N-acetylcysteine reduces oxidative stress in sickle cell patients (original) (raw)
Abstract
Oxidative stress is of importance in the pathophysiology of sickle cell disease (SCD). In this open label randomized pilot study the effects of oral _N_-acetylcysteine (NAC) on phosphatidylserine (PS) expression as marker of cellular oxidative damage (primary end point), and markers of hemolysis, coagulation and endothelial activation and NAC tolerability (secondary end points) were studied. Eleven consecutive patients (ten homozygous [HbSS] sickle cell patients, one HbSβ0-thalassemia patient) were randomly assigned to treatment with either 1,200 or 2,400 mg NAC daily during 6 weeks. The data indicate an increment in whole blood glutathione levels and a decrease in erythrocyte outer membrane phosphatidylserine exposure, plasma levels of advanced glycation end-products (AGEs) and cell-free hemoglobin after 6 weeks of NAC treatment in both dose groups. One patient did not tolerate the 2,400 mg dose and continued with the 1,200 mg dose. During the study period, none of the patients experienced painful crises or other significant SCD or NAC related complications. These data indicate that _N_-acetylcysteine treatment of sickle cell patients may reduce SCD related oxidative stress.
Similar content being viewed by others
Introduction
Oxidative stress plays a role of major importance in the development of organ damage in sickle cell disease (SCD) [1–4]. Oxidative stress in SCD results from factors such as the unstable auto-oxidative sickle hemoglobin (HbS) [5, 6], chronic intravascular hemolysis [7–9], recurrent ischemia reperfusion injury [4], and low grade inflammation [10]. Increased levels of reactive oxygen species (ROS) lead to further acceleration of hemolysis [8], hypercoagulability [11, 12], decreased nitric oxide (NO) bio-availability [13], and endothelial damage [14].
Given the fact that oxidative stress is a likely major contributing factor in the development of both acute and chronic complications in SCD, the potential of anti-oxidants as therapeutics for SCD should be explored. A major intracellular anti-oxidant is the reduced form of the amino-thiol glutathione (GSH) [15, 16]. Due to increased consumption by excessive levels of ROS, sickle cell patients have decreased levels of plasma and erythrocyte total glutathione and the ratio of GSH to its oxidized form glutathione disulfide (GSSG) is reduced [17, 18]. _N_-acetylcysteine (NAC), the rate limiting substrate for GSH generation, is an important antioxidant with pleiotropic effects on inflammation and vasomotor function [19]. NAC readily enters cells and within the cytoplasm it is converted to l-cysteine, which is a precursor to GSH [20]. Treatment of sickle cell patients with NAC has been demonstrated to have an inhibitory effect on the formation of dense red cells [21]. Augmenting the antioxidant capacity in sickle red blood cells by NAC may reduce oxidative red cell membrane damage and reduce its many down-stream pathophysiological effects, such as hemolysis, endothelial damage, and activation, activation of the coagulation cascade, and the decrease of NO bio-availability [13, 22, 23]. In this randomized open label pilot study, the effects of oral NAC treatment on markers of oxidative stress and hemolysis in sickle cell patients were investigated.
Patients and methods
Study population
Consecutive adult (age ≥18 years) homozygous sickle cell anemia (HBSS) or HbSβ0-thalassemia outpatients (high performance liquid chromatography (HPLC) confirmed), at the Academic Medical Center (AMC), Amsterdam, The Netherlands, were eligible for the study. Exclusion criteria were painful crisis and blood transfusion in the preceding 4 weeks and 4 months respectively, pregnancy, or the desire to get pregnant in the following 3 months, calculated glomerular filtration rate of <90 ml/min (Cockcroft and Gault formula), known gastric/duodenal ulcers, active infections, auto-immune inflammatory diseases and use of hydroxyurea, vitamin K antagonists or other oral anticoagulants and contraindications for NAC use. Ten race and age-matched healthy volunteers were included as controls for base-line values. All participants received verbal and written explanation of the objectives and procedure of the study and subsequently provided written informed consent. The study was approved by the AMC Medical Ethical Commission and experiments were performed in accordance with the Declaration of Helsinki. The study was registered in the Dutch Trial Registry (www.trialregister.nl; trial ID number NTR1013).
End point
The primary end point of the study was reduction of erythrocyte phosphatidylserine (PS) expression as a direct indicator of erythrocyte membrane (oxidative) damage. Changes in markers of hemolysis (hemoglobin, reticulocytes, lactate dehydrogenase (LDH), and bilirubin) hypercoagulability, endothelial activation, and inflammation, and tolerability of oral NAC were secondary end points.
