Photodynamic therapy-generated vaccine for cancer therapy (original) (raw)
Introduction
In photodynamic therapy (PDT), a clinically established modality for treatment of cancerous and other lesions, the targeted lesion is destroyed by localized generation of reactive oxygen species mediated by the transfer to molecular oxygen of energy absorbed by light-activated drugs (photosensitizers) [10, 11]. A wide range of photooxidative lesions produced in the membrane and cytoplasm of cancer cells, tumor vasculature and other stromal elements results in rapidly induced massive damage in PDT-treated tumors that threatens local homeostasis. This prompts a strong host reaction whose primary purpose is to contain the disrupted homeostasis, remove the dead tissue and promote tissue healing at the affected site [19]. This PDT-induced host-protecting mechanism, manifested as the inflammatory reaction, acute phase response and immune response, was demonstrated to have an important role in the therapy outcome [11].
The PDT-induced host response is instigated and promoted by an extensive release/expression of various pro-inflammatory mediators from the treated site including complement proteins, heat shock proteins, cytokines and chemokines, and arachidonic acid metabolites [6, 12, 19, 23]. The key elements of innate immune system, the complement system and Toll-like receptors, become engaged in sensing PDT-generated altered self-danger signals and propagating the ensuing inflammatory and immune responses [18, 23]. The innate immune effectors participating in tissue-destructive action in PDT-treated tumors include the components of activated complement system (opsonins and membrane attack complex), neutrophils, mast cells, macrophages and natural killer cells [6, 11, 15, 19]. The activity of these elements of innate immunity culminates in the orchestration of the development of acquired (adaptive) immune response based on the recognition of antigens of PDT-treated tumors [11]. This is evidenced by the reduced or nonexistent curability of tumors growing in immunocompromised mice by PDT (which can be restored by bone marrow transplant or T cell transfer from immunocompetent mice) and the recovery of immune memory cells from distant lymphoid sites underlying the existence of long-lasting systemic immunity raised against even poorly immunogenic PDT-treated tumors [20, 22].
The ability of PDT to induce an antitumor immune response associated with efficient recognition of tumor antigens has prompted the speculation that PDT can be exploited for the generation of an anticancer vaccine. This was confirmed by the demonstration of the efficacy of both preventive and therapeutic cancer vaccines generated by PDT [13, 19]. Gollnick et al. [13] were successful in providing protection against EMT6 tumor challenge by vaccinating naïve mice with supernatants of PDT-treated EMT6 cells. They showed that PDT-generated tumor cell lysates have the capacity to stimulate both the phenotypic and functional maturation of dendritic cells (DC), and to induce a cytotoxic T cell response. Our initial results in developing PDT-generated therapeutic cancer vaccine demonstrated that the growth of established subcutaneous poorly immunogenic mouse tumors can be retarded by lesion-localized injection of tumor cells of the same origin pretreated in vitro by Photofrin-based PDT [19]. This work was further pursued in the experiments described in the present report, which were designed to optimize the protocol for the generation of PDT-based cancer vaccine and examine the mechanisms underlying its action.
Materials and methods
Tumor model
Poorly immunogenic mouse squamous cell carcinoma SCCVII [30] and Lewis lung carcinoma [29] were maintained in vivo by serial transplantation into syngeneic mouse strains (C3H/HeN and C57BL/6, respectively) and in vitro by cell culture in alpha minimal essential medium (αMEM, Sigma Chemical Co., St. Louis, MO, USA) supplemented with 10% fetal bovine serum (HyClone Laboratories Inc., Logan, UT, USA). For experiments, SCCVII or Lewis lung tumors were implanted by injecting subcutaneously 1 million cells in the lower dorsal region. Lewis lung tumors were also inoculated into B6.129S4-C3tm1Crr mice, which are complement C3 knockouts (C3KO), where their growth rate was similar to that in wild-type mice. Vaccine treatment, performed as a single peritumoral injection, was done 5 or 6 days postimplant when the tumors reached 5 mm in largest diameter. Subsequent changes in tumor size were determined by measuring the lesion’s three orthogonal diameters with a caliper. Each treatment group consisted of six mice. The experimental procedures with mice were approved by the Animal Care Committee of the University of British Columbia.
