of low dietary sodium intake, we were unable to determine the effect of dietary sodium intake on urine cytokines/chemokines. We also scheduled studies in female participants to coincide with the early follicular phase of the menstrual cycle to avoid confounding effects of oestrogen on vascular function. Although plasma oestrogen levels were higher than expected and more consistent with the mid-to-late follicular phase, oestrogen concentrations were similar in the three groups studied. In addition, we decreased variability by using a study design that allowed each participant to act as his/her own control. Finally, while it is presumed that hyperfiltration may have induced the increase in cytokines/chemokines and thus ultimate renal injury, it is also possible that other factors (i.e. neurohormonal activation) that increase urinary cytokine/chemokine excretion could also induce hyperfiltration. It is therefore important for future studies to clarify the time course and reversibility of urinary biomarkers using existing and investigational agents. Future work should also determine whether urinary cytokine/chemokine excretion in the DM-H group is systemically derived (i.e. ‘spill-over’) or due to renal production.
In conclusion, similar to previous findings in animal models of diabetes, renal hyperfiltration during clamped euglycaemia was associated with increased urinary excretion of cytokines/chemokines in patients with uncomplicated type 1 diabetes.
Abbreviations
DM-H:
Diabetes patient with hyperfiltration
DM-N:
Diabetes patient with normofiltration
ERPF:
Effective renal plasma flow
FGF-2:
Fibroblast growth factor-2
GFR:
Glomerular filtration rate
GFRCYSTATIN C :
GFR determined using cystatin C
GFRINULIN :
GFR determined using inulin
GM-CSF:
Granulocyte–macrophage colony-stimulating factor
MCP:
Monocyte chemoattractant protein
MDC:
Macrophage-derived chemokine
MIP-1α:
Macrophage inflammatory protein-1α
PAH:
_para_-Aminohippurate
PDGF:
Platelet-derived growth factor
RANTES:
Regulated on activation, normal T cell expressed and secreted
RBF:
Renal blood flow
RVR:
Renal vascular resistance
sCD40K:
sCD40 ligand
References
- Riser BL, Varani J, Cortes P, Yee J, Dame M, Sharba AK (2001) Cyclic stretching of mesangial cells up-regulates intercellular adhesion molecule-1 and leukocyte adherence: a possible new mechanism for glomerulosclerosis. Am J Pathol 158:11–17
Article PubMed CAS Google Scholar - Sayyed SG, Hagele H, Kulkarni OP et al (2009) Podocytes produce homeostatic chemokine stromal cell-derived factor-1/CXCL12, which contributes to glomerulosclerosis, podocyte loss and albuminuria in a mouse model of type 2 diabetes. Diabetologia 52:2445–2454
Article PubMed CAS Google Scholar - Sayyed SG, Ryu M, Kulkarni OP et al (2011) An orally active chemokine receptor CCR2 antagonist prevents glomerulosclerosis and renal failure in type 2 diabetes. Kidney Int 80:68–78
Article PubMed CAS Google Scholar - Yamagishi S, Inagaki Y, Okamoto T et al (2002) Advanced glycation end product-induced apoptosis and overexpression of vascular endothelial growth factor and monocyte chemoattractant protein-1 in human-cultured mesangial cells. J Biol Chem 277:20309–20315
Article PubMed CAS Google Scholar - Dworkin RH, Malone DC, Panarites CJ, Armstrong EP, Pham SV (2010) Impact of postherpetic neuralgia and painful diabetic peripheral neuropathy on health care costs. J Pain 11:360–368
Article PubMed Google Scholar - Cherney DZ, Scholey JW, Sochett E, Bradley TJ, Reich HN (2011) The acute effect of clamped hyperglycemia on the urinary excretion of inflammatory cytokines/chemokines in uncomplicated type 1 diabetes: a pilot study. Diabetes Care 34:177–180
Article PubMed CAS Google Scholar - Cherney DZI, Scholey JW, Daneman D et al (2012) Urinary markers of renal inflammation in adolescents with type 1 diabetes mellitus and normoalbuminuria. Diabet Med 29:1297–1302
Article PubMed CAS Google Scholar - Wolkow PP, Niewczas MA, Perkins B et al (2008) Association of urinary inflammatory markers and renal decline in microalbuminuric type 1 diabetics. J Am Soc Nephrol 19:789–797