Study protocol
After baseline measurements and randomization to either 1,200 or 2,400 mg of NAC per day, patients started taking NAC (acetylcysteine 600 mg tablets dissolved in water; Pharmachemie B.V. Haarlem, The Netherlands) orally twice daily during 6 weeks followed by another 6 weeks of follow-up after NAC cessation. Both during NAC treatment (visits 0–3) and in the post-treatment period (visits 4–6) patients were seen two-weekly for follow-up visits during which questionnaires pertaining to side effects were completed, weight, blood pressure, and pulse were measured and a blood sample was drawn via venipuncture. Patients kept a daily pain score diary (visual analogue scale pain score). Patient compliance was monitored by history taking and pill counts. If possible, NAC treatment would not be discontinued in case of an (hospital admission due to) acute vaso-occlusive pain crisis or other clinical event.
Blood samples
Standard blood counts were performed in EDTA anti-coagulated blood (Cell-Dyn 4000; Abbott, Illinois, USA). LDH and total and direct bilirubin levels were measured in heparinized plasma with spectrophotometry (P800 Modular; Roche, Basel, Switzerland). Citrate, serum, and EDTA samples were centrifuged immediately for 15 min at 3,000 rpm (4°C). Aliquots were stored at −80°C until further analysis. l-arginine (immediate precursor of NO), asymmetric dimethylarginine (ADMA), and symmetric dimethylarginine (SDMA) levels were measured in EDTA plasma using reversed-phase HPLC as described elsewhere [24, 25]. NT-proBNP levels were measured in EDTA plasma with electrochemiluminescence immunoassay (Roche Diagnostics). Ultra-sensitive C-reactive protein (US-CRP) was measured with ELISA according to the manufacturer's instructions (Biokit, Barcelona, Spain). Prothrombin fragment 1 + 2 (F1 + 2) and thrombin–antithrombin (TAT) complexes were determined using sandwich enzyme-linked immunosorbent assay (ELISA; Enzygnost, Dade Behring Marburg GmbH, Germany). Plasma levels of von Willebrand Factor antigen (vWF-ag) were determined in citrate plasma with in an in-house ELISA. Soluble vascular adhesion molecule-1 (sVCAM-1) levels were determined in serum (R&D Systems; Minneapolis, MN, USA).
Glutathione levels
Concentrations of GSH and GSSG were determined according to Tietze et al. and Sacchetta et al., respectively [26, 27]. After measurement in EDTA full blood, the glutathione values were adjusted for erythrocyte numbers and mean corpuscular volume (MCV). GSH is the difference between total glutathione and GSSG concentrations.
Phosphatidylserine exposure
For measurement of PS expression, first 5 μl blood (EDTA) was washed in a HEPES buffer (10 mM HEPES, 150 mM NaCl, 5 mM KCl, 1.8 mM CaCl2, 2.1 mM MgCl2, pH 7.4), centrifuged at 3,650 rpm for 90 s and after adding 5 μl annexin V antibodies (Annexin V-FITC; IQP-120F, IQ products) incubated for 10–15 min at 4°C in the dark. After the incubation period, the erythrocytes were washed, resuspended in 500 μl HEPES buffer, and erythrocyte PS expression was measured by flow cytometry using FACSCalibur (BD Biosciences, CA, USA). Assuming that especially mature old sickle erythrocytes have higher levels of external PS expression leading to their removal, we also analyzed the percentage of sickle erythrocytes having external PS expression more than one log greater than the PS negative erythrocytes.
Cell-free hemoglobin
Plasma levels of cell-free hemoglobin were determined in citrate plasma with a spectrophotometer (Shimadzu UV-2401 PC) according to the methods of Kahn et al. [28] which adjusts for hyperbilirubinemia and lipemia.
Advanced glycation end products
Two different advanced glycation end products (AGEs) were measured (i.e., pentosidine and N ε-(carboxy-methyl)lysine (CML)) at baseline and after the 6 weeks of NAC treatment. Pentosidine and CML were measured in EDTA plasma using single-column HPLC with fluorescence detection and ultra performance liquid chromatography-tandem mass spectrometry respectively, as previously described [29, 30].
Statistical analysis
Friedman test for repeated measures of non-parametric data was used for comparisons between different time points within a treatment groups. The Wilcoxon signed rank test was used for comparisons between two related samples within a treatment group. The Mann–Whitney U test was used for comparisons between two groups. Continuous data are presented as medians with corresponding inter quartile ranges (IQR), unless stated otherwise. P < 0.05 was considered statistically significant (SPSS 16.0, Chicago, IL, USA).
