The Scottish Structural Proteomics Facility: targets, methods and outputs (original) (raw)
Abstract
The Scottish Structural Proteomics Facility was funded to develop a laboratory scale approach to high throughput structure determination. The effort was successful in that over 40 structures were determined. These structures and the methods harnessed to obtain them are reported here. This report reflects on the value of automation but also on the continued requirement for a high degree of scientific and technical expertise. The efficiency of the process poses challenges to the current paradigm of structural analysis and publication. In the 5 year period we published ten peer-reviewed papers reporting structural data arising from the pipeline. Nevertheless, the number of structures solved exceeded our ability to analyse and publish each new finding. By reporting the experimental details and depositing the structures we hope to maximize the impact of the project by allowing others to follow up the relevant biology.
Similar content being viewed by others
Introduction
Structural biology is firmly embedded as a crucial tool for the molecular biologist, whether the interest is academic or commercial. The genome sequencing revolution of the 1990s transformed the biological landscape and to remain relevant to the scale of this new information, structural biology had to accelerate its pace of discovery. The Scottish Structural Proteomics Facility (SSPF) comprised of four partner institutions: the Universities of St Andrews, Dundee, Glasgow and Warwick. The Structural Proteomics of Rational Targets (SPoRT) laboratory situated at the University of St Andrews was established with the focus of developing small scale high throughput structural biology and to facilitate collaboration with other universities. Much larger international efforts have focussed on productivity and throughput as exemplified by the NIH Protein Structure Initiative in the USA, Protein 3000 in Japan, Structural Proteomics in Europe (SPINE) [1] and the international Wellcome Trust Structural Genomics Consortium (SGC) [2]. Our remit was to generate structures, publications and to facilitate access for “non-structural” labs to structural biology. The “high throughput” lab commenced work in 2004 and focussed on development of efficient strategies for target selection, cloning, protein production, crystallization and crystal structure determination. Due to demands for higher throughput, automated equipment was employed at several key stages in the pipeline.
We chose targets against the following criteria: obtaining a structure was likely to be tractable (in essence this meant expression in Escherichia coli); the protein was likely to have a novel structure; the structure had the potential to either underpin antibiotic development or illuminate some defined aspect of biology or biochemistry. As we discuss, one of the significant challenges was to balance choice of “publishable” target work against the work of running the pipeline and the tractability of target (including any subsequent biochemical investigation). Target selection thus differed from many other efforts which focus on folds predicted to be “novel” (exemplified by the NIH efforts) or on comprehensive coverage of a functional class (such as the focus on short chain dehydrogenases and kinases of the SGC).
During the build up of the SPoRT pipeline, we operated structure determination in essentially a traditional manner, in that projects were processed without automation in a sequential manner. This was because the funding imposed a number of output milestones which had to be met, thus there are “high throughput” structures and “low throughput” structures. The St Andrews laboratory was funded by a combination of sources and its staff complement consisted primarily of five post-doctoral research associates (PDRA) and two technicians. In 4 years, we have cloned more than 350 genes resulting in the production of 165 proteins from which 42 have yielded crystal structures. This represents about 25% of purified proteins that made it to crystal structure. Another measure of efficiency is that in its lifetime the pipeline delivered over two structures per PDRA per year. Given that the pipeline only became operative from the second year, the efficiency metrics by the end of the project were significantly higher than the beginning. However, the success of the pipeline outstripped our ability to analyse and follow up the structures. In this report, we provide a detailed account of the pipeline, the experimental structural biology and highlight some of the issues we faced. This strategy allows others to reproduce the experiments and to pursue the biological implications of our work.
Materials and methods
Bioinformatics analyses for target selection
Proteins were submitted to the pipeline as cohorts of targets; collection of genes with a common theme or source. Proteins were then analysed using a standard suite of analyses: SignalP 3.0 to detect signal peptides which indicate the protein would otherwise normally be exported from the cytoplasm [3]; TMHMM2 to detect transmembrane regions which may reduce protein solubility [4]; and RONN and PONDR to analyze the sequence for predicted disordered regions which are believed to interfere with crystallization [5, 6]. As a result of these analyses, truncations were made in a few cases where the prediction suggested significant disorder at the C- or N- termini, signal peptides or transmembrane regions. BLAST searches of the PDB were carried out to establish the originality and potential impact of the structure. Targets were also analysed using the pI versus GRAVY crystallization predictor plot as described in [7]. This work was initially carried out manually or by basic PERL scripts. However, a core part of the SSPF was to develop improved techniques for target selection, so in order to streamline and improve the ease and reliability of target selection we developed an automated system for carrying out sequence analysis called TarO [8]. TarO runs a suite of analyses on a target sequence including orthology detection and returns a summarised view of the findings as well as an annotated multiple sequence alignment that assists in the identification of domain boundaries and disordered regions. The system is web-accessible and has been used extensively within the SSPF as well as by external users world-wide. In addition to TarO, three techniques for crystallisation propensity prediction were also developed. The OB-Score [9] combines hydrophobicity and predicted pI within a statistical framework, the ParCrys algorithm combines more features of the sequence [10] while XANNpred (manuscript submitted) is a machine-learning technique that includes a broad range of properties including predicted secondary structure. TarO presents results sorted by ParCrys and the OB-Score to highlight proteins which may be more amenable to processing in the SSPF high-throughput pipeline. While TarO and the crystallisation predictors were not applied in all structures output by SSPF, they were input to decision making processes in the later years of the project.
Genome-wide scale target selection tools
We also developed bioinformatics protocols for large-scale target selection. A round of target selection was conducted to select tractable prokaryotic proteins expected to be relevant to human biology. The 657,391 proteins in the CMR (‘Omniome’) database [11] was the starting point for this work. The progress of these proteins through the target selection filters is summarised in Table 1. Proteins predicted to be amenable to producing diffraction-quality crystals (OB-Score ≥ 5) were searched against Ensembl human [12] with PSIBLAST [13], which identified 55,405 matches with expected structural similarity according to published thresholds [14]. In order to explore novel structure space, proteins with a match to the PDB [15] were excluded, leaving 4,461 proteins. Uniprot [16] identifiers were inferred to provide a mechanism for obtaining pre-calculated annotations, particularly Pfam [17] and Gene Ontology (GO) [18] data. Proteins were excluded by the criteria of one or more predicted TMHMM2 transmembrane regions, sequence length outwith 60–600 amino acids, and more than 20% predicted SEG low-complexity [19]. A total of 344 Pfam families were inferred for 3,008 proteins that passed the above filters. The total set of sequences from the 344 Pfam families were searched against the PDB (BLASTP, 1E-6, 90% identify, 90% query coverage) to exclude proteins where a homology modelling template was available. The final filtered set contained 1,329 proteins from 143 Pfam families. These proteins were then ranked according to OB-Score, and a custom scoring system ‘SOFA’ (see below).