Article PubMed CAS Google Scholar - Cherney DZ, Scholey JW, Miller JA (2008) Insights into the regulation of renal hemodynamic function in diabetic mellitus. Curr Diabetes Rev 4:280–290
Article PubMed CAS Google Scholar - Magee GM, Bilous RW, Cardwell CR, Hunter SJ, Kee F, Fogarty DG (2009) Is hyperfiltration associated with the future risk of developing diabetic nephropathy? A meta-analysis. Diabetologia 52:691–697
Article PubMed CAS Google Scholar - Cherney DZ, Reich HN, Jiang S et al (2012) Hyperfiltration and the effect of nitric oxide inhibition on renal and endothelial function in humans with uncomplicated type 1 diabetes mellitus. Am J Physiol Regul Integr Comp Physiol 303:R710–R718
Article PubMed CAS Google Scholar - Cherney DZ, Sochett EB (2011) Evolution of renal hyperfiltration and arterial stiffness from adolescence into early adulthood in type 1 diabetes. Diabetes Care 34:1821–1826
Article PubMed CAS Google Scholar - Cherney DZ, Miller JA, Scholey JW et al (2008) The effect of cyclooxygenase-2 inhibition on renal hemodynamic function in humans with type 1 diabetes. Diabetes 57:688–695
Article PubMed CAS Google Scholar - Davidson WD, Sackner MA (1963) Simplification of the anthrone method for the determination of inulin in clearance studies. J Lab Clin Med 62:351–356
PubMed CAS Google Scholar - Jung K, Klotzek S, Schulze BD (1990) Refinements of assays for low concentrations of inulin in serum. Nephron 54:360–361
Article PubMed CAS Google Scholar - Florijn KW, Barendregt JN, Lentjes EG et al (1994) Glomerular filtration rate measurement by "single-shot" injection of inulin. Kidney Int 46:252–259
Article PubMed CAS Google Scholar - Cherney DZ, Miller JA, Scholey JW et al (2010) Renal hyperfiltration is a determinant of endothelial function responses to cyclooxygenase 2 inhibition in type 1 diabetes. Diabetes Care 33:1344–1346
Article PubMed CAS Google Scholar - Perkins BA, Sochett EB, Cherney DZ (2012) Ability of cystatin C to detect changes in glomerular filtration rate after ACE inhibition in patients with uncomplicated type 1 diabetes. Clin Exp Hypertens 34:606–611
Article PubMed CAS Google Scholar - Cherney DZ, Sochett EB, Dekker MG, Perkins BA (2010) Ability of cystatin C to detect acute changes in glomerular filtration rate provoked by hyperglycaemia in uncomplicated type 1 diabetes. Diabet Med 27:1358–1365
Article PubMed CAS Google Scholar - Tesch GH (2008) MCP-1/CCL2: a new diagnostic marker and therapeutic target for progressive renal injury in diabetic nephropathy. Am J Physiol Renal Physiol 294:F697–F701
Article PubMed CAS Google Scholar - Takebayashi K, Suetsugu M, Matsumoto S, Aso Y, Inukai T (2009) Effects of rosuvastatin and colestimide on metabolic parameters and urinary monocyte chemoattractant protein-1 in type 2 diabetic patients with hyperlipidemia. South Med J 102:361–368
Article PubMed Google Scholar - Ye SD, Zheng M, Zhao LL et al (2009) Intensive insulin therapy decreases urinary MCP-1 and ICAM-1 excretions in incipient diabetic nephropathy. Eur J Clin Invest 39:980–985
Article PubMed CAS Google Scholar - Beck RW, Tamborlane WV, Bergenstal RM, Miller KM, Dubose SN, Hall CA (2012) The T1D Exchange Clinic Registry. J Clin Endocrinol Metab 97:4383–4389
Article PubMed CAS Google Scholar - Sasson AN, Cherney DZ (2012) Renal hyperfiltration related to diabetes mellitus and obesity in human disease. World J Diabetes 3:1–6
Article PubMed Google Scholar - Vallon V, Wead LM, Blantz RC (1995) Renal hemodynamics and plasma and kidney angiotensin II in established diabetes mellitus in rats: effect of sodium and salt restriction. J Am Soc Nephrol 5:1761–1767
PubMed CAS Google Scholar - Montanari A, Biggi A, Cabassi A et al (2012) Renal hemodynamic response to L-arginine in uncomplicated, type 1 diabetes mellitus: the role of buffering anions and tubuloglomerular feedback. Am J Physiol Renal Physiol 303:F648–F658
Article PubMed CAS Google Scholar - Bank N, Aynedjian HS (1993) Role of EDRF (nitric oxide) in diabetic renal hyperfiltration. Kidney Int 43:1306–1312
Article PubMed CAS Google Scholar - Vallon V, Thomson SC (2012) Renal function in diabetic disease models: the tubular system in the pathophysiology of the diabetic kidney. Annu Rev Physiol 74:351–375