Vaccine generation
Sufficient numbers of SCCVII cells were expanded in vitro and frozen in liquid nitrogen. Generation of the vaccine was started by incubating the required numbers of thawed cells in serum-free αMEM with benzoporphyrin derivative monoacid ring A (BPD or verteporfin , a lipid-formulated photosensitizer provided by QLT, Inc., Vancouver, BC, Canada) for 60 min at 37°C. The concentration of BPD used was 0.4 μg/ml except that they were noted differently. The incubation was carried out in 50 ml polypropylene tubes (Falcon 2070) which enabled maintaining the cells nonadherent. The cells were then pelleted by centrifugation, washed in phosphate buffered saline (PBS), suspended in 1 ml PBS and transferred into a 3 cm diameter Petri dish that was placed into a beam of 690±1 nm light for the exposure of 1 J/cm2 (15 mW/cm2) while kept on ice. The light source was a 250 mW diode laser (SDL7422-H1; Spectra Diode Labs, San Jose California), delivering 690 nm light for monodirectional illumination through a liquid light guide, model 77638 (8-mm core diameter, Oriel Instruments, Stratford, CT, USA). In most experiments, the cells were then exposed to X-ray treatment (60 Gy). The X-ray source was Philips RT 250 (250 kV, 0.5 mm Cu, dose rate 3.26 Gy/min). Finally, between 5 and 50 million cells (volume 0.2 ml) were injected peritumorally per mouse. Control groups included the treatment with lysates obtained from equal numbers of SCCVII cells subjected to three cycles of the freeze–thaw procedure. Tumor mismatched control was the PDT-vaccine generated from in vitro expanded FsaR fibrosarcoma cells, a model also syngeneic to C3H/HeN mice.
Flow cytometry
The impact of PDT-vaccine treatment was investigated by flow cytometry-based analysis of cells obtained from inguinal lymph nodes draining vaccinated tumors, and of cells retrieved from the vaccination site one hour after the vaccine injection consisting of 2×106 PDT-treated SCCVII cells. In the latter case, SCCVII cells were, in some experiments, surface biotinylated before being used for the vaccine generation or for X-ray-only-treated vaccine control. This was achieved by incubating SCCVII cell suspension on ice for 30 min with a biotin conjugate with sulfosuccinimidyl ester supplied in FluoReporter Cell-Surface Biotinylation kit (Molecular Probes, Eugene, OR, USA) following the manufacturer’s instructions. These cells were later identified in flow cytometry analysis by staining with streptavidin-Cy-Chrome conjugate (PharMingen, BD Biosciences, Mississauga, ON, Canada). In other experiments, vaccine cells were not pre-biotinylated and they were identified by the fluorescence of BPD in the red spectral region as shown in our previous report [21]. The antibodies used for flow cytometry were against the following mouse cell surface antigens: CD3, CD4, CD8, CD44, CD45RB, and CD45R/B220 (all obtained from PharMingen); they were conjugated with FITC, PE or Cy-Chrome chromophore which facilitated their use in a three-color analysis. Also used were FITC-conjugated rat anti-mouse CD205 (Serotec Ltd., Oxford, UK), chicken anti-mouse heat shock protein 70 (HSP70) produced by Santa Cruz Biotechnology Inc. (Santa Cruz, CA, USA), FITC-conjugated goat anti-mouse C3 (Cappel, ICN Pharmaceuticals Inc., Aurora, OH, USA), and rat anti-mouse C3b/iC3b/C3c (mAb 3/26 generously provided by Dr. A. Erdei). Secondary, FITC-conjugated antibodies were goat anti-chicken IgY and chicken anti-rat IgG (both from Gallus Immunotech Inc., Fergus, ON, Canada) for visualizing HSP70 and C3b/iC3b/C3c, respectively. Isotype control staining included FITC/PE-conjugated or nonconjugated ChromePure goat IgG, rat IgG and chicken IgY (Jackson ImmunoResearch Laboratories, West Grove, PA, USA). Flow cytometry analysis was performed with a Coulter Epics Elite ESP (Coulter Electronics, Hialeah, FL, USA).
Statistical analysis
The data are presented as mean with standard deviation. Unpaired Student’s t test was applied for testing the difference between means and the differences were considered significant when P<0.05.
Results
A cohort of mice with subcutaneously growing SCCVII tumors was divided into five treatment groups (six mice per group). The first group were controls that were left untreated, while the mice in other groups were injected peritumorally with 1×107 SCCVII cells that were pretreated in vitro using different protocols. Cell lysate obtained by the freeze–thaw procedure was given to the second group, the mice in the third group received cells treated with a lethal dose of X-rays (60 Gy), while those in the fourth and fifth groups were injected with cells that were in vitro treated first by BPD-based PDT and then with X-rays (60 Gy). The BPD concentration in the PDT protocol was either 0.4 μg/ml (vaccine-1, fourth group) or 20 μg/ml (vaccine-2, fifth group). The PDT treatment used for the vaccine-1 was lethal to >50% of cells and that for the vaccine-2 was supralethal. Monitoring of tumor growth after the therapy revealed no significant differences in the growth rate between the untreated controls and the groups receiving either lysed or X-ray treated cells (Fig. 1). In contrast, the rate of tumor growth was considerably slower in the groups that received PDT-generated vaccines. For instance, at 13 days’ posttherapy the average tumor size in both vaccine-1 and vaccine-2 groups was significantly smaller than in the control group (P<0.005). There was, however, no significant difference between the efficacies of the two PDT vaccines. Hence, the vaccine-1 protocol was chosen for the remaining experiments in this study.