Table 1 CMR target selection summary
A Gene Ontology-based scoring system (Specificity Of Functional Annotation, ‘SOFA’) was implemented to estimate the extent of available functional annotation for candidate targets, because functional annotation is often important for interpretation of new protein structures. The GO term with the least number of children was the starting point for calculating a protein’s SOFA score. From this term, the ratio of the parent to child terms was calculated; higher scores indicated more parents and therefore estimated more knowledge was available about the protein’s function. Where proteins had leaf-node GO term(s) the SOFA score was always higher than for proteins with no leaf-node GO term. Also, a greater number of leaf node terms corresponded to higher scores.
Further large-scale target selection was conducted with the aim of identifying tractable proteins relevant to Staphylococcus aureus therapeutics development. A set of 253 identifiers were taken from the literature [20, 21], indicating genes essential for infectivity in mouse models and/or essential for viability for growth in rich media. These were mapped to 1637 S. aureus proteins in CMR, corresponding to 1 MSSA (MSSA476) and 5 MRSA (MRSA252, Mu50, COL, MW2, and N315) strains. These proteins were subject to a series of filters similar to those described above. The filtering criteria were OB-Score ≥ 3, <2 TMHMM2 transmembrane regions, ≤20% SEG low-complexity, sequence length 60–600 amino acids. PSIBLAST matches to PDB and Ensembl human proteins were excluded according to published thresholds. Matches to human proteins were excluded because these were considered to be less attractive therapeutic targets. Pfam matches were inferred, leaving 51 proteins from 36 Pfam families and 50 proteins without a Pfam match. SOFA scoring was applied to these 101 proteins and further analyses of these proteins including TarO and literature searching proposed a final set of 20 targets from 11 functional categories. This final set of 20 S. aureus proteins were submitted to the pipeline for production.
Cloning
With the exception of purified proteins provided by collaborators, open reading frames (ORFs) of all targets were cloned using a modified version of the Gateway cloning system. Each target was cloned with an N-terminal TEV protease cleavable Hisx6 tag, with the BP recombination site relocated out of the cloning sequence. Target genes were amplified using one common and two gene-specific primers. The common primer, encoding the attB1 recombination site, RBS, ATG start codon, six histidine residues and TEV protease site (Fig. 1) was generated by PCR. The primers used for the above process were 5′-GGGG_ACAAGTTTGTACAAAAAAGCAGGCT_TCGAAGGAGATATACATATG-3′ (designated ATTF; attB1 site is in italics) and 5′-GCCCTGAAAATACAGGTTTC-3′ (designated ATTR) with template of NdeI/NcoI-DNA fragments released from pEHISTEV plasmid DNA [22]. PCR products thus generated were purified by ethanol precipitation and resuspended to a final concentration of 4 μM. Target genes were amplified in PCR reactions consisting of 5 μl of each pair of gene-specific primers (10 μM), 2.5 μl of the common primers, 5 μl of dNTP mixture (200 μM each), 10 μl of thermo polymerase buffer (5×) containing 2 mM MgSO4 and 4% DMSO, 2 units of Pfu DNA polymerase and 10 ng of DNA template in a total volume of 50 μl. Two-stage PCR amplifications were carried out in a 96-well formatted PCR plate. After denaturing the template at 95°C for 5 min, amplifications were carried out at 95°C for 1 min, Tm-5 for 1 min and 72°C for 4 min for 5 cycles and then followed by another 25 cycles using the same procedure except that an annealing temperature of 62°C was employed instead of Tm-5 to increase the specificity of the amplification. PCR products were cleaned using the PCR cleaning kit (Promega) and diluted to a concentration of 50 ng/μl.
Fig. 1
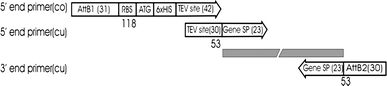
Bar presentation of primers used for gene amplifications at SSPF. The 5′ end common primer (co) is a double-stranded primer generated by PCR and used for cloning genes with the N-terminal TEV protease cleavable 6× His tag. The 5′ end custom (gene-specific) primer (cu) contains overlap sequence of 30 bp with the common primer and the gene specific sequence of 23 bp. The 3′ end custom (gene-specific) primer (cu) contains the gene-specific sequence of 23 bp and attB2 recombination site of 30 bp. AttB: BP recombination sites, RBS: ribosome binding site, ATG: start codon, 6× His: six histidine tag, TEV site: TEV site: sequence coding TEV protease cleavage site and the spacer. Numbers indicate the length of the primers (bp)
BP recombination was carried out as described in the Gateway cloning instruction manual using pDONR221 as donor vector. The recombination reaction consisted of 100 ng of _att_B-PCR products, 100 ng of pDONR221 vector and 1 μl of BP clonase II enzyme mix in TE buffer to a total volume of 10 μl. The mixture was incubated at 25°C for 1 h and then further incubated at 37°C for 15 min following the addition of 2 μl proteinase K. E. coli DH5α chemical competent cells were transformed with 2 μl reaction mix and the transformed cells were spread onto L-agar plates containing 50 μg/ml kanamycin. Plasmid DNA was prepared by picking two colonies and cultivating in separate 10 ml L-broth media containing 50 μg/ml kanamycin, prior to insert verification by agarose gel electrophoresis.
LR recombination was carried out using pDEST14 as the destination vector with two verified pDONR221 clones. The recombination reaction contained 100 ng of entry clone pDONR221 DNA, 100 ng of pDEST14 vector, 1 μl of LR clonase II enzyme mix in TE buffer to a total volume of 10 μl. The reaction mixture was incubated for 1 h at 25°C and then incubated at 37°C for 15 min after adding 2 μl of proteinase K. A total volume of 2 μl of each BP reaction was transformed into 50 μl of DH5α chemical competent cells and selected for ampicillin resistance on an L-agar plate. Two clones were picked and the plasmid DNA isolated for each LR reaction. The insertion sequence of each clone was verified and the prepared DNA was stored at −80°C for expression trials.