Article PubMed CAS Google Scholar - Thomas MC, Moran JL, Harjutsalo V et al (2012) Hyperfiltration in type 1 diabetes: does it exist and does it matter for nephropathy? Diabetologia 55:1505–1513
Article PubMed CAS Google Scholar - Noh H, Ha H, Yu MR et al (2002) High glucose increases inducible NO production in cultured rat mesangial cells. Possible role in fibronectin production. Nephron 90:78–85
Article PubMed CAS Google Scholar - Sasaki M, Shikata K, Okada S et al (2011) The macrophage is a key factor in renal injuries caused by glomerular hyperfiltration. Acta Med Okayama 65:81–89
PubMed CAS Google Scholar - Ichinose K, Maeshima Y, Yamamoto Y et al (2005) Antiangiogenic endostatin peptide ameliorates renal alterations in the early stage of a type 1 diabetic nephropathy model. Diabetes 54:2891–2903
Article PubMed CAS Google Scholar - Merchant ML, Perkins BA, Boratyn GM et al (2009) Urinary peptidome may predict renal function decline in type 1 diabetes and microalbuminuria. J Am Soc Nephrol 20:2065–2074
Article PubMed CAS Google Scholar - Gilbert RE, Kim SA, Tuttle KR et al (2007) Effect of ruboxistaurin on urinary transforming growth factor-beta in patients with diabetic nephropathy and type 2 diabetes. Diabetes Care 30:995–996
Article PubMed CAS Google Scholar - Andersen S, van Nieuwenhoven FA, Tarnow L et al (2005) Reduction of urinary connective tissue growth factor by losartan in type 1 patients with diabetic nephropathy. Kidney Int 67:2325–2329
Article PubMed CAS Google Scholar - Tashiro K, Koyanagi I, Saitoh A et al (2002) Urinary levels of monocyte chemoattractant protein-1 (MCP-1) and interleukin-8 (IL-8), and renal injuries in patients with type 2 diabetic nephropathy. J Clin Lab Anal 16:1–4
Article PubMed CAS Google Scholar - Cherney DZ, Konvalinka A, Zinman B et al (2009) Effect of protein kinase Cbeta inhibition on renal hemodynamic function and urinary biomarkers in humans with type 1 diabetes: a pilot study. Diabetes Care 32:91–93
Article PubMed CAS Google Scholar - Dzurik R, Pontuch P, Spustova V (1997) Modulators and mediators of kidney disease progression: new targets of prevention and treatment. Bratisl Lek Listy 98:663–666
PubMed CAS Google Scholar - Cohen-Bucay A, Viswanathan G (2012) Urinary markers of glomerular injury in diabetic nephropathy. Int J Nephrol 2012:146987
PubMed Google Scholar - Ruster C, Wolf G (2008) The role of chemokines and chemokine receptors in diabetic nephropathy. Front Biosci 13:944–955
Article PubMed CAS Google Scholar - Mezzano S, Aros C, Droguett A et al (2004) NF-kappaB activation and overexpression of regulated genes in human diabetic nephropathy. Nephrol Dial Transplant 19:2505–2512
Article PubMed CAS Google Scholar - Mezzano S, Droguett A, Burgos ME, et al (2003) Renin-angiotensin system activation and interstitial inflammation in human diabetic nephropathy. Kidney Int Suppl (86):S64–S70
- Sochett EB, Cherney DZ, Curtis JR, Dekker MG, Scholey JW, Miller JA (2006) Impact of renin angiotensin system modulation on the hyperfiltration state in type 1 diabetes. J Am Soc Nephrol 17:1703–1709
Article PubMed CAS Google Scholar - Mauer M, Zinman B, Gardiner R et al (2009) Renal and retinal effects of enalapril and losartan in type 1 diabetes. N Engl J Med 361:40–51
Article PubMed CAS Google Scholar - Halimi JM (2012) The emerging concept of chronic kidney disease without clinical proteinuria in diabetic patients. Diabetes Metab 38:291–297
Article PubMed CAS Google Scholar - Perkins BA, Krolewski AS (2009) Early nephropathy in type 1 diabetes: the importance of early renal function decline. Curr Opin Nephrol Hypertens 18:233–240
Article PubMed CAS Google Scholar - Miller JA (1997) Renal responses to sodium restriction in patients with early diabetes mellitus. J Am Soc Nephrol 8:749–755
PubMed CAS Google Scholar
Acknowledgements
The authors would like to thank P. Yip and J. Cheung-Hum (Special Testing Laboratory, University Health Network, Toronto, Canada) for their invaluable assistance with biochemical assays included in this work. The authors are grateful to the study participants whose time and effort are critical to the success of our research programme.