Results
Eleven patients (10 HbSS and 1 HbS-β0-thalassemia; median age 23 years (range 20–47), 6 male, 5 female) who met eligibility criteria, were included in the study. One patient (P4) discontinued using NAC after 3 weeks and withdrew from the study. Two patients who used <80% of prescribed dosing are shown in gray in the figures. One patient on the 2,400 mg NAC dose had gastro-intestinal complaints that disappeared after switching to 1,200 mg on the second day of treatment which she continued using (P6). No other patient reported adverse events. Levels of hemoglobin, LDH, and bilirubin and reticulocyte and leukocyte counts did not change significantly (Fig. 1). Sickle cell patients had significantly lower whole blood total glutathione and GSH levels as compared to healthy controls (Fig. 2a). Total glutathione levels increased during treatment period in patients of both 1,200 mg (from 136 (100–198) to 169 (121–221) μmol/l) and 2,400 mg (from 150 (136–165) to 163 (142–178) μmol/l) dose groups (Fig. 3, panels a and b), with the differences being statistically significant when analyzing the two dose groups combined (from 150 (113–168) to 167 (142–179) μmol/l, P < 0.05).
Fig. 1
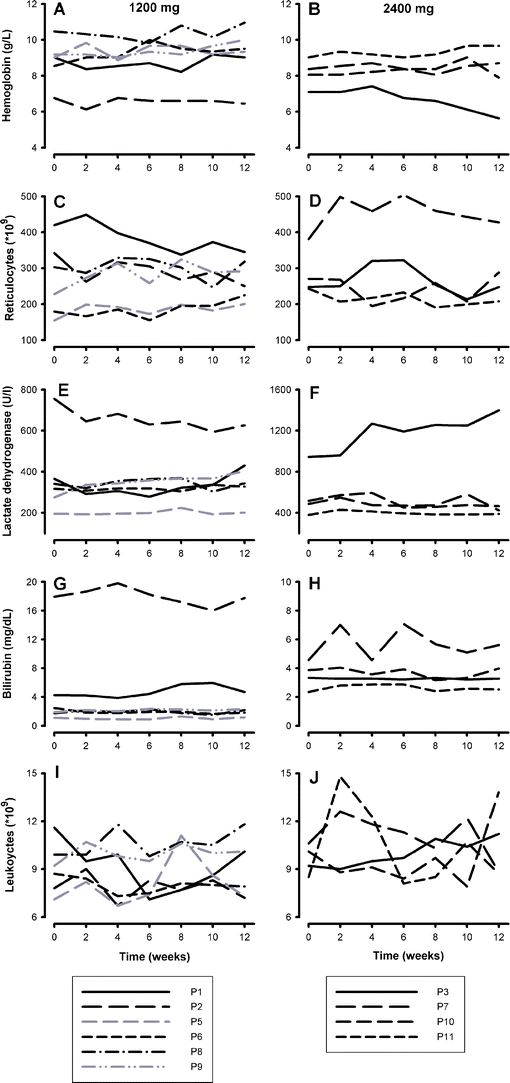
Markers of hemolysis during the 12 weeks of study period. Panels a and b hemoglobin, panels c and d reticulocytes, panels e and f lactate dehydrogenase, panels g and h bilirubin, panels i and j leukocytes. Panels on the left: 1,200 mg and panels on the right 2,400 mg. Patients are numbered in the order of inclusion. Two patients with compliance of <80% are shown in gray. Patient number 4 (P4) discontinued using NAC and withdrew from the study
Fig. 2
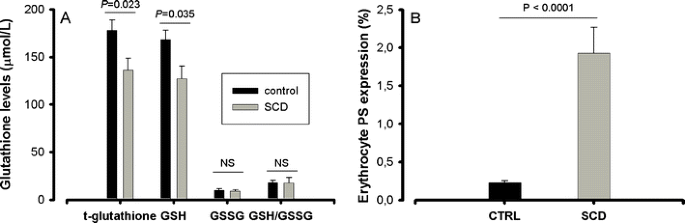
Glutathione levels and erythrocyte phosphatidylserine (PS) expression in sickle cell patients (SCD) as compared to healthy controls (CTRL). a Levels of total glutathione (t-glutathione) and the reduced glutathione (GSH) are significantly lower in sickle cell patients (gray bars) than in race-matched healthy controls (black bars). The oxidized disulfide form of glutathione (GSSG) and GSH/GSSG ratios were comparable between patients and controls. b Sickle erythrocytes (SCD, gray bar) have a significantly higher outer membrane PS expression as compared to erythrocytes of race matched healthy controls (CTRL, black bar)
Fig. 3
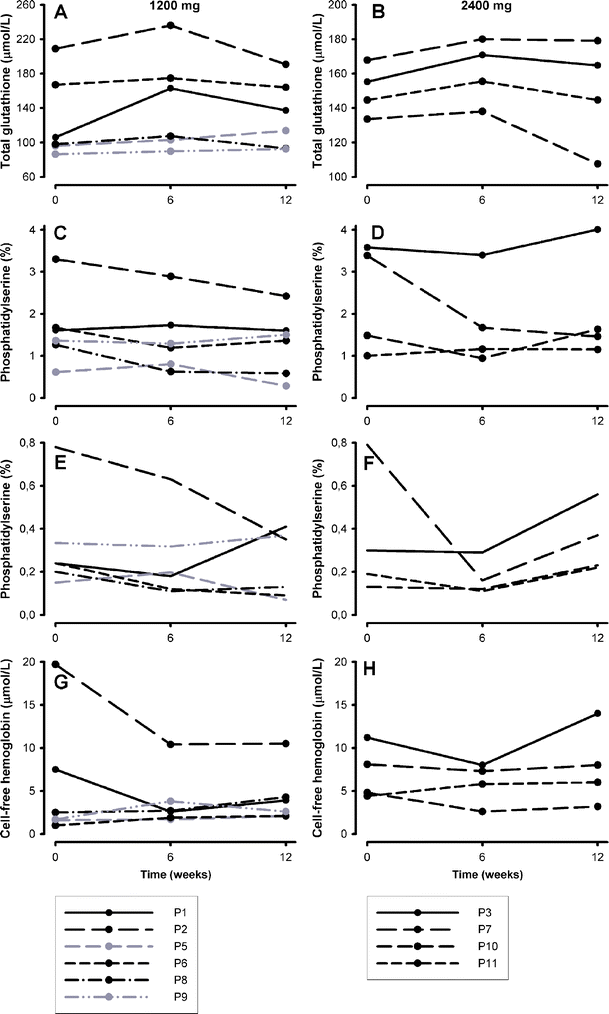
Total glutathione levels, erythrocyte phosphatidylserine (PS) expression, and cell-free hemoglobin levels at baseline, after 6 weeks of NAC treatment and 6 weeks after cessation of NAC. Panels a and b total gluathione, panels c and d erythrocyte PS expression, panels e and f erythrocytes with very positive external PS expression (>1 log greater than the PS negative erythrocytes) and panels g and h cell-free hemoglobin. Panels on the left, 1,200 mg NAC; panels on the right, 2,400 mg
