Rapid, reliable, and reproducible molecular sub-grouping of clinical medulloblastoma samples (original) (raw)
Abstract
The diagnosis of medulloblastoma likely encompasses several distinct entities, with recent evidence for the existence of at least four unique molecular subgroups that exhibit distinct genetic, transcriptional, demographic, and clinical features. Assignment of molecular subgroup through routine profiling of high-quality RNA on expression microarrays is likely impractical in the clinical setting. The planning and execution of medulloblastoma clinical trials that stratify by subgroup, or which are targeted to a specific subgroup requires technologies that can be economically, rapidly, reliably, and reproducibly applied to formalin-fixed paraffin embedded (FFPE) specimens. In the current study, we have developed an assay that accurately measures the expression level of 22 medulloblastoma subgroup-specific signature genes (CodeSet) using nanoString nCounter Technology. Comparison of the nanoString assay with Affymetrix expression array data on a training series of 101 medulloblastomas of known subgroup demonstrated a high concordance (Pearson correlation r = 0.86). The assay was validated on a second set of 130 non-overlapping medulloblastomas of known subgroup, correctly assigning 98% (127/130) of tumors to the appropriate subgroup. Reproducibility was demonstrated by repeating the assay in three independent laboratories in Canada, the United States, and Switzerland. Finally, the nanoString assay could confidently predict subgroup in 88% of recent FFPE cases, of which 100% had accurate subgroup assignment. We present an assay based on nanoString technology that is capable of rapidly, reliably, and reproducibly assigning clinical FFPE medulloblastoma samples to their molecular subgroup, and which is highly suited for future medulloblastoma clinical trials.
Similar content being viewed by others
Introduction
Currently, patients enrolled on clinical trials for medulloblastoma are stratified on the basis of clinical variables such as age, extent of resection, presence of metastases, and histology. Recently, several publications have reported that the histological entity known as medulloblastoma comprises several distinct molecular variants or subgroups [1, 6, 12, 17]. Despite variation in the number and nomenclature of the subgroups identified, the current consensus is that medulloblastoma comprises four core subgroups (i.e., WNT, SHH, Group C, and Group D), with mounting evidence for clinically relevant substructure (subtypes within the subgroups) [1, 11]. Each subgroup exhibits distinct demographics, transcriptomics, genomics, and clinical outcomes [1, 12]. While some subgroups are well treated, or debatably even over-treated using current protocols (i.e., WNT medulloblastomas), others have a very poor outcome (i.e., Group C medulloblastomas). Additionally, as the subgroups have very different molecular genetic profiles, any successful strategies for targeted therapy will likely be subgroup specific (i.e., SMO inhibitors for SHH subgroup tumors). Although the retrospective classification of various medulloblastoma cohorts into molecular subgroups has been scientifically insightful, medulloblastoma sub-grouping has not yet been applied in the setting of a prospective clinical trial for either patient stratification or patient selection for targeted therapy.
There is currently no well-accepted gold standard test for medulloblastoma subgroup assignment. The methodology used in most of the published literature on medulloblastoma subgroups has been the analysis of high-quality RNA from flash-frozen samples that were analyzed using genome-wide transcriptional microarrays. Although an excellent tool for retrospective research studies, gene expression microarray profiling is likely inappropriate and inadequate for routine clinical use or for clinical trials due to the need for large amounts of high-quality RNA (from frozen tumor tissue), lot-to-lot variability of microarrays, bioinformatic complexity, and relatively high cost. Specifically, RNA isolated from formalin-fixed paraffin embedded (FFPE) medulloblastoma samples is fragmented, and not suitable for hybridization to expression microarrays. In both routine clinical settings and clinical trials, a rapid test completion time is critical, making microarray platforms an inefficient diagnostic tool.
In contrast, medulloblastoma subgroup assignment using immunohistochemistry (IHC) performed on FFPE cases has shown recent promise. We recently reported a four-antibody protocol for classification of medulloblastomas, and applied this method to a large series (n = 294) of FFPE medulloblastomas on tissue microarrays (TMAs), effectively classifying ~98% of samples [12]. Ellison et al. [3] recently reported an IHC-based assay for classifying medulloblastomas into WNT, SHH, and non-WNT/SHH subgroups using a distinct set of antibodies. Challenges in bringing IHC to the clinic for subgroup assignment remain due to lot-to-lot variability of antibodies, inter-institutional differences in tissue fixation and embedding, technical variations of IHC, and inter and intra-observer variability in image interpretation. The inclusion of IHC markers for subgroup ascertainment in future clinical trials would likely be complimented by another orthogonal technology, to confirm subgroup affiliation as identified by IHC, and provide treating clinicians with confidence that the correct subgroup has been assigned. By its very nature, IHC is likely limited to one or two markers per subgroup, and these markers must be proteins excluding the use of non-coding RNAs as markers. While some of the described antibodies will likely become widely used as clinical tests, complimentary and confirmatory technologies may be required in the setting of a clinical trial.
To develop and optimize a more rapid, reliable, reproducible, and economical method for medulloblastoma classification, we have taken advantage of the recently described nanoString nCounter System, a non-enzymatic multiplexed assay that uses sequence-specific probes to digitally measure target abundance (i.e., mRNA) within a given sample [5, 7, 9]. Based on nanoString technology, and using information from existing gene expression array data [12], we designed a custom CodeSet (i.e., probe library) consisting of interrogating probes against 22 medulloblastoma subgroup-specific signature genes. We tested our nanoString assay on our own medulloblastoma series of known subgroup affiliation prior to validation of the assay on three non-overlapping medulloblastoma cohorts with known subgroup affiliation. Finally, the assay was applied to a large series of FFPE medulloblastomas to establish its applicability in the classification of routine clinical samples as would be encountered in the setting of a prospective clinical trial.
Patients and methods
Patient samples
All samples were obtained in accordance with the Research Ethics Board at the Hospital for Sick Children (Toronto, Canada). Primary medulloblastomas comprising the training series for nanoString (n = 101) have been previously described [10, 12, 13]. Samples contributing to the validation series (n = 130) have been previously described and were obtained as total RNA extracted from fresh-frozen tissue from the DKFZ (Heidelberg, Germany; Remke series, n = 55), the Dana-Farber Cancer Institute (Boston, USA; Cho series, n = 39) [1], and the Academic Medical Center (Amsterdam, the Netherlands; Kool series, n = 36) [6]. Formalin-fixed paraffin embedded (FFPE) cases (n = 84) were obtained as paraffin sections from the Hospital for Sick Children (Toronto, Canada; n = 34), Johns Hopkins University (Baltimore, USA; n = 25), and the DKFZ (Heidelberg, Germany; n = 25).