Fig. 1
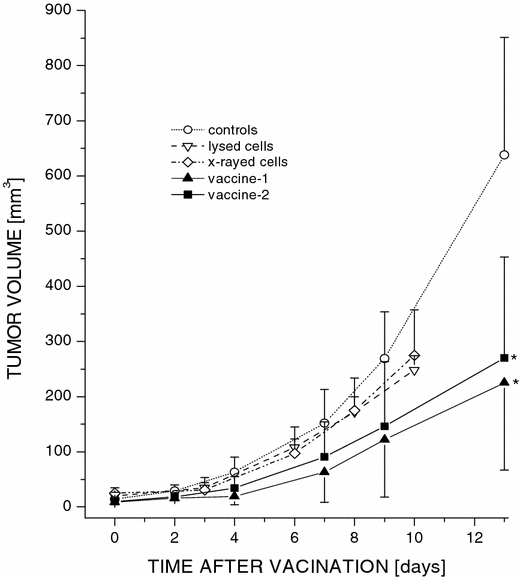
The effect of PDT-generated vaccine on growth of established SCCVII tumors. In vitro cultured SCCVII cells were incubated with BPD, either 0.4 μg/ml (vaccine-1) or 20 μg/ml (vaccine-2), in serum-free medium for 1 h at 37°C, then treated by light (690 nm; 1 J/cm2), followed by the exposure to X-rays (60 Gy). Mice bearing subcutaneously growing SCCVII tumors received each 1×107 of these vaccine cells by peritumoral injection. Response to the vaccine treatment was determined by subsequent tumor size measurement. Also shown is the growth of control untreated tumors, and tumors in control groups injected with 1×107 X-ray-only-treated or lysed SCCVII cells. Omitted for clarity are the day 13 data points for lysed and X-rayed cells (583±77 and 828±153 mm3, respectively). Each treatment group consisted of six mice. Bars are SD. *Significant difference compared to the untreated controls at day 13 postvaccination (P<0.05)
For further optimization of the PDT-vaccine protocol, we examined its efficacy depending on the number of SCCVII cells used in the vaccine injection. In this testing, 5, 10, 20 or 50 million of vaccine cells were injected per mouse. The vaccine consisting of 2×107 cells showed a greatest retardation of tumor growth (Fig. 2). The data presented as means of tumor size for each of the treatment groups at various time intervals after vaccination are depicted in Fig. 2a. The dynamics of growth of individual tumors is also shown for the three treatment groups (Fig. 2b), which illustrates the variations typically observed within the same treatment group in response to the PDT-vaccine treatment. Within the group receiving 2×107 cells in the vaccine injection, one tumor regressed and became impalpable, one tumor remained palpable with a minimal increase in size, while the remaining four tumors continued to grow but at a reduced rate compared to the control group.
Fig. 2
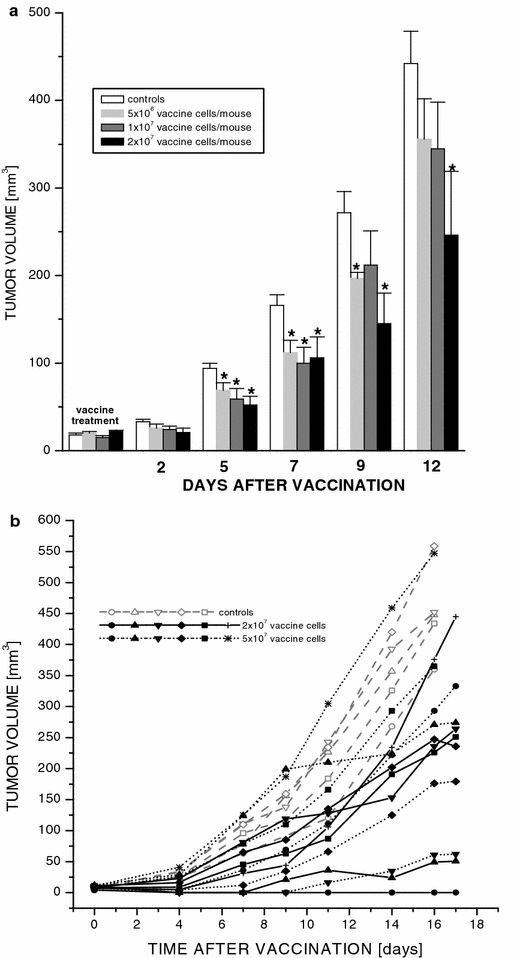
The impact of cell number used in PDT-generated vaccine on its effectiveness in treating subcutaneous SCCVII tumors. The vaccine was generated as described for the vaccine-1 in Fig. 1. The cell number used per vaccination by peritumoral injection of mice bearing SCCVII tumors was varied in different treatment groups from 5×106 to 5×107. The postvaccination tumor growth is shown either as a means of tumor size (+ SD) per treatment groups (_N_=6) receiving 0 (nonvaccinated controls), 5×106, 1×107, or 2×107 vaccine cells, or b tumor size values for individual tumors receiving 0 (controls), 2×107 or 5×107 vaccine cells. *Significant difference compared to the nonvaccinated controls (P<0.05)
To verify the specificity of the PDT-generated vaccine, mice bearing SCCVII tumors were treated with the vaccine prepared from the same tumor cell line or from FsaR cells (fibrosarcoma syngeneic to the same mouse strain). It can be seen that, in contrast to the PDT-vaccine from the same tumor cell line, the PDT-vaccine from the mismatched tumor cell line was not effective (Fig. 3).