Red blood cell PS expression was significantly higher in sickle cell patients as compared to healthy controls (Fig. 2b) and decreased after 6 weeks of NAC treatment in patients of both 1,200 mg (from 1.64 (1.35–2.89)% to 1.46 (0.76–2.60)%) and 2,400 mg (from 2.44 (1.12–3.53)% to 1.42 (1.00–2.97)%) NAC groups (Fig. 3, panels c and d) reaching statistical significance when analyzing both dose groups together (from 1.64 (1.32–3.37)% to 1.43 (1.00–2.6)%), P = 0.036). Also, the percentage of sickle erythrocytes with very high external PS expression (more than one log greater than the PS negative erythrocytes) decreased during the treatment and returned towards baseline values after cessation of NAC treatment (Fig. 3, panels e and f). The decrease was statistically significant in the 2,400 mg group (P = 0.039) and when analyzing both dose groups together (P = 0.030).
Plasma levels of cell-free hemoglobin decreased after NAC treatment and returned towards baseline levels after cessation of the treatment in patients of both 1,200 (from 5.0 (1.4–16.7) to 2.7 (2.1–8.5) μmol/l) and 2,400 mg (from 6.5 (4.5–10.4) μmol/l to 6.5 (3.4–7.8) μmol/l) groups, though the differences were not statistically significant (Fig. 3, panels g and h). Both pentosidine and CML were significantly higher in sickle cell patients at baseline as compared to healthy controls, and CML decreased significantly after 6 weeks of treatment when analyzing the two dose groups together (Fig. 4). None of the other measured parameters changed during the treatment period (Table 1).
Fig. 4
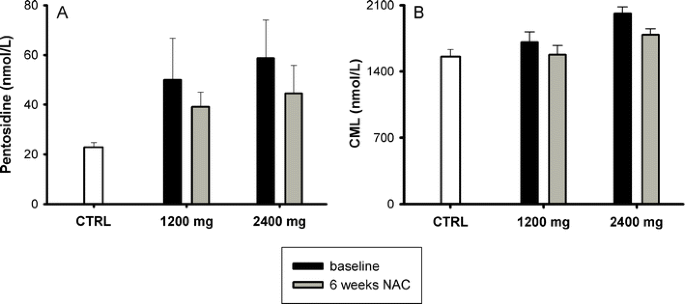
Plasma levels of AGEs (pentosidine and N ε-(carboxy-methyl)lysine (CML)) in controls (CTRL; white bar) and sickle cell patients (SCD) at baseline (black bar) and after 6 weeks (gray bar) _N_-acetylcysteine (NAC) treatment. a Baseline pentosidine levels were higher in sickle cell patients than in controls (P < 0.0001). Pentosidine decreased after 6 weeks NAC treatment in both 1,200 and 2,400 mg groups, though the differences were not statistically significant. b CML levels at baseline were also higher in sickle cell patients than in controls (P = 0.019) and decreased after 6 weeks NAC treatment in both groups. Means ± SEM
Table 1 Markers of endothelial and coagulation activation and NO bioavailability before and after NAC treatment
During the treatment period, none of the study patients was admitted with SCD related complications. The daily pain score did not change during treatment (data not shown).