NanoString CodeSet design and expression quantification
Signature genes for each medulloblastoma subgroup were included in the CodeSet on the basis of their observed subgroup-specific expression, as previously determined by Affymetrix exon array analysis [10, 12]. Specifically, conventional t test statistics restricted on the proportion of false discoveries (FDR) were employed to compare each subgroup to the remaining three subgroups in order to identify the most highly significant, differentially expressed genes. The CodeSet was designed to consist of a total of 25 genes with 5–6 signature genes included for each subgroup: WNT (WIF1, TNC, GAD1, DKK2, EMX2), SHH (PDLIM3, EYA1, HHIP, ATOH1, SFRP1), Group C (IMPG2, GABRA5, EGFL11, NRL, MAB21L2, NPR3), Group D (KCNA1, EOMES, KHDRBS2, RBM24, UNC5D, OAS1). Three housekeeping genes (ACTB, GAPDH, and LDHA) were also included in the CodeSet for biological normalization purposes. Probe sets for each gene in the CodeSet were designed and synthesized at nanoString Technologies.
Total RNA (100 ng) from fresh-frozen tissue and FFPE material was analyzed using the nanoString nCounter Analysis System at the University Health Network Microarray Centre (Toronto, Canada), the Oncogenomics Core Facility at the University of Miami (Miami, USA), and the Frontiers in Genetics Facility at the University of Geneva (Geneva, Switzerland). All procedures related to mRNA quantification including sample preparation, hybridization, detection, and scanning were carried out as recommended by nanoString Technologies.
Sample processing
Total RNA was extracted from fresh-frozen tissue using the Trizol method (Invitrogen) according to the manufacturer’s instructions. For FFPE samples, ~3–5 paraffin sections per sample were first deparaffinized with xylene prior to RNA extraction using the RNeasy FFPE kit (Qiagen) as directed by the manufacturer. RNA concentration was measured using a Nanodrop 1000 instrument (Nanodrop) and RNA integrity was assessed using an Agilent 2100 bioanalyzer at The Centre for Applied Genomics at the Hospital for Sick Children (Toronto, Canada).
NanoString data processing and class prediction analysis
Raw nanoString counts for each gene within each experiment were subjected to a technical normalization using the counts obtained for positive control probe sets prior to a biological normalization using the three housekeeping genes included in the CodeSet. Normalized data was log2-transformed and then used as input for class prediction analysis.
A series of medulloblastomas with known subgroup affiliation (n = 101) were used to establish a training dataset for subsequent class prediction analysis of independent cohorts utilized in the study. Various class prediction algorithms were assessed by a tenfold cross-validation scheme, using a set of scoring indices to establish a pipeline for prediction of medulloblastoma subgroups using nanoString data derived from the training series. Based on superior performance in cross-validation analysis, the PAM method [18] was selected for all downstream class prediction analyses.
All class prediction analyses were performed in the R statistical programming environment (v2.13). Implementations of the class prediction algorithms were imported from the following R packages: MASS v7.3 (linear discriminant analysis; LDA), class v7.3 (k-nearest neighbor; KNN), e1071 v1.5 (support vector machine; SVM), nnet v7.3 (multinomial log-linear model; MULT), and pamr v1.51 (prediction analysis for microarrays; PAM) [18]. During cross-validation, the training set of 101 samples was randomly split into 10 partitions. Each class predictor was trained on nine of the partitions, and the performance of the predictor was subsequently tested on the one remaining partition. Each of the 10 partitions was used as the testing set in turn for a round of cross-validation, for a total of 10,000 rounds of cross-validation, which was repeated three times with reproducible results.
The scoring indices used during testing were accuracy, Jaccard similarity index, Rand index, adjusted Rand index, and Fowlkes–Mallows index. The latter four indices are different indices for determining the similarity between two groupings, which are the known and predicted classifications of samples in the current analysis. These indices serve as more stringent measures of accuracy in multi-class prediction. Aside from the accuracy measures (validity), the reliabilities of the predictors were also determined using Shannon entropy as a measure of uncertainty. Predictors with varying predicted classes for the same sample across the cross-validation rounds have higher entropy values, and are hence less reliable.
Since the model parameters for SVM can affect the prediction performance, these parameters were optimized by a grid search in a separate round of cross-validation. The ranges of searched parameter values were: [2−5, 215] for C; [2−15, 23] for gamma; [2, 8] for degree; [−1, 1] for coef0. Further, SVM using different kernels (linear, radial basis, polynomial, and sigmoid) were assessed, and the kernel with the best performance was selected. Similarly for KNN, the best model was selected from models with different k.
Regression analysis of prediction accuracy
Cumulative prediction accuracy was modeled as a function of FFPE sample age. The prediction accuracies were first calculated for each sample age year-group. The cumulative accuracies were then determined by calculating the cumulative sum of the accuracies, weighted by the size of each year-group. The data were fitted using a 5-parameter logistic regression model, as implemented in the drc v2.1 R package. The maximum asymptote parameter (D) was constrained at 1 in order to reflect the high accuracy the predictor achieved with recent FFPE samples.
RNA integrity assessment
RNA derived from FFPE material was subjected to Agilent Bioanalyzer analysis to determine RNA integrity. Smear analysis was performed using the Agilent 2100 expert software to determine the proportion of RNA ≥300 nucleotides (nt) within a given sample.