Fig. 3
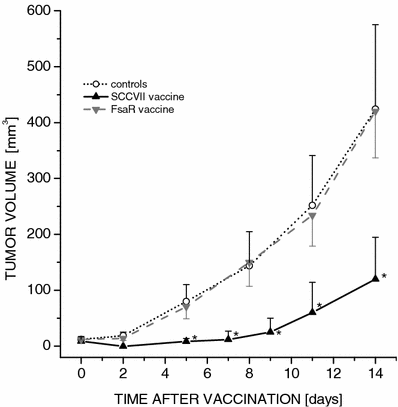
The effect of PDT-vaccines generated from SCCVII or FsaR tumor cells on growth of SCCVII tumors. The vaccine was generated from either SCCVII or FsaR cells as described for the vaccine-1 in Fig. 1, and 2×107 cells were injected peritumorally per SCCVII tumor-bearing mouse. The growth of tumors after SCCVII cell-based vaccine or mis-matched (FsaR cell-based) vaccine and nonvaccinated control tumors is depicted by showing means of tumor size per treatment group. Bars are SD; _N_=6. *Significant difference compared to the nonvaccinated controls (P<0.05)
In order to obtain an insight into the host response induced by the PDT-vaccine treatment, we first examined the cell populations contained in the tumor-draining lymph nodes that were excised at 4 days’ postvaccination. The results show that there was a marked increase in cell numbers within all major lymph node populations in vaccinated mice compared to nonvaccinated tumor-bearing mice (Fig. 4). This increase ranged from 5–6-fold for T-lymphocyte populations, over 10-fold for DC populations, to over 17-fold for B-lymphocytes. In addition, among T cells there was an increased percentage of cells bearing the CD44+CD45RB– memory phenotype (Fig. 4 insert). Control mice vaccinated with X-ray-only-treated (PDT-untreated) cells showed no significant increase in lymph node cell numbers (not shown).
Fig. 4
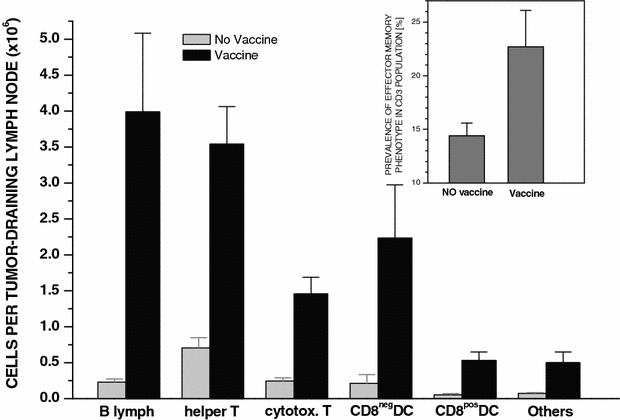
Size of immune cell populations found in lymph nodes draining vaccinated or nonvaccinated SCCVII tumors. Four days after PDT-vaccine treatment as described for the vaccine-1 in Fig. 1 using 2×107 cells per vaccination by peritumoral injection, mice bearing vaccinated SCCVII tumors were sacrificed and inguinal lymph node cells were collected. Antibody staining of these cells followed by flow cytometry enabled their classification and the determination of total number of cells per lymph node for major immune cell populations. The data obtained with lymph node cells collected from SCCVII tumor-bearing control (nonvaccinated) mice are shown for comparison. The insert depicts the levels of T-lymphocytes bearing memory phenotype (CD44+CD45RB–). Bars are SD; _N_=4. The values for all populations in the vaccine group are significantly higher than the values for the equivalent populations in the no vaccine group (P<0.05)
In further experiments, we focused on the analysis of the cells retrieved from the vaccine injection site. The samples retrieved at 1 h after vaccination revealed large numbers of DCs co-localized with the vaccine cells (Fig. 5). The vaccine cells in the retrieved samples (identified by the photosensitizer fluorescence) exhibited a positive surface staining with antibodies against complement C3 protein (Fig. 6). This was documented with both polyclonal goat anti-mouse C3 (Fig. 6a, c) and monoclonal rat anti-mouse C3 (Fig. 6b), the latter reacting preferentially with cleaved C3 fragments [25]. The retrieved vaccine cells were also found to express HSP70 on their surface (Fig. 6, insert), which is in accordance with our recently published findings with in vitro PDT-treated cells [23]. The finding from our previous in vitro studies that the binding of C3 to PDT-treated cells can be blocked by anti-HSP70 antibodies [7] was also verified with the retrieved vaccine cells. In this case, anti-HSP70 (40 μg/mouse) or its immunoglobulin isotype control was injected together with SCCVII vaccine cells into SCCVII tumor-bearing mice. This resulted in the abrogation of C3 fluorescence on the retrieved vaccine cells that was not evident in the samples with the antibody isotype control (Fig. 6c).