Funding
This work was supported by operating grants from the Canadian Institutes of Health Research, Heart Stroke Foundation (to D. Z. I. Cherney), a University of Toronto Dean’s Fund Grant and a grant from the Juvenile Diabetes Research Foundation to D. Daneman, F. H. Mahmud and E. B. Sochett. D. Z. I. Cherney was also supported by a Kidney Foundation of Canada Scholarship, a Canadian Diabetes Association-KRESCENT Program Joint New Investigator Award and a University of Toronto Dean’s Fund Award. R. Har is supported by a Banting and Best Fellowship and an Institute of Medical Science Open Fellowship Award. J. W. Scholey is the CIHR/AMGEN Canada Kidney Research Chair at the University Health Network, University of Toronto.
Duality of interest
The results presented in this paper have not been published previously in whole or in part. M. L. Fritzler is the owner of Eve Technologies Corporation, who performed the cytokine assays for this project. All other authors declare that there is no duality of interest associated with their contribution to this manuscript.
Contribution statement
DZIC, VL and RH were involved in study design and acquisition and interpretation of data. JWS, DD, FM, RD, YE, ES, HNR and MLF were involved in interpretation of data. All authors revised the manuscript and approved the final version.
DZIC is the guarantor of this work and, as such, had full access to all the data in the study and takes responsibility for the integrity of the data and the accuracy of the data analysis.
Author information
Authors and Affiliations
- Division of Nephrology, Toronto General Hospital, University Health Network, Banting and Best Diabetes Centre, University of Toronto, 585 University Ave, 8N-845, Toronto, ON, Canada, M5G 2N2
R. Har & D. Z. I. Cherney - Division of Nephrology, Toronto General Hospital, University Health Network, Toronto, ON, Canada
J. W. Scholey, V. Lai & H. N. Reich - Division of Pediatric Endocrinology, Hospital for Sick Children, University of Toronto, Toronto, ON, Canada
D. Daneman, F. H. Mahmud, R. Dekker, Y. Elia & E. B. Sochett - Eve Technologies, Calgary, AB, Canada
M. L. Fritzler
Authors
- R. Har
You can also search for this author inPubMed Google Scholar - J. W. Scholey
You can also search for this author inPubMed Google Scholar - D. Daneman
You can also search for this author inPubMed Google Scholar - F. H. Mahmud
You can also search for this author inPubMed Google Scholar - R. Dekker
You can also search for this author inPubMed Google Scholar - V. Lai
You can also search for this author inPubMed Google Scholar - Y. Elia
You can also search for this author inPubMed Google Scholar - M. L. Fritzler
You can also search for this author inPubMed Google Scholar - E. B. Sochett
You can also search for this author inPubMed Google Scholar - H. N. Reich
You can also search for this author inPubMed Google Scholar - D. Z. I. Cherney
You can also search for this author inPubMed Google Scholar
Corresponding author
Correspondence toD. Z. I. Cherney.
Additional information
D. Z. I. Cherney and H. N. Reich contributed equally to this study.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article
Cite this article
Har, R., Scholey, J.W., Daneman, D. et al. The effect of renal hyperfiltration on urinary inflammatory cytokines/chemokines in patients with uncomplicated type 1 diabetes mellitus.Diabetologia 56, 1166–1173 (2013). https://doi.org/10.1007/s00125-013-2857-5
- Received: 21 November 2012
- Accepted: 23 January 2013
- Published: 15 February 2013
- Issue Date: May 2013
- DOI: https://doi.org/10.1007/s00125-013-2857-5