Results
Establishment of a nanoString assay for medulloblastoma subgroup identification
To identify a set of genes that would facilitate the prospective classification of medulloblastomas into the four core subgroups of the disease, we re-analyzed gene expression array data from a large series of primary cases of known subgroup [10, 12]. Supervised analysis identified signature genes that demonstrate subgroup-specific expression patterns across the four molecular subgroups (Table 1). From this data, we selected 5–6 highly significant signature genes per subgroup for inclusion in a custom nanoString CodeSet (probe library) for downstream classification analysis (Supplementary Figure 1). As an initial evaluation of our nanoString CodeSet’s capacity to measure the gene expression level of medulloblastoma signature genes, we first profiled a series of 101 primary cases (training series) for which we also had matching Affymetrix exon array data [10, 12]. Comparison of the newly generated nanoString data with the published exon array data for this training series showed remarkable similarity between the two datasets (Fig. 1a; Supplementary Figure 2). Pearson correlation analysis confirmed a high degree of concordance (r = 0.86) between the two platforms (Fig. 1b; Supplementary Figure 3). Using a series of well-described class prediction algorithms, we evaluated the capacity of our custom CodeSet to accurately classify samples within the training series into the correct subgroup. Following 10,000 iterations in cross-validation analysis, the PAM algorithm [18] exhibited the most accurate class prediction results, reliably assigning 98/101 (~97%) cases in the training series (Fig. 1c, d; Supplementary Figure 4). In this cross-validation exercise, PAM outperformed other commonly used class prediction methods such as Support Vector Machine (SVM) and K-Nearest Neighbor (KNN). Isolation of tumor RNA, performance of the nanoString assay, and bioinformatic analysis required 3–4 days for completion, at a cost of approximately $60 USD per sample. These data suggest that we have generated an assay capable of predicting medulloblastoma subgroup affiliation with high accuracy that is additionally both rapid and economical.
Fig. 1
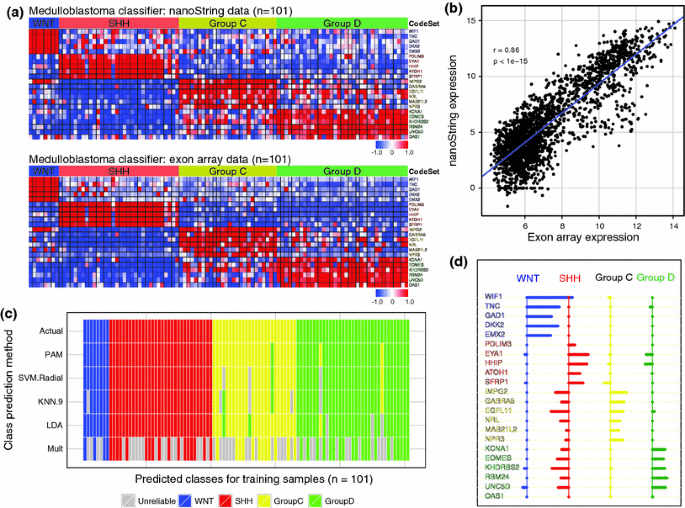
A nanoString CodeSet for medulloblastoma subgroup assignment. a Expression heatmaps for 22 medulloblastoma signature genes in a series of 101 primary medulloblastomas (training series) profiled by both the nanoString nCounter System (upper panel) and by Affymetrix exon array (lower panel). The 22 signature genes comprise the nanoString CodeSet used throughout the study. b Pearson correlation analysis of nanoString expression data versus Affymetrix expression data for the 22 signature genes shown in a across the training series of 101 medulloblastomas. r = Pearson correlation. c Virtual heatmap depicting results of cross-validation analysis for multiple class prediction algorithms evaluated on the training series for medulloblastoma subgroup prediction accuracy. Samples are ordered horizontally according to their known subgroup affiliation (‘Actual’). Results represent the consensus subgroup assignment following 10,000 iterations and discordant cases are labeled according to the subgroup in which they were erroneously classified. Samples labeled in grey represent those in which a single subgroup could not be reliably assigned. d Centroid plot for the nanoString CodeSet as determined by the PAM algorithm. Genes are grouped according to the subgroup for which they exhibit a positive centroid value
Validation of the nanoString classifier on multiple published medulloblastoma cohorts
To validate our nanoString CodeSet approach to medulloblastoma subgroup prediction, we obtained three independent validation cohorts (n = 130) with well-documented molecular profiles including subgroup affiliation as determined in recently published gene expression array studies [1, 6]. NanoString profiling of the 130 cases comprising this validation series followed by implementation of PAM resulted in an impressively high concordance between known and predicted molecular subgroup affiliations across the three independent cohorts (Fig. 2a–d; Supplementary Table 1; concordance = 127/130, ~98%). There was only a single subgroup misclassification in the Cho series (1/39), two misclassified cases in the Kool series (2/36), and none in the Remke series (0/55). Distribution of validation cases appeared to faithfully represent the relative subgroup frequencies that have been reported by multiple studies in the literature (Fig. 2d) [1, 6, 12]. For sample misclassifications, there was no apparent bias towards one particular subgroup, although it is important to note that all Group C medulloblastomas in this series (n = 28) were accurately classified using our assay (Fig. 2d). As Group C medulloblastomas have an inferior prognosis [1, 12], it is encouraging that cases of this affiliation were classified with high accuracy. Although there is no true gold standard for subgroup assignment as discussed above, we conclude that our nanoString assay is highly reliable for determination of subgroup affiliation.