Fig. 5
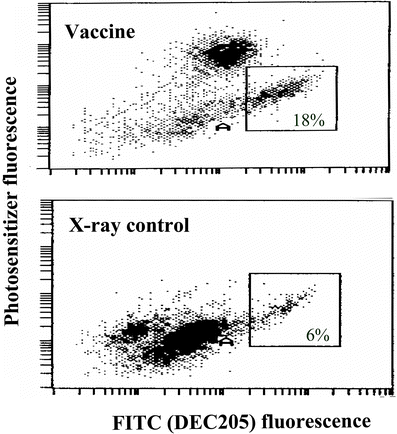
Sequestration of dendritic cells to the site of PDT-vaccine injection. The mice with SCCVII tumors were sacrificed 1 hour after receiving PDT vaccine cells (2×107 SCCVII cells treated by the vaccine-1 protocol described for Fig. 1) or the same number of X-ray-only-treated SCCVII cells. The cell samples retrieved from the vaccine injection site were stained with FITC-conjugated DEC205 antibody for flow cytometry-based identification of dendritic cells. The vaccine cells were identified by BPD fluorescence. Pre-biotinylation (as described in Materials and Methods) of SCCVII cells used in X-ray-only protocol enabled their identification by subsequent staining with a chromophore-conjugated streptavidin. Dot plot graphs of representative samples are shown
Fig. 6
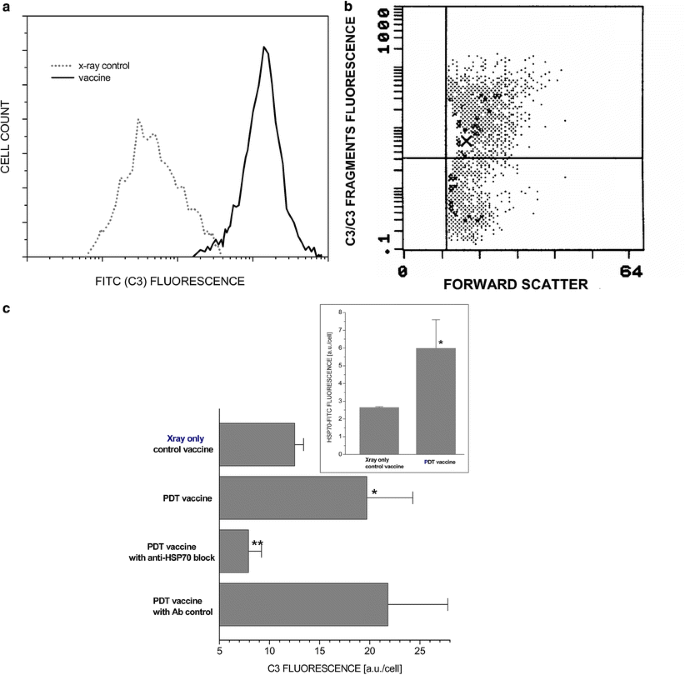
Complement opsonization of administered PDT-vaccine cells and the role of HSP70. Following the same protocol for PDT-vaccine generation using SCCVII cells and the retrieval from the vaccination site at 1 hour after vaccination of SCCVII tumor-bearing mice as described for Fig. 5, the collected cells were stained with goat anti-mouse C3 antibody or rat anti-mouse C3/C3b/iC3b/C3c antibody. a C3-associated fluorescence intensity in cells retrieved in PDT-vaccine and X-ray-only treatment groups, and b dot plot graph of C3/C3b/iC3b/C3c fluorescence in a representative sample from the PDT-vaccine group. c the impact of co-administration with the PDT-vaccine of HSP70-blocking antibody (40 μg/mouse) or its isotype control immunoglobulin on C3-associated fluorescence on the retrieved cells. The insert in this graph shows the levels of surface HSP70 expression (following staining with anti-HSP70 antibody) on the retrieved cells. Bars are SD; _N_=4. *Significant difference compared to X-ray-only group (P<0.05); **Significant difference compared to PDT vaccine group with no anti-HSP70 treatment (P<0.05)
The relevance of C3 deposition on PDT-vaccine cells was demonstrated by the results comparing the effectiveness of PDT-vaccine in wild-type and C3KO mice. In this experiment, Lewis lung carcinoma model was used that is syngeneic to C57BL/six mice since the available C3KO mice have this genetic background. The PDT-vaccine, generated from in vitro cultured Lewis lung cells, was effective against Lewis lung tumors growing in wild-type C57BL/6 mice but exhibited no significant benefit against the same tumors growing in C3-deficient mice (Table 1).