Small-scale expression
The E. coli competent cells BL21 (DE3), C43 (DE3), and BL21 (DE3)-CodonPlus (Strategene) were transformed with the pDEST14 expression vectors. Small-scale expression trials were carried out in Lysogeny Broth (LB; 10 g Tryptone, 5 g Yeast Extract, 10 g NaCl), Tryptone phosphate broth (TPB; 20 g Tryptone, 2 g K2HPO4, 2 g KH2PO4, 5 g NaCl) and auto-induction media, prepared in-house using the recipe from [23] or purchased as ‘Magic Media’ (Invitrogen). For LB and TPB cultures, starter cultures were prepared by inoculating LB supplemented with ampicillin (final concentration 100 μg/ml) with freshly-transformed colonies on LB/ampicillin plates, and incubating overnight at 37°C, 200 rpm. Alternatively, the transformation mix was used directly as inoculum for starter cultures. Growth media (5 ml) in 50 ml Falcon tubes was inoculated with overnight starter culture (1:100 dilution factor) prior to incubation at 37°C, 200 rpm. At mid-log growth phase (OD600 ~ 0.6–0.8), protein expression was induced with 0.4 mM IPTG. For LB cultures, incubation continued for a further 3 h at 37°C for one set of cultures, whilst another set was incubated at 25°C overnight. TPB cultures were incubated overnight at 25°C and 15°C post-induction. For protein expression in auto-induction media, freshly transformed colonies were used to inoculate 3-ml auto-induction media supplemented with 100 μg/ml ampicillin (two colonies per target protein). Cultures were then incubated at 37°C, 300 rpm until cultures turned slightly turbid at which point one set was further incubated at 37°C and the other set at 25°C, both for 42 h. Cultures were harvested by centrifugation and pellets were stored at −20°C. Cell lysis was achieved either chemically using Bugbuster HT solution (Novagen) or mechanically by sonication. Whole cell lysates were analysed for target protein expression by sodium dodecyl sulphate polyacrylamide gel electrophoresis (SDS–PAGE; [24] using pre-cast gels (Invitrogen). For target protein localization, cell lysates were spun down and the supernatant was analysed for His6-tagged soluble protein expression using the BioSprint 15 workstation according to manufacturer’s instructions (Qiagen). The resulting eluates were analysed by SDS-PAGE. Soluble protein expression was scored on the basis of whether protein bands commensurate with the estimated molecular weight were identified on SDS-PAGE gels. Band intensities were used to estimate the amount of soluble protein expressed. For uniformity, protein expression at <5 mg/l (low expression) was classified as 1S, 5–10 mg/l (medium expression) as 2S, and >10 mg/l (high expression) as 3S.
Large-scale expression of target proteins
Protein identities were verified by mass spectrometry prior to scale-up. Overnight starter cultures were used to inoculate 1–6 l (depending on expression levels obtained from small-scale solubility screening experiments) LB, TPB or auto-induction media supplemented with ampicillin. Optimal growth conditions from small-scale expression trials were replicated for large scale expression. Prior to harvesting (centrifugation at 2,400×g, for 30 min at 4°C), 1-ml aliquots were analyzed for protein expression and to estimate final yields. Cell pellets were stored at −80°C until required for purification.
Where appropriate, selenomethionine-labelled proteins were produced using the method of [25]. Essentially, freshly transformed BL21(DE3) or B834(DE3) cells served as inoculum for starter cultures. After overnight incubation, starter cultures were pelleted and washed 3 times with phosphate buffer (1 g NH4Cl, 3 g KH2PO4, 6 g Na2HPO4 · 7H2O/l) prior to inoculation (1:20) of selenomethionine-incorporation media prepared as follows: 100 ml glucose solution (20% glucose, 0.3% MgSO4, 0.01% Fe2(SO4)3, 0.01% thiamine) was added to 900 ml phosphate buffer (same as above). The pH of the media was adjusted to 7.4 prior to addition of ampicillin and 50 mg/l l-(+) Selenomethionine. Cells were cultivated at 37°C until OD600 was between 0.8 and 1.0, at which point IPTG was added to a final concentration of 0.2 mM. Cultures were incubated at 25°C overnight, harvested and stored as described above.
Preparation of His6-tagged tobacco etch virus (TEV) protease
The E. coli recombinant strain BL21 (DE3)-RIL/pRK793 harbouring a mutant (S219V) of the catalytic domain of TEV protease was kindly donated by Partho Ghosh (Department of Chemistry and Biochemistry, University of California). This construct expresses as a maltose-binding protein (MBP) fusion protein that auto-cleaves in vivo, resulting in the TEV protease catalytic domain with uncleavable N-terminal His6 and C-terminal polyarginine tags [26]. Following protein expression at 25°C, the His6-TEV protease was purified by nickel-immobilized metal affinity chromatography (Ni2+-IMAC), followed by desalting chromatography. Aliquots (5 mg/ml) of TEV protease were prepared and stored at −80°C.
Purification
Cell pellets were resuspended in buffer A (see below for all buffer constituents) supplemented with Dnase1 (Sigma) and EDTA-free protease inhibitor cocktail tablets (Roche) prior to cell lysis on ice using the One Shot cell disruptor (Constant Systems Ltd), continuously cooled with cold tap water. Cell lysate was clarified by centrifugation at 39,000×g for 1 h at 4°C and the supernatant was filtered using a 0.45 μm filter (Millipore) prior to purification. Fully-automated purification was carried out on the AKTAxpress chromatography system using pre-packed columns (GE Healthcare). The purification procedure comprised of (1) Ni2+- immobilized metal affinity chromatography (Ni2+-IMAC) using buffers B and C for the wash and elution steps respectively (2) Desalting chromatography (DS) in buffer D for buffer exchange and removal of imidazole, (3) incubation with TEV protease for cleavage of the His6 tag, (4) capture of the His6-tagged TEV protease on a second Ni2+-IMAC column and (5) Gel filtration chromatography (GF) in buffer E to separate out contaminants and aggregates from the target proteins. A schematic representation of the fully-automated version is shown in Fig. 2a. To facilitate on-line off-column cleavage of the His6-tag, a 50-ml superloop preloaded with His6-TEV protease was incorporated into the chromatography system. This arrangement facilitated the direct loading of the DS eluate into the TEV protease-loaded superloop. For manual purification, the Ni2+-IMAC and TEV cleavage steps were carried out on the bench, and the AKTAxpress system was used for DS and GF. All proteins were purified at room temperature and analysed by SDS-PAGE. Aliquots of purified proteins were transferred into thin-walled PCR tubes and flash frozen in liquid nitrogen prior to storage at −80°C.