Fig. 2
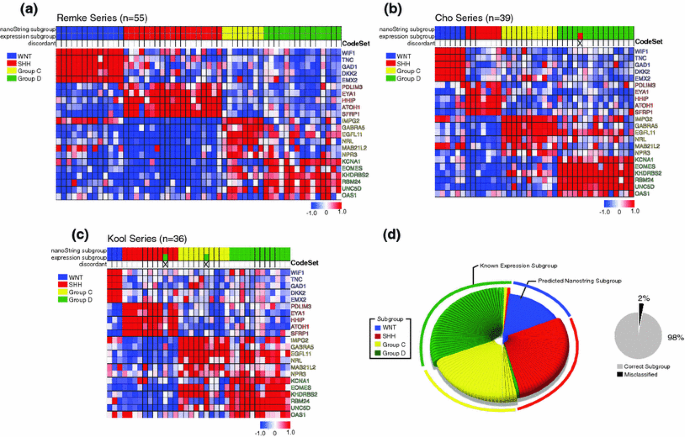
Validation of nanoString assay on multiple published medulloblastoma cohorts with known subgroup affiliation. a–c Expression heatmaps of nanoString class-predicted medulloblastomas of known subgroup status as published by Remke et al. (a), Cho et al. [1] (b), and Kool et al. [6] (c). Samples were sorted according to subgroup prediction as determined by nanoString. nanoString predicted subgroup, known expression subgroup affiliation, and erroneously classified cases are marked above the heatmap. d Left chart Pie chart showing the known subgroup distribution of medulloblastomas from the three independent cohorts analyzed in a–c (n = 130) and the class-predicted subgroup assignments as determined by nanoString profiling. Misclassified cases are marked within each pie segment according to the subgroup in which they were erroneously classified. Right chart Pie chart showing the class prediction success rate (~98%, 127/130) for the validation series
Reproducibility and cross-site validation of the nanoString CodeSet
An ideal clinical test should be consistent and reproducible across different geographic locales and in different laboratories. To determine whether our custom nanoString assay could reliably produce comparable data when performed at different technical sites, we shipped 48 samples from our training series to two additional nanoString-equipped genomic facilities at the University of Miami (USA) and the University of Geneva (Switzerland). Randomization of samples and masking of their associated subgroup status allowed us to perform class prediction analysis on the data generated in Miami and Geneva. All cases analyzed at these two international facilities were assigned to the correct molecular subgroup (Fig. 3a–c; Supplementary Table 2). Inspection of expression heatmaps from data generated at the independent nanoString facilities shows that the results are virtually indistinguishable (Fig. 3a–c). Scatterplot analysis of the Toronto data versus the data from the two validation sites established an extremely high degree of correlation (Toronto/Miami, r = 0.97; Toronto/Geneva, r = 0.98). We conclude that our nanoString approach to medulloblastoma subgroup assignment is highly reproducible across different centers, making it highly suitable in the context of a clinical trial.
Fig. 3
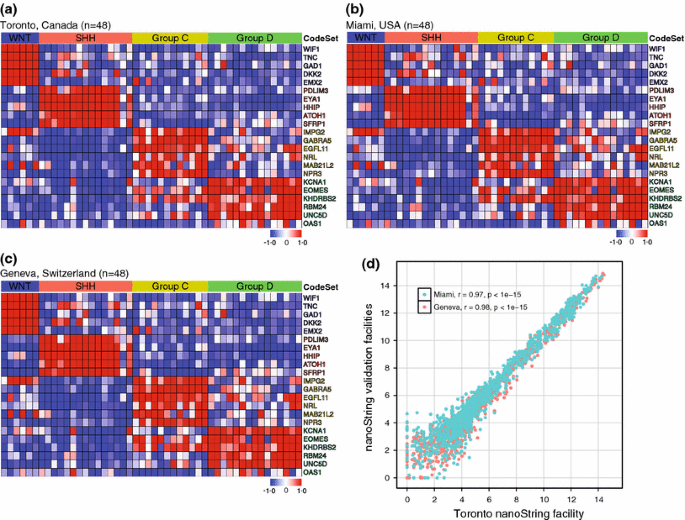
Cross-site validation of medulloblastoma classification using the nanoString CodeSet. a–c Forty-eight primary medulloblastomas of known subgroup affiliation were analyzed using the nanoString CodeSet at three independent facilities: Toronto, Canada (a), Miami, USA (b), and Geneva, Switzerland (c). Class prediction analysis of the data generated at the three independent nanoString facilities resulted in 100% sample classification accuracy. Heatmaps of the normalized nanoString data for the 48 cases are shown. d Scatterplot showing correlation of nanoString expression data generated in Toronto versus that generated at the two international validation sites (Miami, USA and Geneva, Switzerland). r = Pearson correlation
Accurate classification of archival formalin-fixed paraffin embedded (FFPE) medulloblastomas
Most conventional technologies employed for quantification of mRNA abundance (i.e., gene expression arrays, q-RT-PCR, RNA-Seq) require high-quality RNA that exhibits little to no degradation. Nucleic acid (including RNA) extracted from tissue stored as FFPE material is typically highly degraded and fragmented, and therefore not suitable for most molecular profiling platforms. As nanoString relies on relatively short pairs of 50mer probes [5], it exhibits robust performance on RNA extracted from FFPE material with results comparable to those obtained with RNA from fresh-frozen tissue [15].
We applied our nanoString assay to a series of 84 FFPE cases obtained from three independent institutions in three different countries: Hospital for Sick Children (Toronto, Canada), Johns Hopkins University (Baltimore, USA) and the DKFZ (Heidelberg, Germany). For each of these cohorts, molecular subgroup affiliation had been previously determined using either gene expression array profiling of matched frozen tissue, or IHC-based classification. Bioanalyzer analysis of FFPE-derived RNA confirmed all samples had highly degraded RNA (Supplementary Table 3, median RNA Integrity Number (RIN) value = 2.4). NanoString profiling followed by PAM class prediction resulted in accurate classification of 57/84 (~68%) FFPE cases (Supplementary Table 3), a success rate significantly lower than that achieved for fresh-frozen cases (~98%). In order to identify cases in which RNA extracted from FFPE was not of sufficient quality for nanoString to confidently assign subgroup, we established a minimal PAM probability threshold, proceeding only with those FFPE samples where PAM provided a high confidence subgroup assignment. Class prediction accuracy of validation samples decreases with the PAM probability score at two distinct stages (Fig. 4a). The initial decline in accuracy can be explained by inclusion of poor-quality samples, whereas the second decline can be attributed to deterioration of class prediction performance by PAM (Supplementary Figure 5a). The initial probability threshold was therefore set at the point just prior to the second decline in accuracy (PAM probability = 0.75). After filtering based on this probability threshold, the effect of FFPE sample age on class prediction performance was assessed in order to determine an age cut-off for FFPE samples. After limiting our analysis to FFPE cases from the last 8 years, PAM accurately classified ≥95% of cases (Fig. 4b; Supplementary Table 3).