Discussion
In this pilot study, 6 weeks of NAC treatment seemed to reduce oxidative stress in SCD, as reflected by reduced red cell membrane PS expression. Also, plasma levels of AGEs and cell-free hemoglobin seemed to decrease during NAC treatment. None of the other primary end points changed during the treatment period.
Through enzymatic reactions PS is normally restricted to the inner monolayer of the cell membrane [31]. Increased intracellular generation of ROS leads to (per-)oxidative damage to the erythrocyte inner membrane and proteins responsible for maintaining normal PS asymmetry, resulting in abnormal PS externalization [32]. NAC treatment seemed to result in a decrease of surface membrane PS expression with both 1,200 and 2,400 mg dose groups. With a reduction in red cell membrane damage, hemolysis would be expected to be reduced. While the standard markers of hemolysis did not change, there was a decrease in plasma levels of cell-free hemoglobin, though the differences were not statistically significant [28]. Downstream events of pathophysiological PS exposure such as thrombin generation and increased expression of sVCAM-1 did not change [33, 34]. Given large variability of such parameters, we cannot rule out that a potential ameliorating effect of NAC supplementation on endothelial and coagulation activation has been missed due to the small number of included patients.
Increased production and tissue accumulation of AGEs due to oxidative stress are associated with disease severity and organ complications in diabetes and inflammatory diseases [35–37]. AGE interaction with intra- and extracellular tissue structures leads to distortion of normal tissue architecture [23], resulting in (micro-)vasculature basement membrane thickening with reduced vascular wall elasticity, reduced filtration rate across the vessel lumen and diminished arteriolar vasodilatory response all contributing to tissue ischemia [37, 38]. Furthermore, the interaction of AGEs with their receptor (RAGE) enhances the production of pro-inflammatory cytokines, adhesion molecules, and more oxidants, inducing a pro-inflammatory response and further increasing oxidative stress [35]. Steady state plasma AGEs are increased in both children and adults with SCD, and we have recently demonstrated AGEs to be significantly associated with both the degree of hemolysis and the presence of hemolysis-related organ complications in adult sickle cell patients [18]. Somjee et al. [18] reported a strong inverse correlation between plasma AGEs and GSH levels in sickle erythrocytes. Decreased levels of GSH may play an important role in the increased formation of AGEs since GSH is an essential cofactor for glyoxalase 1, the enzyme that detoxifies the major AGE precursor methylglyoxal to _S_-d-lactoyl-glutathione [39]. The decrease in AGE levels during NAC treatment could suggest that production and tissue accumulation of AGEs and thus oxidative tissue damage can be reduced by enhancing GSH production with NAC treatment.
In contrast to the study by Pace et al. [21] in which glutathione increments were only observed in patients treated with 2,400 mg NAC during 6 weeks, increases in glutathione levels in this study were comparable in both 1,200 and 2,400 mg dose groups. A possible explanation for this discrepancy could be of methodological nature. The comparable glutathione increments with concurrent reductions in erythrocyte PS expression and plasma AGEs in both dose groups may suggest that higher dose than 1,200 mg NAC per day may not be needed. This is an important finding as gastro-intestinal side effects may be dose-limiting (as observed in one patient in the current study).
The obvious limitation of our study is the small number of patients in a pilot study lacking a control (placebo) group as well as the short duration of treatment. Nonetheless, the presented data seem to indicate gradual decrease in red cell surface PS expression, plasma levels of AGEs and cell-free hemoglobin with subsequent increments after NAC cessation. Obviously, these findings need confirmation in larger randomized placebo-controlled clinical trials. One of the 11 patients had HbS-β0-thalassemia. The excess of alpha-globin might influence the degree of oxidative stress in this patient. However, both sickle cell and beta-thalassaemia erythrocytes are associated with oxidative stress [40, 41]. Furthermore, disease severity in patients with HbS-β0-thalassemia is comparable with those in HbSS [42–44]. _N_-acetylcysteine amide, an amide form of _N_-acetylcysteine has been shown to reduce oxidative stress in beta thalassemia red blood cells [45]. Exclusion of this patient does not alter the results (data not shown).