- Buffer A: 20 mM sodium phosphate pH 7.4, 500 mM NaCl, 10% glycerol, 10–30 mM imidazole
- Buffer B: 20 mM sodium phosphate pH 7.4, 500 mM NaCl, 10% glycerol, 30–50 mM imidazole
- Buffer C: 20 mM sodium phosphate pH 7.4, 500 mM NaCl, 10% glycerol, 300 mM imidazole
- Buffer D: 20 mM Tris pH 7.5, 500 mM NaCl, 10% glycerol
- Buffer E: 10 mM Tris pH 7.5, 150 mM or 500 mM NaCl, 10% glycerol (optional)
Fig. 2
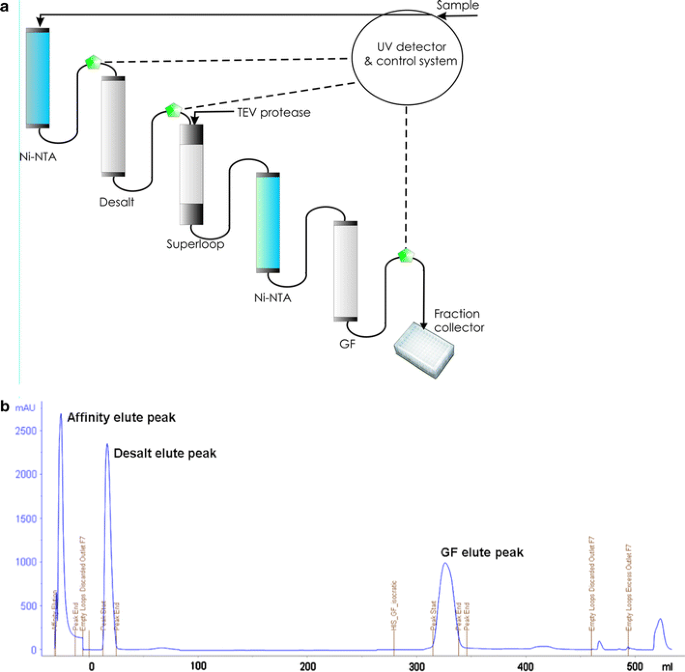
Protein purification in the SPoRT laboratory. a Schematic representation of the fully automated purification. b Chromatogram of fully automated purification showing the Ni-NTA, desalting and gel filtration peaks
Crystallization
The pre-crystallization test (PCT) (Hampton Research) was used to determine the optimal protein concentration (OPC) for crystallization trials. All screening experiments were set up as sitting drops in 96-well crystallization plates at 2 or 3 protein concentrations using either the Cartesian Honeybee X8 + 1 in the Hamilton-Rhombix-Thermo integrated crystallization and imager system or an offline Cartesian Honeybee 963. Drop sizes consisting of 0.15 μl protein + 0.15 μl precipitant (protein concentrations: 1× OPC and 2× OPC) and 0.3 μl protein + 0.15 μl precipitant (protein concentration: 1× OPC) were employed. Initially 4 commercial crystallization screens chosen from JCSG+, Classics, Pegs, pHClear, Anions, Cations (Qiagen) and Wizard I & II (Emerald Biosystems) were used to screen all proteins. Eventually, this practice was phased out in favour of 3 stochastic screens [27] prepared in-house alongside the commercial JCSG+ screen. All screening experiments were incubated at 20°C and imaged at regular intervals with a Rhombix-Thermo imager.
The traditional grid screen approach around the initial hit condition(s) was used primarily for crystal optimization purposes. If this failed, a stochastic approach to optimization was often able to generate suitable single crystals. In both cases, 24 and 96 well grid/stochastic screens based on the original mother liquor were designed and built using a Hamilton Microlab Star and crystallization screens were then set either as before on one of the robots using nanoliter volumes or manually using larger μl drop sizes.
Data collection and structure solution
Crystals deemed to be large enough for diffraction screening were harvested and frozen in liquid nitrogen. Crystals were characterized in house using a Rigaku Micromax™—007HF Cu anode with VariMax optics alongside a Rigaku Saturn 944+ CCD detector and at various synchrotron beamlines including BM14, ID14-1, -2, -4, ID29 and ID 23-1 and -2 at the ESRF and IO2 and IO3 at the Diamond light source Oxfordshire. HKL2000 [28], MOSFLM [29], SCALA [30] and XDS [31] were all used at various times to index and scale data. Structures were phased by whatever method provided the fastest route to structure and included molecular replacement, sulfur-single wavelength anomalous diffraction, selenomethionine or heavy atom anomalous diffraction. SHELXC/D/E [32], SOLVE [33], RESOLVE [34], PHENIX [35] and CNS [36] and programs implemented under the CCP4 package were used to phase and refine structures [37]. COOT [38] and X-Fit [39] were used for model building. CCP4 REFMAC5 [40] was mainly used for refinement, although some structures were refined with PHENIX. The quality of all structures was checked with MOLPROBITY [41] and STAN (http://xray.bmc.uu.se/usf/www.html).
Protein information management system (PIMS)
Given the large volume of data produced by the project and shared between multiple researchers, a database system was required to store and process the information. A companion SPoRT funded project, Protein Information Management System (PIMS), was initiated at the same time as the SSPF to provide a laboratory information system for the project and other similar projects within the UK. The PIMS database was used to store and share experimental results within the project.
Results and discussion
Protein production
All targets have been divided into four groups on the basis of their origins. (1) Targets from Sulfolobus solfataricus and Thermoproteus tenax constitute the Archaea group (2) ORFs from the archaeal Pyrobaculum spherical virus (PSV) and Sulfolobus islandicus rudivirus (SIRV), along with ORFs from Mycobacterium and Streptomyces bacteriophages were classified as archaeal viruses and bacteriophages (3) several pathogenic bacteria make up the bacterial group; and (4), the eukaryote group consisted of proteins of human, frog and trypanosome origins. Statistics for all target groups at various stages of the pipeline are presented in Table 2. Work was stopped on a few targets for two main reasons: structure of target or ortholog was determined in our laboratory or elsewhere, or bioinformatics analysis or publications suggested that the targets were either insoluble or membrane-associated.
Table 2 SPoRT laboratory pipeline scoreboard
The Gateway cloning system was adopted and modified by the SPoRT laboratory. This cloning strategy is independent of restriction enzymes, thus permitting standardized conditions for all of our cloning needs. We found our modified version to be very efficient for our experiments. Although PCR reactions using the double-stranded common and gene-specific primers yielded sufficient PCR product with our standardized cycles, amplification of GC rich sequences predictably yielded less PCR product. The amplified PCR fragments were successfully used for BP recombination with the protocol described. Transformation of E. coli cells with 2 μl of the recombination reaction produced more than 30 colonies on all plates with >99% of the isolated pDEST14 clones containing the right inserts. Intermittent sequencing of cloned genes showed that there was no significant increase in the rate of mutation during the cloning process. Overall, we found this strategy to be very robust as over 350 targets were transferred successfully to the vector, irrespective of the origin of the gene.