Fig. 4
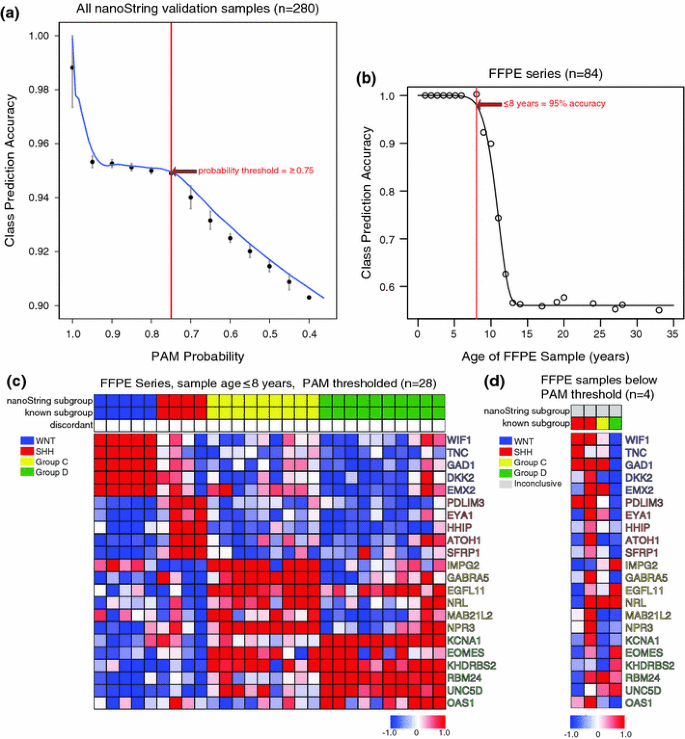
Compatibility of nanoString classification assay with formalin-fixed paraffin embedded (FFPE) material from archival samples. a Plot of nanoString class prediction accuracy for all 280 validation samples according to PAM probability score. Vertical red line denotes threshold at which PAM classification becomes unreliable. b Plot of nanoString class prediction accuracy according to sample age of archival medulloblastomas stored as FFPE material (n = 84). Samples obtained within the past 8 years exhibit an accuracy of ≥95%, as demarcated by the red vertical line on the plot. Sample age range for the FFPE series was 1–33 years, with a median sample age of 10 years. c Heatmap of nanoString data showing class prediction results for FFPE cases ≤8 years of age confidently predicted by PAM (n = 28). 28/28 cases (100%) meeting the threshold were assigned to the correct subgroup. Samples were sorted according to subgroup prediction as determined by nanoString. d NanoString data for FFPE cases ≤8 years of age that failed to meet the PAM probability threshold for subgroup assignment (n = 4). 2/4 cases that failed to meet the PAM threshold, and were not assigned to the correct subgroup
As the threshold for accurate subgroup assignment varied by subgroup, probability thresholds were re-established in a subgroup-specific manner using cases from the last 8 years (Supplementary Figure 5b). The new probability thresholds were chosen to maintain a near 100% class prediction accuracy in high-quality samples (WNT = 0.7, SHH = 0.5, Group C = 0.5, Group D = 0.5). In recent FFPE samples (≤8 years, n = 32), PAM confidently predicted subgroups for 28/32 cases (87.5%) (Fig. 4c, d). In 4/32 (12.5%) cases PAM was unable to provide a high confidence subgroup assignment, suggesting that our current nanoString assay is incapable of sub-grouping them, and that alternative methods would be necessary (Fig. 4d). Notably, 2/4 cases that failed to meet the PAM threshold were in fact accurately classified by our nanoString assay (Supplementary Table 3). For those FFPE cases in which the PAM threshold was exceeded, 28/28 (100%) were assigned to the correct subgroup (Fig. 4c). Multiple logistic regression analysis established that sample age was a more reliable predictor of class prediction accuracy than measures of RNA integrity (i.e., RIN and RNA size) (Supplementary Figure 6). These results confirm the compatibility of our custom nanoString CodeSet with recent FFPE-derived material, and strongly suggest that our nanoString assay for medulloblastoma classification is well suited to the clinical trial setting in which recent FFPE samples are readily available.
Discussion
Current criteria for risk stratification of medulloblastoma patients include patient age, metastatic status, and extent of surgical resection. Patients over the age of three with non-metastatic disease that is gross totally resected are considered average-risk, and all others deemed high-risk. This current stratification scheme fails to account for the extensive prognostic variability that exists between molecular subgroups. Therefore, the next generation of prospective clinical trials for medulloblastoma will almost certainly include molecular subgroup assignment for both patient stratification, and patient selection for targeted therapies. In particular, modulation of the intensity of therapy in a subgroup-specific manner is a very attractive approach in order to improve outcomes for patients. For example, WNT subgroup medulloblastomas are rarely metastatic and have progression-free and overall survival rates of >90% [1, 2, 4, 12, 14]; in contrast, patients with Group C medulloblastoma have a dismal prognosis [1, 12]. Molecular subgroup-based risk stratification will permit a more rational and personalized approach to patient treatment. Furthermore, targeted therapies against activated signaling pathways such as those that attenuate SHH pathway activation currently being evaluated in clinical trials [8] will benefit from subgroup-based stratification as they will likely only be effective in one of the four subgroups.
We describe a novel molecular classification method for medulloblastoma that relies on the nanoString nCounter System. This technology requires minimal RNA input (~100 ng), does not involve any enzymatic amplification, and produces expression data that are highly correlative with data generated by expression arrays. The nanoString-mediated subgroup assay described in this report was significantly more cost effective than performing the equivalent classification using an array-based approach, averaging ~$60 USD per sample for our nanoString assay compared to ~$425 USD per sample for a modern Affymetrix expression array. Using the expression pattern of only 22 medulloblastoma subgroup-specific signature genes we have established an assay that effectively assigns fresh-frozen medulloblastomas to the correct subgroup with ~98% accuracy as confirmed using three independent validation cohorts. Schwalbe et al. [16] recently described a 13-gene multiplex qPCR-based expression assay to classify medulloblastomas into either WNT, SHH, or non-WNT/SHH subgroups. Unsupervised analyses were used in this study to establish the ability of the 13-gene signature to recapitulate subgroup data previously determined in multiple published gene expression cohorts. Although this method proved capable of placing samples into WNT, SHH, and non-WNT/SHH categories, the technique was not directly evaluated on samples belonging to the published cohorts, nor did the assay attempt to make the important distinction between Group C and Group D medulloblastomas, confirmed in multiple recent studies to be both genetically and clinically distinct [1, 6, 12]. In the current study, we have obtained a subset of the same fresh-frozen RNA samples that were used in three independent microarray-based medulloblastoma sub-grouping studies and validated our nanoString assay directly on these templates (n = 130). Class prediction analysis confirmed the accuracy of our assay in ~98% of cases establishing the validity of our protocol. For samples that were misclassified, it is difficult to verify the source of the discrepancy regarding subgroup assignment, although possible explanations could be related to erroneous results of our nanoString assay, potential sample mix-ups, or erroneous classification in the original gene expression array profiling.