In conclusion, _N_-acetylcysteine treatment of sickle cell patients seems to reduce erythrocyte PS expression, plasma AGEs, and cell-free hemoglobin levels. Given the wide availability, safety and low cost of NAC and the findings in this pilot study, we are of the opinion that potential of NAC as a “supportive care therapeutic” in SCD deserves further study.
References
- Nur E, Brandjes DP, Schnog JJ et al (2010) Plasma levels of advanced glycation end products are associated with haemolysis-related organ complications in sickle cell patients. Br J Haematol 151:62–69
Article CAS PubMed Google Scholar - Klings ES, Farber HW (2001) Role of free radicals in the pathogenesis of acute chest syndrome in sickle cell disease. Respir Res 2:280–285
Article CAS PubMed PubMed Central Google Scholar - Morris CR, Suh JH, Hagar W et al (2008) Erythrocyte glutamine depletion, altered redox environment, and pulmonary hypertension in sickle cell disease. Blood 111:402–410
Article CAS PubMed PubMed Central Google Scholar - Nath KA, Grande JP, Croatt AJ et al (2005) Transgenic sickle mice are markedly sensitive to renal ischemia-reperfusion injury. Am J Pathol 166:963–972
Article CAS PubMed PubMed Central Google Scholar - Sheng K, Shariff M, Hebbel RP (1998) Comparative oxidation of hemoglobins A and S. Blood 91:3467–3470
Article CAS PubMed Google Scholar - Hebbel RP, Eaton JW, Balasingam M et al (1982) Spontaneous oxygen radical generation by sickle erythrocytes. J Clin Investig 70:1253–1259
Article CAS PubMed Google Scholar - Nagababu E, Fabry ME, Nagel RL et al (2008) Heme degradation and oxidative stress in murine models for hemoglobinopathies: thalassemia, sickle cell disease and hemoglobin C disease. Blood Cells Mol Dis 41:61–66
Article CAS Google Scholar - Fibach E, Rachmilewitz E (2008) The role of oxidative stress in hemolytic anemia. Curr Mol Med 8:609–619
Article CAS PubMed Google Scholar - Jeney V, Balla J, Yachie A et al (2002) Pro-oxidant and cytotoxic effects of circulating heme. Blood 100:879–887
Article CAS PubMed Google Scholar - Akohoue SA, Shankar S, Milne GL et al (2007) Energy expenditure, inflammation, and oxidative stress in steady-state adolescents with sickle cell anemia. Pediatr Res 61:233–238
Article CAS PubMed Google Scholar - Ataga KI, Orringer EP (2003) Hypercoagulability in sickle cell disease: a curious paradox. Am J Med 115:721–728
Article CAS PubMed Google Scholar - Helley D, Eldor A, Girot R et al (1996) Increased procoagulant activity of red blood cells from patients with homozygous sickle cell disease and beta-thalassemia. Thromb Haemost 76:322–327
Article CAS PubMed Google Scholar - Beckman JS, Koppenol WH (1996) Nitric oxide, superoxide, and peroxynitrite: the good, the bad, and ugly. Am J Physiol 271:C1424–C1437
Article CAS PubMed Google Scholar - Landburg PP, Teerlink T, Muskiet FA et al (2008) Plasma concentrations of asymmetric dimethylarginine, an endogenous nitric oxide synthase inhibitor, are elevated in sickle cell patients but do not increase further during painful crisis. Am J Hematol 83:577–579
Article CAS PubMed Google Scholar - Jones DP (2002) Redox potential of GSH/GSSG couple: assay and biological significance. Methods Enzymol 348:93–112
Article CAS PubMed Google Scholar - Beutler E (1989) Nutritional and metabolic aspects of glutathione. Annu Rev Nutr 9:287–302
Article CAS PubMed Google Scholar - Reid M, Jahoor F (2001) Glutathione in disease. Curr Opin Clin Nutr Metab Care 4:65–71
Article CAS PubMed Google Scholar - Somjee SS, Warrier RP, Thomson JL et al (2005) Advanced glycation end-products in sickle cell anaemia. Br J Haematol 128:112–118
Article CAS PubMed Google Scholar - Zafarullah M, Li WQ, Sylvester J et al (2003) Molecular mechanisms of _N_-acetylcysteine actions. Cell Mol Life Sci 60:6–20
Article CAS PubMed Google Scholar - Faintuch J, Aguilar PB, Nadalin W (1999) Relevance of _N_-acetylcysteine in clinical practice: fact, myth or consequence? Nutrition 15:177–179
Article CAS PubMed Google Scholar - Pace BS, Shartava A, Pack-Mabien A et al (2003) Effects of _N_-acetylcysteine on dense cell formation in sickle cell disease. Am J Hematol 73:26–32
Article CAS PubMed Google Scholar - Frenette PS (2002) Sickle cell vaso-occlusion: multistep and multicellular paradigm. Curr Opin Hematol 9:101–106
Article PubMed Google Scholar - Goldin A, Beckman JA, Schmidt AM et al (2006) Advanced glycation end products: sparking the development of diabetic vascular injury. Circulation 114:597–605
Article CAS PubMed Google Scholar - Teerlink T, Nijveldt RJ, de Jong S et al (2002) Determination of arginine, asymmetric dimethylarginine, and symmetric dimethylarginine in human plasma and other biological samples by high-performance liquid chromatography. Anal Biochem 303:131–137
Article CAS PubMed Google Scholar - de Jong S, Teerlink T (2006) Analysis of asymmetric dimethylarginine in plasma by HPLC using a monolithic column. Anal Biochem 353:287–289
Article PubMed CAS Google Scholar - Tietze F (1969) Enzymic method for quantitative determination of nanogram amounts of total and oxidized glutathione: applications to mammalian blood and other tissues. Anal Biochem 27:502–522.