Our expression strategy involved the use of three different variables comprising expression strain, growth medium and temperature. At the outset of the project, three E. coli (DE3) expression strains (BL21, C43 and Codon Plus) were screened for expression. The rationale for including E. coli BL21(DE3) Codon Plus was to overcome the anticipated codon bias of archaeal and eukaryotic genes. However, preliminary results from small scale expression trials using this strain showed that expression levels were not improved, and therefore further usage was discontinued. The C43(DE3) strain is particularly useful for expression of toxic proteins in E. coli [42, 43]. Initial expression trials comparing C43(DE3) and BL21(DE3) cells revealed that the former was particularly beneficial for expression of the archaeal viral proteins. Overall, 67% of targets scaled-up were expressed in C43(DE3) and the rest in BL21(DE3). Growth media employed in our facility consisted of LB, TPB and auto-induction media. We found the auto-induction media to be particularly useful for small-scale expression screening as it facilitated parallel screening by eliminating the need to measure cell densities prior to induction. However, since most of the targets were equally soluble using either TPB or LB, we opted for these relatively inexpensive and simpler media at the scale-up stage. Consequently, only 4% of proteins were scaled-up using auto-induction media, with the rest shared equally between LB and TPB. Targets scaled-up using auto-induction media and TPB were those that either expressed at the 1S level or a minority that did not express at all in LB media. Overall 72% (190/264) of all expressed targets were soluble (Table 2), a success rate attributable to the different expression systems and conditions utilized by the SPoRT laboratory. Although protein targets of prokaryotic origins constitute most of the soluble proteins (63%; 120/190), it is not certain if this is due to the larger number of prokaryotic targets compared to the others or the use of E. coli for expression. The majority of soluble targets expressed sufficiently: 27% at the 3S level, 60% at the 2S level, and 13% at the 1S level. It was tempting to rescue insoluble proteins using fusion proteins such as MBP and thioredoxin, but we were not convinced that this would significantly increase the overall number of soluble targets. Moreover, this would require more manpower and resources. All soluble proteins scaled-up successfully, although the volume of media had to be adjusted depending on the expression level. Typically 2 l of culture was grown for 3S proteins, 4 l for 2S proteins and 6 l for 1S proteins.
Initially, small-scale cell lysis was carried out using Bugbuster for convenience. Eventually, this was replaced by sonication for two main reasons. Firstly, we observed that higher protein yields of poorly-expressing proteins were recovered by sonication compared to Bugbuster. Secondly, we observed that in certain cases, protein yields were inconsistent between estimated and final yields from large-scale cultures, presumably due to the detergents in Bugbuster. Although not convenient for a high-throughput approach, we adopted sonication in order to eliminate any ambiguities. A consistent and practical plan was adopted for the expression process in order to feed the pipeline continuously with proteins for processing. Typically, up to twenty targets were chosen per week for expression trials. Concurrently, proteins judged to be soluble from the previous week and confirmed by mass spectrometry were prepared in large quantities. The BioSprint 15 workstation, which can perform 15 nickel pull-down experiments in about 30 min, expedited sample processing times. Additionally, the Nanodrop Spectrophotometer was very beneficial as it provided a quick, easy and efficient means for not only measuring optical densities of cultures, but also spectra of DNA and proteins. As it was never the case that all twenty proteins were soluble, up to ten proteins were scaled up per week and passed on for purification. All protein expression experiments were routinely carried out on a weekly basis by one technician, with one PDRA trouble-shooting problems such as conflicting expression and mass spectrometry results. Such problems were usually resolved by repeating the trials and/or gene sequencing.
Affinity tags are ideal for HTP protein purification because a single chromatography method can be adopted. The advantages of using the His6-tag are well-documented. Using either manual or fully-automated purification methods, we obtained >90% protein purity in sufficient amounts for about 88% (167/190) of all soluble proteins. A typical elution profile for proteins purified using the fully-automated version is shown in Fig. 2b, with clearly identifiable Ni2+-IMAC, DS and GF peaks. The fully-automated method, incorporating on-line off-column TEV cleavage, was made possible by the inclusion of the 50-ml superloop in-series, as described in Materials and Methods. This method required no user-intervention once the sample was applied to the system. Also, the fully-automated method was less time-consuming, eliminating all manual handling steps such as sample loading and pooling between each chromatographic step. TEV cleavage using the fully-automated method was just as efficient as the manual method. Up to ten proteins were routinely purified per week by one technician. However, in several cases, it was difficult to control the final volume of eluates from all columns leading up to the GF column, which is limited to a maximum permissible volume of 5 ml. Elution volumes from chromatography steps prior to GF were typically larger than this volume. The outcome of this was that only 50% of the purified protein eventually made it to GF. In the latter stages of the project, after several unsuccessful attempts were made to rectify the situation, we simply purified all our targets manually. Consequently, the number of proteins purified weekly reduced to about six.
Crystallization and crystal structures
All purified protein samples were screened successfully using the nanolitre-scale Rhombix-Hamilton-Thermo integrated system. This technology not only reduced protein production costs (final protein yield was not always important), but allowed for immediate setting up of crystal trials soon after purification. As shown in Table 2, 61 of the 167 purified proteins (37%) formed diffraction-quality crystals. Despite the nanolitre-scale size of the crystallization drops, most of the crystals obtained were robust enough to provide complete datasets suitable for structural determination. As shown in Table 2, 42 structures have been solved thus far, representing 25% (42/167) of the targets that entered crystallization trials. Different phasing methods were used for structure determination with two-thirds of the structures using methods other than molecular replacement (see Table 3). This outcome was anticipated since most of the SPoRT target sequences did not display significant homology (>20%) with protein sequences in all databases at the outset of the project.
Table 3 Summary of all SPoRT laboratory crystal structures showing phasing methods, origins and functions of proteins, and retrospective analysis of all structurally characterized targets using three crystallization predictors
Retrospective analysis of target selection
Protein crystallization propensity predictors such as OB-Score, ParCrys, Xtalpred [44] are widely utilized by structural genomics groups for target selection. At the initial stages of this project, some proteins were prioritized using the pI/GRAVY crystallization predictor, which groups proteins into clusters A to C, with A being the most likely to crystallize and C the least likely. Significantly, proteins in Cluster C are predominantly membrane proteins. Preference was given to proteins that fell into either cluster A or B. In order to gain further insight into the efficacy of these predictors, we have carried out a retrospective analysis of all structures determined in our laboratory. XtalPred uses a scale of one to five, with one being the most likely to crystallize and five being the least. ParCrys predicts sequences to be high-scoring, amenable or recalcitrant. As shown in Table 3, 44% (18 out of 42) of the structures would not have been selected if Xtalpred was used initially to select targets. In the case of ParCrys and pI/GRAVY, this would have been 34% (14 out of 42) and 39% (16 out of 42) respectively. It is noteworthy to mention here that the pI/GRAVY analysis revealed that the targets were grouped into either Clusters A or B but never Cluster C. Consequently, 61% of the targets were exclusively outliers (as denoted in Table 3), and therefore would not have been selected. Although these results suggest that the predictors are not perfect, we observe that the ‘uncrystallizable proteins’ are dominated by the PSV and SIRV ORFs with sequences unlike any other functionally characterized protein. Pairwise comparisons reveal that conflicting outcomes (i.e. one predictor suggests that the protein is crystallizable whereas another suggests otherwise) are predicted for 14 proteins by XtalPred versus ParCrys, 13 proteins by XtalPred versus pI/GRAVY plot and 11 proteins by ParCrys versus pI/GRAVY plot. This discrepancy may be due to different parameters utilized by the predictors namely; size, homologs in PDB, GRAVY index and pI. Therefore, it is reasonable to suggest that at least two predictors should be utilized in order to provide a wider coverage of crystallization space.