We previously introduced an IHC-based classification scheme for sub-grouping medulloblastoma using only four commercially available antibodies [12]. This IHC-based method is very robust in our laboratory; although challenges remain in making the technique generalizable, including variability in antibody batches, sample preparation methods, staining procedures, and inter-observer reliability. We would suggest that in the future, IHC-based methods could be used in concert with a nanoString-based assay to provide clinicians with a high confidence assignment of subgroup for clinical medulloblastoma samples. The two methods are orthogonal, and highly complimentary.
To test the reproducibility of our nanoString-based classification assay across different centers, we analyzed a series of 48 cases at nanoString facilities in Toronto, Miami, and Geneva. The expression data generated at the three international sites were virtually indistinguishable, and produced correlation coefficients of ≥0.97. This impressive level of reproducibility achieved using the nanoString technology suggests that our assay could produce identical results at any institute equipped with the nanoString nCounter System, or that RNA samples from centers around the world could be studied at a central location.
Pathologists have long stored tumor biopsies as FFPE material in order to preserve as much cellular and structural integrity of the original tumor specimen as possible, making samples amenable to study for decades. A significant drawback associated with this preservation technique is that DNA and RNA extracted from FFPE material is typically highly degraded, and therefore of limited use in molecular studies. The nanoString technology has known compatibility with degraded RNA isolated from FFPE cases [15], largely due to the usage of relatively short 50mer probes [5]. In a large series of 84 FFPE medulloblastomas from three independent pathology labs, our nanoString assay could assign subgroup with high confidence in 87.5% of cases from the last 8 years. Of those FFPE cases with a high confidence subgroup assignment, 100% were accurately classified as compared to the gold standard of expression profiling. Although 2/4 FFPE cases that failed to meet the PAM threshold were assigned to the correct subgroup, we suggest that higher specificity at the expense of sensitivity is necessary for a biomarker in the setting of a clinical trial.
In conclusion, we have developed, optimized, and validated a novel assay for medulloblastoma sub-grouping that is compatible with conditions common to current clinical trial settings. Future incorporation of this or similar molecular classification pipelines into prospective clinical trials will enhance our current understanding of the biological and prognostic significance of medulloblastoma subgroups, and we anticipate that this information will lead to improved care and outcomes for our patients.
References
- Cho YJ, Tsherniak A, Tamayo P, Santagata S, Ligon A, Greulich H, Berhoukim R, Amani V, Goumnerova L, Eberhart CG, Lau CC, Olson JM, Gilbertson RJ, Gajjar A, Delattre O, Kool M, Ligon K, Meyerson M, Mesirov JP, Pomeroy SL (2011) Integrative genomic analysis of medulloblastoma identifies a molecular subgroup that drives poor clinical outcome. J Clin Oncol 29:1424–1430
Article PubMed Google Scholar - Clifford SC, Lusher ME, Lindsey JC, Langdon JA, Gilbertson RJ, Straughton D, Ellison DW (2006) Wnt/Wingless pathway activation and chromosome 6 loss characterize a distinct molecular sub-group of medulloblastomas associated with a favorable prognosis. Cell Cycle 5:2666–2670
Article PubMed CAS Google Scholar - Ellison DW, Dalton J, Kocak M, Nicholson SL, Fraga C, Neale G, Kenney AM, Brat DJ, Perry A, Yong WH, Taylor RE, Bailey S, Clifford SC, Gilbertson RJ (2011) Medulloblastoma: clinicopathological correlates of SHH, WNT and non-SHH/WNT molecular subgroups. Acta Neuropathol 121:381–396
Article PubMed CAS Google Scholar - Gajjar A, Chintagumpala M, Ashley D, Kellie S, Kun LE, Merchant TE, Woo S, Wheeler G, Ahern V, Krasin MJ, Fouladi M, Broniscer A, Krance R, Hale GA, Stewart CF, Dauser R, Sanford RA, Fuller C, Lau C, Boyett JM, Wallace D, Gilbertson RJ (2006) Risk-adapted craniospinal radiotherapy followed by high-dose chemotherapy and stem-cell rescue in children with newly diagnosed medulloblastoma (St Jude Medulloblastoma-96): long-term results from a prospective, multicentre trial. Lancet Oncol 7:813–820
Article PubMed Google Scholar - Geiss GK, Bumgarner RE, Birditt B, Dahl T, Dowidar N, Dunaway DL, Fell HP, Ferree S, George RD, Grogan T, James JJ, Maysuria M, Mitton JD, Oliveri P, Osborn JL, Peng T, Ratcliffe AL, Webster PJ, Davidson EH, Hood L, Dimitrov K (2008) Direct multiplexed measurement of gene expression with color-coded probe pairs. Nat Biotechnol 26:317–325