Table 1 The effect of PDT-generated vaccine on the growth of subcutaneous Lewis lung tumors in wild-type (C57BL/6) and C3KO mice
The results of the examination of the cells from lymph nodes draining PDT-vaccine-treated tumors excised at 24-h postvaccination also support the role of C3. Compared to the cells from nonvaccinated controls or X-ray-only vaccine controls (the latter not shown), the surface-localized C3 was clearly elevated on B cells and even more dramatically on DCs, while no significant change was detected on T cells (Fig. 7).
Fig. 7
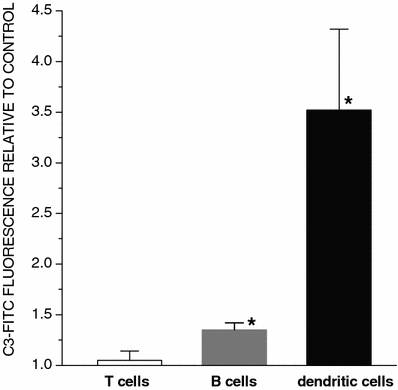
Presence of bound C3 on cells in tumor-draining lymph nodes after PDT-vaccine treatment. The vaccine, generated as described for the vaccine-1 in Fig. 1, was given to SCCVII tumor-bearing mice (2×107 vaccine cells per mouse peritumorally). The mice were sacrificed 20 h later, tumor-draining lymph nodes excised and the collected cells stained with antibodies against mouse C3, CD3 (T cells), CD19 (B-cells) and DEC205 (DCs) for flow cytometry analysis. The same was done with tumor-draining lymph node cells obtained from untreated SCCVII tumor-bearing mice (controls). The results are presented as values relative to those in corresponding populations of the control group. Bars are SD; _N_=4. *Significant difference compared to the same population in the control group
Discussion
This study demonstrates that PDT can be exploited for the generation of effective therapeutic cancer vaccines. It is shown that growth of established subcutaneous poorly immunogenic tumors can be inhibited with a vaccine generated from in vitro PDT-treated tumor cells of the same origin. While in their successful application of PDT vaccine for the prophylactic treatment (protecting against subsequent tumor challenge) Gollnick et al. [13] used lysates of PDT-treated cells, in the present study we employed whole cells collected immediately after they were PDT treated and exposed to a lethal X-ray dose. Although photosensitizer Photofrin has proven to be effective for generating the PDT-vaccine in our initial experiments [19], as described in this report, we have switched to BPD because with this photosensitizer the incubation can be reduced to 1 hour with the cells already prepared as a suspension.
In order to identify the optimal vaccine protocol, we examined the effect of vaccines prepared by employing a moderate or high PDT dose (by changing the BPD concentration) and the impact of varying the number of PDT-treated cells injected per vaccination. While there was no significant difference in the results with the two different PDT doses used for the vaccine preparation (Fig. 1), the number of cells employed in the vaccination turned out to represent an important parameter. The efficacy of PDT vaccine increased with increasing the number of used cells, reaching a maximum benefit with 2×107 cells/vaccination. A further increase to 5×107 cells/vaccination proved counterproductive, due presumably to a self-regulatory downregulation of host response in the presence of excessive antigen load [8].
The unique advantage of PDT for the generation of cancer vaccines is evidenced by the fact that comparable treatments with lysed cells or X-ray treated cells showed no significant benefit (Fig. 1). The treatment with a lethal X-ray dose was included as the last step in the PDT-vaccine generation procedure to simulate clinically acceptable safety restriction protocols for excluding the risk of secondary tumor generation. Tumor specificity of PDT-generated vaccines, documented by the negative results obtained with mismatched tumors (Fig. 3) [13], is a firm indication that an induced tumor-specific immune response is underlying the observed antitumor effect. Indeed, vital manifestations of such immune response are documented by the findings of a massive sequestration of DCs at the vaccination site (Fig. 5) as well as of a dramatic increase in the numbers of DCs, helper and cytotoxic T cells (memory cells in particular), and B cells in tumor-draining lymph nodes (Fig. 4).
We have identified two elements that seem relevant for the unique capacity of PDT for the cancer vaccine generation: surface expression of HSP70 and binding of complement proteins on PDT-treated cells. In related studies, we have reported on the induction by PDT of HSP70 expression on the surface of treated cells [23], as well as on the fixation of complement proteins on these cells [7]. In accordance with these findings, we detected high levels of both HSP70 and complement proteins on the surface of PDT-vaccine cells retrieved from the vaccination site at 1 hour after their tumor-localized administration (Fig. 6). We have suggested earlier that the surface exposure of HSP70 facilitates the opsonization of C3 or its fragments on PDT-treated cells [7]. This is supported by the results showing that the binding of C3/C3 fragments on vaccine cells was inhibited by the co-administration of HSP70-blocking antibodies at the time of vaccination (Fig. 6). Complement proteins may be opsonizing peptides associated with surface-expressed HSP70 recognized as altered self-targets by the complement system, or they could bind HSP70 directly as shown by Prohászka et al. [28]. Relevance of complement in the efficacy of PDT-vaccine is demonstrated by the lack of therapeutic benefit against tumors growing in C3-deficient mice (Table 1).