Scientific highlights
Reports describing the crystal structures and biochemical analyses of 10 SPoRT-generated targets have been published in peer-reviewed journals (see Table 3 for references). Experimental details of crystal structures yet to be published are presented in Supplementary information. Below, we summarize a few examples to highlight the scientific impact of a few of our accomplishments.
Iron is an essential nutrient and microorganisms synthesize low molecular weight high affinity iron chelators (siderophores) in order to sequester environmental or host iron. The synthesis of these compounds is both a potential antibacterial target and of interest biochemically. We have reported the crystal structure and biochemical analysis of AcsD, an enzyme involved in the biosynthesis of achromobactin from the Gram negative plant pathogen Pectobacterium chrysanthemi [45] and the structure of AlcC, a new member of the superfamily, will be reported in due course. Other antimicrobial targets included those selected from Pseudomonas aeruginosa, an opportunistic antibiotic-resistant human pathogen. The Pseudomonas quinolone signal (PQS) is implicated in both pathogenesis and formation of biofilms and was of particular interest. In total eight P. aeruginosa structures have been determined. Methicillin-resistant Staphylococcus aureus (MRSA) is the cause of considerable concern in hospitals. We employed comparative two-dimensional gel analysis and mass spectrometry [46, 47] to identify proteins that were either upregulated or downregulated in MRSA compared to methicillin-sensitive S. aureus (MSSA), and therefore considered to be essential with potentially novel folds. Seven structures have been determined from this target.
Archaeal proteins are frequently used as tools to understand equivalent proteins in eukaryotes, most particularly those involved in DNA replication and repair. Of the 61 archaeal targets selected, 60 expressed, about half were soluble and nine structures were determined. These included crystal structures of two helicases implicated in DNA repair: XPD from S. tokodaii [48] and hel308 from S. solfataricus [49], a RadA paralog [50] and a heterotrimeric PCNA sliding clamp [51].
Sulfolobus islandicus rudivirus (SIRV) and Pyrobaculum spherical virus (PSV) are dsDNA viruses that infect archaea and their proteins show little or no detectable homology to any proteins of known function [52, 53]. We reasoned that structural data from these viruses could help elucidate viral protein function and improve our understanding of viral evolution. The virus genomes (48 ORFs from SIRV and 45 ORFs from PSV) were of a suitable size for us to attempt complete coverage. We were able to determine eight structures having cloned and attempted to express all genes.
Biological research is of course very broad but we were able to contribute to a number of local projects by providing structural data. The most striking was a novel protein harvested from the foam nests of Polypedates leucomystax, a tropical frog species from Malaysia. The crystal structure of the protein, named ranasmurfin, revealed intra- and inter-molecular cross-links featuring the lysine-tyrosine quinone (LTQ) co-factor. At 1.16 Å resolution, the amino acid sequence was identified directly from the electron density map and verified by mass spectrometry to be a novel protein [54].
Conclusions
It is clear from many studies that structure determination can be largely automated and the efficiency greatly improved. What is more, our effort demonstrates that this can be carried out on a relatively small scale and be focused to deliver results to the scientific community. The ability to run such a pipeline appears to be a skill in itself. Our own efficiency improved year on year as new working practices and approaches developed with experience. Despite this success, we have not been able to reach the point of guaranteeing a structure for every target nor in being able a priori to identify which targets will give structures. Target selection is improving and new tools are emerging that should continue to drive up the target to structure ratio (reviewed in [55]), although this is not the same as a structure for every biological target. A small group can gain efficiency by being able to adjust and alter procedures. For example, we found the use of heavy atoms a very efficient first attempt to solve structures rather than always proceeding to selenomethionine.
Our experience is that a structural biology laboratory within a collaborative centre, bringing the techniques of modern structural biology to bear on novel problems, has some advantages. The pipeline has the capacity to move very quickly to respond to a scientific need. Personnel who run the pipeline improved in efficiency over the time of the project. In a collaboration, the expertise of those carrying out the structural biology can be leveraged in the analysis of the structure, design and interpretation of subsequent experiments.
Efficiency and automation require significant intellectual resources. We observe that in this lies an issue for the future. Even in our relatively small scale effort, we were unable to match the productivity of the pipeline at generating structures with publication. Only in cases where targets were carefully chosen in advance or the structure was immediately scientifically significant, were we able to follow this more traditional path to publications. With collaborations, we were able to perform significant biochemical analysis for some proteins. However, as we report, many structures have been determined with very limited or no analysis. By fully reporting the experimental detail and depositing the data, we aim to prompt others to perform this analysis.