Article CAS PubMed Google Scholar - Sacchetta P, Di CD, Federici G (1986) Alkaline hydrolysis of N-ethylmaleimide allows a rapid assay of glutathione disulfide in biological samples. Anal Biochem 154:205–208.
Article CAS PubMed Google Scholar - Kahn SE, Watkins BF, Bermes EW Jr (1981) An evaluation of a spectrophotometric scanning technique for measurement of plasma hemoglobin. Ann Clin Lab Sci 11:126–131
CAS PubMed Google Scholar - Scheijen JL, van de Waarenburg MP, Stehouwer CD et al (2009) Measurement of pentosidine in human plasma protein by a single-column high-performance liquid chromatography method with fluorescence detection. J Chromatogr B Anal Technol Biomed Life Sci 877:610–614
Article CAS Google Scholar - Teerlink T, Barto R, Ten Brink HJ et al (2004) Measurement of nepsilon-(carboxymethyl)lysine and nepsilon-(carboxyethyl)lysine in human plasma protein by stable-isotope-dilution tandem mass spectrometry. Clin Chem 50:1222–1228
Article CAS PubMed Google Scholar - Zwaal RF, Schroit AJ (1997) Pathophysiologic implications of membrane phospholipid asymmetry in blood cells. Blood 89:1121–1132
Article CAS PubMed Google Scholar - Banerjee T, Kuypers FA (2004) Reactive oxygen species and phosphatidylserine externalization in murine sickle red cells. Br J Haematol 124:391–402
Article CAS PubMed Google Scholar - Manodori AB, Barabino GA, Lubin BH et al (2000) Adherence of phosphatidylserine-exposing erythrocytes to endothelial matrix thrombospondin. Blood 95:1293–1300
Article CAS PubMed Google Scholar - Setty BN, Kulkarni S, Stuart MJ (2002) Role of erythrocyte phosphatidylserine in sickle red cell-endothelial adhesion. Blood 99:1564–1571
Article CAS PubMed Google Scholar - Ahmed N (2005) Advanced glycation endproducts—role in pathology of diabetic complications. Diabetes Res Clin Pract 67:3–21
Article CAS PubMed Google Scholar - Miyata T, Ishiguro N, Yasuda Y et al (1998) Increased pentosidine, an advanced glycation end product, in plasma and synovial fluid from patients with rheumatoid arthritis and its relation with inflammatory markers. Biochem Biophys Res Commun 244:45–49
Article CAS PubMed Google Scholar - Greenwald SE (2007) Ageing of the conduit arteries. J Pathol 211:157–172
Article CAS PubMed Google Scholar - Aronson D (2003) Cross-linking of glycated collagen in the pathogenesis of arterial and myocardial stiffening of aging and diabetes. J Hypertens 21:3–12
Article CAS PubMed Google Scholar - Brouwers O, Niessen PM, Haenen G et al (2010) Hyperglycaemia-induced impairment of endothelium-dependent vasorelaxation in rat mesenteric arteries is mediated by intracellular methylglyoxal levels in a pathway dependent on oxidative stress. Diabetologia 53:989–1000
Article CAS PubMed PubMed Central Google Scholar - Amer J, Ghoti H, Rachmilewitz E et al (2006) Red blood cells, platelets and polymorphonuclear neutrophils of patients with sickle cell disease exhibit oxidative stress that can be ameliorated by antioxidants. Br J Haematol 132:108–113
Article CAS PubMed Google Scholar - Amer J, Goldfarb A, Fibach E (2004) Flow cytometric analysis of the oxidative status of normal and thalassemic red blood cells. Cytom A 60:73–80
Article Google Scholar - Morris CR (2008) Mechanisms of vasculopathy in sickle cell disease and thalassemia. Hematol Am Soc Hematol Educ Prog 177–185
Article Google Scholar - van den Tweel XW, van der Lee JH, Heijboer H et al (2010) Development and validation of a pediatric severity index for sickle cell patients. Am J Hematol 85:746–751
Article PubMed Google Scholar - Powars DR, Hiti A, Ramicone E et al (2002) Outcome in hemoglobin SC disease: a four-decade observational study of clinical, hematologic, and genetic factors. Am J Hematol 70:206–215
Article CAS PubMed Google Scholar - Amer J, Atlas D, Fibach E (2007) _N_-acetylcysteine amide (AD4) attenuates oxidative stress in beta-thalassemia blood cells. Biochim Biophys Acta 1780:249–255
Article PubMed CAS Google Scholar
Acknowledgements
None.