Both complement and heat shock proteins are potent mediators of innate immune response and were also demonstrated to have a role in the orchestration of adaptive immunity [14, 16, 26, 27]. At present, the understanding of heat shock protein-mediated immunity remains fragmentary and obscured by the diversity of putative functions, cellular receptors, multiplicity of chaperons and artifacts associated with endotoxin contamination. However, peptides bound to HSP70 were directly demonstrated to generate a peptide-specific CTL response more efficient by four orders of magnitude than the peptide alone, which suggest a physiological role for HSP70-bound peptide in accessing the class I presentation pathway [17]. The uncovered HSP70 segment on the surface of PDT-treated cells contains the domain involved in the interaction with its chaperoned peptides [4, 23], and in this way these tumor antigen peptides could become exposed to immune surveillance elements. Various immune cells including DCs, macrophages, NK cells, neutrophils and B lymphocytes have specific receptors for HSPs and for complement proteins [2, 3] that can be expected to promote the interaction of these immune effectors with the PDT-vaccine cells having HSP70 and complement protein on their surface. On the other hand, binding of complement proteins to tumor antigens is known to enhance the capture, processing, and presentation of these antigens to T and B lymphocytes [1, 5, 9]. Our finding of elevated levels of C3/C3 fragments on DCs and B cells localized in the tumor-draining lymph nodes treated by the PDT-vaccine (Fig. 7) is in accordance with the concept that tumor antigens captured within the complexes between complement and tumor proteins/peptides could, after binding to complement receptors on antigen-presenting cells that are subsequently migrating to the draining lymph nodes, become recognized by the effectors of adaptive immunity [5]. The interplay between tumor peptides, heat shock and complement proteins responsible for generating potent antitumor response following PDT-vaccine remains to be further dissected and more directly characterized by further investigation.
The PDT-generated vaccine belongs to the category of target tumor-derived whole cancer cell-based vaccines. Despite the identification of increasing numbers of tumor-specific antigens, this vaccine category remains a viable option in cancer vaccine development. Antigenic repertoire contained in whole tumor cell-derived vaccines allows for circumventing the problems associated with shedding or downregulation of specific antigens as well as with major histocompatibility (MHC)-restricted epitope identification for individual patients. Moreover, targeting tumor antigens unique to individual patients generated by random mutation in cancer cells may be important for securing an effective therapeutic response [16, 24].
Preliminary evidence from our ongoing studies indicates that further improvements can be achieved in the optimization of the protocols for the generation of PDT-generated cancer vaccine that would exhibit even greater efficacy of tumor control with high cure rates. Prospects for clinical use of PDT-generated vaccines include their use in conjunction with established modalities such as surgery or radiotherapy. Initial testing reveals that the response of SCCVII tumors to X-ray therapy can be improved by an additional vaccination with PDT-treated SCCVII cells (M. Korbelik and J. Sun, unpublished results).
References
- Arvieux J, Yssel H, Colomb MG (1988) Antigen-bound C3b and C4b enhances antigen-presenting cell function in activation of human T-cell clones. Immunology 65:229–235
PubMed CAS Google Scholar - Asea A (2003) Chaperone-induced signal transduction pathways. Exerc Immunol Rev 9:25–33
PubMed Google Scholar - Bohana-Kashtan O, Ziporen L, Donin L, Kraus S, Fishelson Z (2004) Cell signals transduced by complement 41:583–597
CAS Google Scholar - Bolzer C, Li G, Issels RD, Multhoff G (1998) Definition of extracellular localized epitopes of Hsp70 involved in an NK immune response. Cell Stress Chaperones 3:6–11
Article Google Scholar - Carroll MC (2004) The complement system in regulation of adaptive immunity. Nat Immunol 5:981–986
Article PubMed CAS Google Scholar - Cecic I, Korbelik M (2002) Mediators of peripheral blood neutrophilia induced by photodynamic therapy of solid tumors. Cancer Lett 183:43–51
Article PubMed CAS Google Scholar - Cecic I, Korbelik M (2005) Deposition of complement proteins on cells treated by photodynamic therapy in vitro. J Environ Pathol Toxicol Oncol 25 (in press)