Abbreviations
BLAST:
Basic local alignment search tool
PDB:
Protein data bank
PDRA:
Post-doctoral research associates
PONDR:
Predictor of natural disordered regions
RONN:
Regional order neural network
SGC:
Structural genomics consortium
SPINE:
Structural proteomics in Europe
SPoRT:
Structural proteomics of rational targets
SSPF:
Scottish Structural Proteomics Facility
References
- Banci L, Bertini I, Cusack S, de Jong RN, Heinemann U, Jones EY, Kozielski F, Maskos K, Messerschmidt A, Owens R, Perrakis A, Poterszman A, Schneider G, Siebold C, Silman I, Sixma T, Stewart-Jones G, Sussman JL, Thierry JC, Moras D (2006) First steps towards effective methods in exploiting high-throughput technologies for the determination of human protein structures of high biomedical value. Acta Crystallogr D Biol Crystallogr 62(Pt 10):1208–1217
Article CAS PubMed Google Scholar - Lee WH, Atienza-Herrero J, Abagyan R, Marsden BD (2009) SGC—structural biology and human health: a new approach to publishing structural biology results. PLoS One 4(10):e7675
Article PubMed Google Scholar - Bendtsen JD, Nielsen H, von Heijne G, Brunak S (2004) Improved prediction of signal peptides: SignalP 3.0. J Mol Biol 340(4):783–795
Article PubMed Google Scholar - Krogh A, Larsson B, von Heijne G, Sonnhammer EL (2001) Predicting transmembrane protein topology with a hidden Markov model: application to complete genomes. J Mol Biol 305(3):567–580
Article CAS PubMed Google Scholar - Li X, Romero P, Rani M, Dunker AK, Obradovic Z (1999) Predicting protein disorder for N-, C-, and internal regions. Genome Inform Ser Workshop Genome Inform 10:30–40
CAS PubMed Google Scholar - Yang ZR, Thomson R, McNeil P, Esnouf RM (2005) RONN: the bio-basis function neural network technique applied to the detection of natively disordered regions in proteins. Bioinformatics 21(16):3369–3376
Article CAS PubMed Google Scholar - Canaves JM, Page R, Wilson IA, Stevens RC (2004) Protein biophysical properties that correlate with crystallization success in Thermotoga maritima: maximum clustering strategy for structural genomics. J Mol Biol 344(4):977–991
Article CAS PubMed Google Scholar - Overton IM, van Niekerk CA, Carter LG, Dawson A, Martin DM, Cameron S, McMahon SA, White MF, Hunter WN, Naismith JH, Barton GJ (2008) TarO: a target optimisation system for structural biology. Nucleic Acids Res 36(Web Server issue):W190–W196
Google Scholar - Overton IM, Barton GJ (2006) A normalised scale for structural genomics target ranking: the OB-score. FEBS Lett 580(16):4005–4009
Article CAS PubMed Google Scholar - Overton IM, Padovani G, Girolami MA, Barton GJ (2008) ParCrys: a Parzen window density estimation approach to protein crystallization propensity prediction. Bioinformatics 24(7):901–907
Article CAS PubMed Google Scholar - Peterson JD, Umayam LA, Dickinson T, Hickey EK, White O (2001) The comprehensive microbial resource. Nucleic Acids Res 29(1):123–125
Article CAS PubMed Google Scholar - Hubbard TJ, Aken BL, Beal K, Ballester B, Caccamo M, Chen Y, Clarke L, Coates G, Cunningham F, Cutts T, Down T, Dyer SC, Fitzgerald S, Fernandez-Banet J, Graf S, Haider S, Hammond M, Herrero J, Holland R, Howe K, Johnson N, Kahari A, Keefe D, Kokocinski F, Kulesha E, Lawson D, Longden I, Melsopp C, Megy K, Meidl P, Ouverdin B, Parker A, Prlic A, Rice S, Rios D, Schuster M, Sealy I, Severin J, Slater G, Smedley D, Spudich G, Trevanion S, Vilella A, Vogel J, White S, Wood M, Cox T, Curwen V, Durbin R, Fernandez-Suarez XM, Flicek P, Kasprzyk A, Proctor G, Searle S, Smith J, Ureta-Vidal A, Birney E (2007) Ensembl 2007. Nucleic Acids Res 35(Database issue):D610–D6177
Google Scholar - Altschul SF, Madden TL, Schaffer AA, Zhang J, Zhang Z, Miller W, Lipman DJ (1997) Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res 25(17):3389–3402
Article CAS PubMed Google Scholar - Rost B (1999) Twilight zone of protein sequence alignments. Protein Eng 12(2):85–94
Article CAS PubMed Google Scholar - Berman H, Henrick K, Nakamura H, Markley JL (2007) The worldwide protein data bank (wwPDB): ensuring a single, uniform archive of PDB data. Nucleic Acids Res 35(Database issue):D301–D303
Google Scholar - Apweiler R, Bairoch A, Wu CH, Barker WC, Boeckmann B, Ferro S, Gasteiger E, Huang H, Lopez R, Magrane M, Martin MJ, Natale DA, O’Donovan C, Redaschi N, Yeh LS (2004) UniProt: the universal protein knowledgebase. Nucleic Acids Res 32(Database issue):D115–D119
Google Scholar - Bateman A, Coin L, Durbin R, Finn RD, Hollich V, Griffiths-Jones S, Khanna A, Marshall M, Moxon S, Sonnhammer EL, Studholme DJ, Yeats C, Eddy SR (2004) The Pfam protein families database. Nucleic Acids Res 32(Database issue):D138–D141
Google Scholar - Harris MA, Clark J, Ireland A, Lomax J, Ashburner M, Foulger R, Eilbeck K, Lewis S, Marshall B, Mungall C, Richter J, Rubin GM, Blake JA, Bult C, Dolan M, Drabkin H, Eppig JT, Hill DP, Ni L, Ringwald M, Balakrishnan R, Cherry JM, Christie KR, Costanzo MC, Dwight SS, Engel S, Fisk DG, Hirschman JE, Hong EL, Nash RS, Sethuraman A, Theesfeld CL, Botstein D, Dolinski K, Feierbach B, Berardini T, Mundodi S, Rhee SY, Apweiler R, Barrell D, Camon E, Dimmer E, Lee V, Chisholm R, Gaudet P, Kibbe W, Kishore R, Schwarz EM, Sternberg P, Gwinn M, Hannick L, Wortman J, Berriman M, Wood V, de la Cruz N, Tonellato P, Jaiswal P, Seigfried T, White R (2004) The gene ontology (GO) database and informatics resource. Nucleic Acids Res 32(Database issue):D258–D261
- Wootton JC, Federhen S (1996) Analysis of compositionally biased regions in sequence databases. Methods Enzymol 266:554–571
Article CAS PubMed Google Scholar - Forsyth RA, Haselbeck RJ, Ohlsen KL, Yamamoto RT, Xu H, Trawick JD, Wall D, Wang L, Brown-Driver V, Froelich JM, Kedar GC, King K, McCarthy M, Malone C, Misiner B, Robbins D, Tan Z, Zhu ZY, Carr G, Mosca DA, Zamudio C, Foulkes JG, Zyskind JW (2002) A genome-wide strategy for the identification of essential genes in Staphylococcus aureus. Mol Microbiol 43(6):1387–1400
Article CAS PubMed Google Scholar - Ji Y, Zhang B, Van SF, Horn WP, Woodnutt G, Burnham MK, Rosenberg M (2001) Identification of critical staphylococcal genes using conditional phenotypes generated by antisense RNA. Science 293(5538):2266–2269