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Author information
Authors and Affiliations
- Department of Internal Medicine, Slotervaart Hospital, Amsterdam, The Netherlands
Erfan Nur, Dees P. Brandjes, Hans-Martin Otten & John-John B. Schnog - Department of Hematology, Academic Medical Center, Amsterdam, The Netherlands
Erfan Nur, Ludo M. Evers & Bart J. Biemond - Department of Clinical Chemistry, VU University Medical Center, Amsterdam, The Netherlands
Tom Teerlink - Tytgat Institute for Liver and Intestinal Research, Academic Medical Center, Amsterdam, The Netherlands
Ronald P. J. Oude Elferink - Pathology and Laboratory Medicine, University Medical Center Groningen, Groningen, The Netherlands
Frits Muskiet - Laboratory of Clinical Thrombosis and Hemostasis, Academic Hospital Maastricht, Maastricht, The Netherlands
Hugo ten Cate - Immunology Laboratory, Red Cross Blood Bank Foundation, Curaçao, Netherlands Antilles
Ashley J. Duits & John-John B. Schnog - Department of Hematology/Medical Oncology, Sint Elisabeth Hospital, Curaçao, Netherlands Antilles
John-John B. Schnog
Authors
- Erfan Nur
You can also search for this author inPubMed Google Scholar - Dees P. Brandjes
You can also search for this author inPubMed Google Scholar - Tom Teerlink
You can also search for this author inPubMed Google Scholar - Hans-Martin Otten
You can also search for this author inPubMed Google Scholar - Ronald P. J. Oude Elferink
You can also search for this author inPubMed Google Scholar - Frits Muskiet
You can also search for this author inPubMed Google Scholar - Ludo M. Evers
You can also search for this author inPubMed Google Scholar - Hugo ten Cate
You can also search for this author inPubMed Google Scholar - Bart J. Biemond
You can also search for this author inPubMed Google Scholar - Ashley J. Duits
You can also search for this author inPubMed Google Scholar - John-John B. Schnog
You can also search for this author inPubMed Google Scholar
Consortia
on behalf of the CURAMA study group
Corresponding author
Correspondence toJohn-John B. Schnog.
Additional information
The CURAMA study group is a collaborative effort studying sickle cell disease in the Netherlands Antilles and the Netherlands. Participating centers: The Red Cross Blood Bank Foundation, Curaçao, Netherlands Antilles; The Antillean Institute for Health Research, Curaçao, Netherlands Antilles, The Department of Internal Medicine, Slotervaart Hospital, Amsterdam, the Netherlands; the Department of Vascular Medicine and the Department of Hematology, Academic Medical Center, Amsterdam, the Netherlands; the Department of Hematology, Erasmus Medical Center, Rotterdam, the Netherlands; Pathology and Laboratory Medicine, University Medical Center Groningen the Netherlands; the Department of Internal Medicine, the Laboratory of Clinical Thrombosis and Hemostasis, and the Cardiovascular Research Institute, Academic Hospital Maastricht, the Netherlands.
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License (https://creativecommons.org/licenses/by-nc/2.0), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Nur, E., Brandjes, D.P., Teerlink, T. et al. _N_-acetylcysteine reduces oxidative stress in sickle cell patients.Ann Hematol 91, 1097–1105 (2012). https://doi.org/10.1007/s00277-011-1404-z
- Received: 11 September 2011
- Accepted: 30 December 2011
- Published: 10 February 2012
- Issue Date: July 2012
- DOI: https://doi.org/10.1007/s00277-011-1404-z