- Critchfield JM, Racke MK, Zuniga-Pflucker JC, Cannella B, Raine CS, Goverman J, Lenardo MJ (1994) T cell depletion in high antigen dose therapy of autoimmune encephalomyelitis. Science 263:1139–1143
Article PubMed CAS Google Scholar - Dempsey PW, Allison ME, Akkaraju S, Goodnow CC, Fearon DT (1996) C3d of complement as a molecular adjuvant: bridging innate and acquired immunity. Science 271:348–350
Article PubMed CAS Google Scholar - Dougherty TJ (2002) An update on photodynamic therapy applications. J Clin Laser Med Surg 20:3–7
Article PubMed Google Scholar - Dougherty TJ, Gomer CJ, Henderson BW, Jori G, Kessel D, Korbelik M, Moan J, Peng Q (1998) Photodynamic therapy. J Natl Cancer Inst 90:889–905
Article PubMed CAS Google Scholar - Gollnick SO, Evans SS, Baumann H, Owczarczak B, Maier P, Wang WC, Ungar E, Henderson BW (2003) Role of cytokines in photodynamic therapy-induced local and systemic inflammation. Br J Cancer 88:1772–1779
Article PubMed CAS Google Scholar - Gollnick SO, Vaughan L, Henderson BW (2002) Generation of effective antitumor vaccines using photodynamic therapy. Cancer Res 62:1604–1608
PubMed CAS Google Scholar - Hawlisch H, Wills-Karp M, Karp CL, Köhl J (2004) The anaphylatoxins bridge innate and adaptive immune responses in allergic asthma. Mol Immunol 41:123–131
Article PubMed CAS Google Scholar - Henderson BW, Gollnick SO (2003) Mechanic principles of photodynamic therapy. In: Vo-Dinh T (ed) Biomedical Photonics Handbook. CRC Press, Boca Raton, pp 36-1–36-27
- Hoos A, Levey DL, Lewis JJ (2004) Autologous heat shock protein-peptide complexes for vaccination against cancer: from bench to bedside. Dev Biol (Basel) 116:109–115
CAS Google Scholar - Javid B, MacAry PA, Oehlmann W, Singh M, Lehner PJ (2004) Peptides complexed with the protein HSP70 generate efficient human cytolytic T-lymphocyte responses. Biochem Soc Trans 32:622–625
Article PubMed CAS Google Scholar - Korbelik M (2004) The role of Toll-like receptors in photodynamic therapy-elicited host response. Proc SPIE 5319:87–95
Article CAS Google Scholar - Korbelik M, Cecic I (2003) Mechanism of tumor destruction by photodynamic therapy. In: Nalwa HS (ed) Handbook of Photochemistry and Photobiology, vol 4. American Scientific Publishers, Stevenson Ranch, pp 39–77
- Korbelik M, Dougherty GJ (1999) Photodynamic therapy-mediated immune response against subcutaneous tumors. Cancer Res 59:1941–1946
PubMed CAS Google Scholar - Korbelik M, Krosl G (1995) Accumulation of benzoporphyrin derivative in malignant and host cell populations of the murine RIF tumor. Cancer Lett 97:249–254
Article PubMed CAS Google Scholar - Korbelik M, Krosl G, Krosl J, Dougherty GJ (1999) The role of host lymphoid populations in the response of mouse EMT6 tumor to photodynamic therapy. Cancer Res 56:5647–5652
Google Scholar - Korbelik M, Sun J, Cecic I (2005) Photodynamic therapy-induced cell surface expression and release of heat shock proteins: relevance for tumor response. Cancer Res 65:1018–1026
PubMed CAS Google Scholar - Lewis JJ (2004) Therapeutic cancer vaccines: using unique antigens. Proc Natl Acad Sci USA 101(Suppl 2):14653–14656
Article PubMed CAS Google Scholar - Mastellos D, Prechl J, László G, Papp K, Oláh E, Argyropoulos E, Franchini S, Tudoran R, Markiewski M, Lambris JD, Erdei A (2004) Novel monoclonal antibodies against mouse C3 interfering with complement activation: description of fine specificity and applications to various immunoassays. Mol Immunol 40:1213–1221
Article PubMed CAS Google Scholar - Morgan BP, Marchbank KJ, Longhi MP, Harris CL, Gallimore AM (2005) Complement: central to innate immunity and bridging to adaptive responses. Immunol Lett 97:171–179
Article PubMed CAS Google Scholar - Prohászka Z, Füst G (2004) Immunological aspects of heat shock proteins—the optimum stress of life. Mol Immunol 41:29–44
Article PubMed Google Scholar - Prohászka Z, Singh M, Nagy K, Lakos G, Duba J, Füst G (2002) Heat shock protein 70 is a potent activator of the human complement system. Cell Stress Chaperones 7:17–22
Article PubMed Google Scholar - Sugiura K, Stock CC (1955) The effect of phosphoramides on the growth of a variety of mouse and rat tumors. Cancer Res 15:38–51
PubMed CAS Google Scholar - Suit HD, Sedlacek RS, Silver G, Dosoretz D (1985) Pentobarbital anesthesia and the response of tumor and normal tissue in the C3Hf/Sed mouse to radiation. Radiat Res 104:47–65
Article PubMed CAS Google Scholar