Article PubMed CAS Google Scholar - Kool M, Koster J, Bunt J, Hasselt NE, Lakeman A, van Sluis P, Troost D, Meeteren NS, Caron HN, Cloos J, Mrsic A, Ylstra B, Grajkowska W, Hartmann W, Pietsch T, Ellison D, Clifford SC, Versteeg R (2008) Integrated genomics identifies five medulloblastoma subtypes with distinct genetic profiles, pathway signatures and clinicopathological features. PLoS One 3:e3088
Article PubMed Google Scholar - Kulkarni MM (2011) Digital multiplexed gene expression analysis using the NanoString nCounter system Curr Protoc Mol Biol Chapter 25:Unit25B 10
Google Scholar - Low JA, de Sauvage FJ (2010) Clinical experience with Hedgehog pathway inhibitors. J Clin Oncol 28:5321–5326
Article PubMed CAS Google Scholar - Malkov VA, Serikawa KA, Balantac N, Watters J, Geiss G, Mashadi-Hossein A, Fare T (2009) Multiplexed measurements of gene signatures in different analytes using the Nanostring nCounter Assay System. BMC Res Notes 2:80
Article PubMed Google Scholar - Northcott PA, Fernandez LA, Hagan JP, Ellison DW, Grajkowska W, Gillespie Y, Grundy R, Van Meter T, Rutka JT, Croce CM, Kenney AM, Taylor MD (2009) The miR-17/92 polycistron is up-regulated in sonic hedgehog-driven medulloblastomas and induced by N-myc in sonic hedgehog-treated cerebellar neural precursors. Cancer Res 69:3249–3255
Article PubMed CAS Google Scholar - Northcott PA, Hielscher T, Dubuc A, Mack S, Shih D, Remke M, Al-Halabi H, Albrecht S, Jabado N, Eberhart CG, Grajkowska W, Weiss WA, Clifford SC, Bouffet E, Rutka JT, Korshunov A, Pfister S, Taylor MD (2011) Pediatric and adult sonic hedgehog medulloblastomas are clinically and molecularly distinct. Acta Neuropathol 122:231–240
Article PubMed Google Scholar - Northcott PA, Korshunov A, Witt H, Hielscher T, Eberhart CG, Mack S, Bouffet E, Clifford SC, Hawkins CE, French P, Rutka JT, Pfister S, Taylor MD (2011) Medulloblastoma comprises four distinct molecular variants. J Clin Oncol 29:1408–1414
Article PubMed Google Scholar - Northcott PA, Nakahara Y, Wu X, Feuk L, Ellison DW, Croul S, Mack S, Kongkham PN, Peacock J, Dubuc A, Ra YS, Zilberberg K, McLeod J, Scherer SW, Sunil Rao J, Eberhart CG, Grajkowska W, Gillespie Y, Lach B, Grundy R, Pollack IF, Hamilton RL, Van Meter T, Carlotti CG, Boop F, Bigner D, Gilbertson RJ, Rutka JT, Taylor MD (2009) Multiple recurrent genetic events converge on control of histone lysine methylation in medulloblastoma. Nat Genet 41:465–472
Article PubMed CAS Google Scholar - Pfister S, Remke M, Benner A, Mendrzyk F, Toedt G, Felsberg J, Wittmann A, Devens F, Gerber NU, Joos S, Kulozik A, Reifenberger G, Rutkowski S, Wiestler OD, Radlwimmer B, Scheurlen W, Lichter P, Korshunov A (2009) Outcome prediction in pediatric medulloblastoma based on DNA copy-number aberrations of chromosomes 6q and 17q and the MYC and MYCN loci. J Clin Oncol 27:1627–1636
Article PubMed Google Scholar - Reis PP, Waldron L, Goswami RS, Xu W, Xuan Y, Perez-Ordonez B, Gullane P, Irish J, Jurisica I, Kamel-Reid S (2011) mRNA transcript quantification in archival samples using multiplexed, color-coded probes. BMC Biotechnol 11:46
Article PubMed CAS Google Scholar - Schwalbe EC, Lindsey JC, Straughton D, Hogg TL, Cole M, Megahed H, Ryan SL, Lusher ME, Taylor MD, Gilbertson RJ, Ellison DW, Bailey S, Clifford SC (2011) Rapid diagnosis of medulloblastoma molecular subgroups. Clin Cancer Res 17:1883–1894
Article PubMed CAS Google Scholar - Thompson MC, Fuller C, Hogg TL, Dalton J, Finkelstein D, Lau CC, Chintagumpala M, Adesina A, Ashley DM, Kellie SJ, Taylor MD, Curran T, Gajjar A, Gilbertson RJ (2006) Genomics identifies medulloblastoma subgroups that are enriched for specific genetic alterations. J Clin Oncol 24:1924–1931
Article PubMed CAS Google Scholar - Tibshirani R, Hastie T, Narasimhan B, Chu G (2002) Diagnosis of multiple cancer types by shrunken centroids of gene expression. Proc Natl Acad Sci USA 99:6567–6572
Article PubMed CAS Google Scholar
Acknowledgments
M.D.T. is supported by a clinician-scientist award from the Canadian Institutes of Health Research. P.A.N. is supported by a Restracomp fellowship at the Hospital for Sick Children. Grant support is acknowledged from The Pediatric Brain Tumor Foundation, Genome Canada, Genome BC, Terry Fox Research Institute, Ontario Institute for Cancer Research, Pediatric Oncology Group Ontario, Funds from ‘The Family of Kathleen Lorette’ and the Clark H. Smith Brain Tumor Centre, Montreal Children’s Hospital Foundation, Hospital for Sick Children: Sonia and Arthur Labatt Brain Tumor Research Centre, Chief of Research Fund, Cancer Genetics Program, Garron Family Cancer Centre, B.R.A.I.N. Child. C.G.E is supported by an NIH R01 operating grant (NS055089). We thank Susan Archer for assistance with technical writing.
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Author information
Authors and Affiliations
- The Arthur and Sonia Labatt Brain Tumour Research Center, Hospital for Sick Children, Toronto, ON, Canada
Paul A. Northcott, David J. H. Shih, Adrian Dubuc, James T. Rutka & Michael D. Taylor - Program in Developmental and Stem Cell Biology, Hospital for Sick Children, Toronto, ON, Canada
Paul A. Northcott, David J. H. Shih, Adrian Dubuc & Michael D. Taylor - Department of Laboratory Medicine and Pathobiology, University of Toronto, Toronto, ON, Canada
David J. H. Shih, Adrian Dubuc, James T. Rutka & Michael D. Taylor - Division Molecular Genetics, German Cancer Research Center (DKFZ), Heidelberg, Germany
Marc Remke, Marcel Kool & Stefan Pfister - Department of Pediatric Oncology, Hematology and Immunology, University of Heidelberg, Heidelberg, Germany
Marc Remke & Stefan Pfister - Department of Neurology, Children’s Hospital Boston, Boston, MA, USA
Yoon-Jae Cho & Scott L. Pomeroy - Department of Pathology, The Hospital for Sick Children, Toronto, ON, Canada
Cynthia Hawkins - Departments of Pathology, Ophthalmology and Oncology, Johns Hopkins University, Baltimore, MD, USA
Charles G. Eberhart - Oncogenomics Core Facility, Sylvester Comprehensive Cancer Center, Miller School of Medicine, University of Miami, Miami, FL, USA
Toumy Guettouche & Yoslayma Cardentey - Hussman Institute for Human Genomics, Center for Genome Technology, Miller School of Medicine, University of Miami, Miami, FL, USA
Toumy Guettouche - Neuro-oncology Program, Division of Haematology/Oncology, The Hospital for Sick Children, Toronto, ON, Canada
Eric Bouffet - Canada’s Michael Smith Genome Sciences Centre, BC Cancer Agency, Vancouver, Canada
Marco Marra - Department of Medical Genetics, University of British Columbia, Vancouver, Canada
Marco Marra - Division of Hematology/Oncology and Genetics and Genome Biology Program, Hospital for Sick Children, Toronto, ON, Canada
David Malkin - Departments of Pediatrics and Medical Biophysics, Faculty of Medicine, University of Toronto, Toronto, ON, Canada
David Malkin - Division of Neurosurgery, Hospital for Sick Children, Toronto, ON, Canada
James T. Rutka & Michael D. Taylor - Department of Neuropathology, University of Heidelberg, Heidelberg, Germany
Andrey Korshunov - Clinical Cooperation Unit Neuropathology, German Cancer Research Center, Heidelberg, Germany
Andrey Korshunov
Authors
- Paul A. Northcott
You can also search for this author inPubMed Google Scholar - David J. H. Shih
You can also search for this author inPubMed Google Scholar - Marc Remke
You can also search for this author inPubMed Google Scholar - Yoon-Jae Cho
You can also search for this author inPubMed Google Scholar - Marcel Kool
You can also search for this author inPubMed Google Scholar - Cynthia Hawkins
You can also search for this author inPubMed Google Scholar - Charles G. Eberhart
You can also search for this author inPubMed Google Scholar - Adrian Dubuc
You can also search for this author inPubMed Google Scholar - Toumy Guettouche
You can also search for this author inPubMed Google Scholar - Yoslayma Cardentey
You can also search for this author inPubMed Google Scholar - Eric Bouffet
You can also search for this author inPubMed Google Scholar - Scott L. Pomeroy
You can also search for this author inPubMed Google Scholar - Marco Marra
You can also search for this author inPubMed Google Scholar - David Malkin
You can also search for this author inPubMed Google Scholar - James T. Rutka
You can also search for this author inPubMed Google Scholar - Andrey Korshunov
You can also search for this author inPubMed Google Scholar - Stefan Pfister
You can also search for this author inPubMed Google Scholar - Michael D. Taylor
You can also search for this author inPubMed Google Scholar
Corresponding author
Correspondence toMichael D. Taylor.
Electronic supplementary material
Below is the link to the electronic supplementary material.
401_2011_899_MOESM1_ESM.eps
Supplementary Figure 1. Subgroup-specific expression of medulloblastoma signature genes comprising the nanoString CodeSet. Boxplot summaries of Affymetrix exon array data for the 22 genes comprising the nanoString CodeSet. Log2-transformed signal intensities for 101 primary medulloblastomas are plotted for each of the CodeSet genes stratified by molecular subgroup (EPS 1818 kb)
401_2011_899_MOESM2_ESM.eps
Supplementary Figure 2. Comparison of exon array expression data with nanoString expression data for medulloblastoma signature genes. Boxplot summaries of Affymetrix exon array data and nanoString expression data for a subset of medulloblastoma signature genes contained within the CodeSet. Log2-transformed signal intensities (exon array) and probe counts (nanoString) for 101 primary medulloblastomas are plotted for each gene stratified by molecular subgroup (EPS 573 kb)
401_2011_899_MOESM3_ESM.eps
Supplementary Figure 3. Pearson correlation analysis of nanoString expression data versus exon array expression data. Scatterplots for each gene in the CodeSet depict the correlation between nanoString expression data and exon array data. r = Pearson correlation (EPS 1363 kb)
401_2011_899_MOESM4_ESM.eps
Supplementary Figure 4. Evaluation of class prediction algorithms for assigning medulloblastoma subgroup affiliation. (a) Virtual heatmap depicting results of cross-validation analysis for all class prediction algorithms evaluated on the training series for medulloblastoma subgroup prediction accuracy. Samples are ordered horizontally according to their known subgroup affiliation (‘Actual’). Results represent the consensus subgroup assignment following 10,000 iterations and discordant cases are labeled according to the subgroup in which they were erroneously classified. Samples labeled in grey represent those in which a single subgroup could not be reliably assigned. (b-f) Bar graphs depicting class prediction algorithm performance. Metrics evaluated included accuracy (b), Rand index (d), adjusted Rand index (d), Jaccard similarity index (e), and Fowlkes-Mallows index (f) (EPS 947 kb)
401_2011_899_MOESM5_ESM.eps
Supplementary Figure 5. Association of PAM probability score and class prediction accuracy. Plots are shown for all fresh-frozen validation samples and FFPE cases ≤ 8 years of age (a), and for each predicted molecular subgroup (b). Data points represent mean accuracies at each prediction probability interval. Vertical red lines denote probability threshold before deterioration in performance. Error bars denote SD (EPS 319 kb)
401_2011_899_MOESM6_ESM.eps
Supplementary Figure 6. Association of class prediction accuracy with sample age and measures of RNA integrity for FFPE-derived RNA. Boxplot summaries comparing sample age (top), RNA sample length (i.e., proportion of RNA sample ≥ 300 nucleotides (nt) in length (middle), and RNA Integrity Numbers (RINs) of accurately classified FFPE samples (Yes) with misclassified FFPE samples (No). Significance for the three predictors was determined by multiple logistic regression analysis (EPS 494 kb)
Supplementary Table 1 (XLS 42 kb)
Supplementary Table 2 (XLS 33 kb)
Supplementary Table 3 (XLS 35 kb)
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License (https://creativecommons.org/licenses/by-nc/2.0), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Northcott, P.A., Shih, D.J.H., Remke, M. et al. Rapid, reliable, and reproducible molecular sub-grouping of clinical medulloblastoma samples.Acta Neuropathol 123, 615–626 (2012). https://doi.org/10.1007/s00401-011-0899-7
- Received: 29 September 2011
- Revised: 19 October 2011
- Accepted: 21 October 2011
- Published: 06 November 2011
- Issue Date: April 2012
- DOI: https://doi.org/10.1007/s00401-011-0899-7