Article CAS PubMed Google Scholar - Liu H, Naismith JH (2009) A simple and efficient expression and purification system using two newly constructed vectors. Protein Expr Purif 63(2):102–111
Article CAS PubMed Google Scholar - Studier FW (2005) Protein production by auto-induction in high density shaking cultures. Protein Expr Purif 41(1):207–234
Article CAS PubMed Google Scholar - Laemmli UK (1970) Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 227(5259):680–685
Article CAS PubMed Google Scholar - Guerrero SA, Hecht HJ, Hofmann B, Biebl H, Singh M (2001) Production of selenomethionine-labelled proteins using simplified culture conditions and generally applicable host/vector systems. Appl Microbiol Biotechnol 56(5–6):718–723
Article CAS PubMed Google Scholar - Kapust RB, Tozser J, Fox JD, Anderson DE, Cherry S, Copeland TD, Waugh DS (2001) Tobacco etch virus protease: mechanism of autolysis and rational design of stable mutants with wild-type catalytic proficiency. Protein Eng 14(12):993–1000
Article CAS PubMed Google Scholar - Rupp B (2003) Maximum-likelihood crystallization. J Struct Biol 142(1):162–169
Article CAS PubMed Google Scholar - Otwinowski Z, Minor W (1997) Processing of X-ray diffraction data collected in oscillation mode. In: Methods in enzymology. Academic Press, New York, pp 307–326
- Leslie AGW (1992) Recent changes to the MOSFLM package for processing film and image plate data. Joint CCP4 + ESF-EAMCB newsletter on protein crystallography, no. 26
- Evans P (2006) Scaling and assessment of data quality. Acta Crystallogr D Biol Crystallogr 62(Pt 1):72–82
PubMed Google Scholar - Kabsch W (1993) Automatic processing of rotation diffraction data from crystals of initially unknown symmetry and cell contants. J Appl Crystallogr 26:795–800
Article CAS Google Scholar - Schneider TR, Sheldrick GM (2002) Substructure solution with SHELXD. Acta Crystallogr D Biol Crystallogr 58(Pt 10 Pt 2):1772–1779
- Terwilliger TC, Berendzen J (1999) Automated MAD and MIR structure solution. Acta Crystallogr D Biol Crystallogr 55(Pt 4):849–861
Article CAS PubMed Google Scholar - Terwilliger TC (2000) Maximum-likelihood density modification. Acta Crystallogr D Biol Crystallogr 56(Pt 8):965–972
Article CAS PubMed Google Scholar - Adams PD, Grosse-Kunstleve RW, Hung LW, Ioerger TR, McCoy AJ, Moriarty NW, Read RJ, Sacchettini JC, Sauter NK, Terwilliger TC (2002) PHENIX: building new software for automated crystallographic structure determination. Acta Crystallogr D Biol Crystallogr 58(Pt 11):1948–1954
Article PubMed Google Scholar - Brunger AT, Adams PD, Clore GM, DeLano WL, Gros P, Grosse-Kunstleve RW, Jiang JS, Kuszewski J, Nilges M, Pannu NS, Read RJ, Rice LM, Simonson T, Warren GL (1998) Crystallography & NMR system: a new software suite for macromolecular structure determination. Acta Crystallogr D Biol Crystallogr 54(Pt 5):905–921
Article CAS PubMed Google Scholar - Collaborative Computational Project N (1994) The CCP4 suite: programs for protein crystallography. Acta Crystallogr D Biol Crystallogr 50(Pt 5):760–763
Article Google Scholar - Emsley P, Cowtan K (2004) Coot: model-building tools for molecular graphics. Acta Crystallogr D Biol Crystallogr 60(Pt 12 Pt 1):2126–2132
- McRee DE (1999) XtalView/Xfit—a versatile program for manipulating atomic coordinates and electron density. J Struct Biol 125(2–3):156–165
Article CAS PubMed Google Scholar - Murshudov GN, Vagin AA, Dodson EJ (1997) Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr D Biol Crystallogr 53(Pt 3):240–255
Article CAS PubMed Google Scholar - Davis IW, Leaver-Fay A, Chen VB, Block JN, Kapral GJ, Wang X, Murray LW, Arendall WB, 3rd, Snoeyink J, Richardson JS, Richardson DC (2007) MolProbity: all-atom contacts and structure validation for proteins and nucleic acids. Nucleic Acids Res 35(Web Server issue):W375–W383
Google Scholar - Dumon-Seignovert L, Cariot G, Vuillard L (2004) The toxicity of recombinant proteins in Escherichia coli: a comparison of overexpression in BL21(DE3), C41(DE3), and C43(DE3). Protein Expr Purif 37(1):203–206
Article CAS PubMed Google Scholar - Miroux B, Walker JE (1996) Over-production of proteins in Escherichia coli: mutant hosts that allow synthesis of some membrane proteins and globular proteins at high levels. J Mol Biol 260(3):289–298
Article CAS PubMed Google Scholar - Slabinski L, Jaroszewski L, Rychlewski L, Wilson IA, Lesley SA, Godzik A (2007) XtalPred: a web server for prediction of protein crystallizability. Bioinformatics 23(24):3403–3405
Article CAS PubMed Google Scholar - Schmelz S, Kadi N, McMahon SA, Song L, Oves-Costales D, Oke M, Liu H, Johnson KA, Carter LG, Botting CH, White MF, Challis GL, Naismith JH (2009) AcsD catalyzes enantioselective citrate desymmetrization in siderophore biosynthesis. Nat Chem Biol 5(3):174–182
Article CAS PubMed Google Scholar - Seetharamappa J, Oke M, Liu H, McMahon SA, Johnson KA, Carter L, Dorward M, Zawadzki M, Overton IM, van Niekirk CA, Graham S, Botting CH, Taylor GL, White MF, Barton GJ, Coote PJ, Naismith JH (2007) Expression, purification, crystallization, data collection and preliminary biochemical characterization of methicillin-resistant Staphylococcus aureus Sar2028, an aspartate/tyrosine/phenylalanine pyridoxal-5′-phosphate-dependent aminotransferase. Acta Crystallogr Sect F Struct Biol Cryst Commun 63(Pt 5):452–456
Article PubMed Google Scholar - Seetharamappa J, Oke M, Liu H, McMahon SA, Johnson KA, Carter L, Dorward M, Zawadzki M, Overton IM, van Niekirk CA, Graham S, Botting CH, Taylor GL, White MF, Barton GJ, Coote PJ, Naismith JH (2007) Purification, crystallization and data collection of methicillin-resistant Staphylococcus aureus Sar2676, a pantothenate synthetase. Acta Crystallogr Sect F Struct Biol Cryst Commun 63(Pt 6):488–491
Article PubMed Google Scholar - Liu H, Rudolf J, Johnson KA, McMahon SA, Oke M, Carter L, McRobbie AM, Brown SE, Naismith JH, White MF (2008) Structure of the DNA repair helicase XPD. Cell 133(5):801–812
Article CAS PubMed Google Scholar - Richards JD, Johnson KA, Liu H, McRobbie AM, McMahon S, Oke M, Carter L, Naismith JH, White MF (2008) Structure of the DNA repair helicase hel308 reveals DNA binding and autoinhibitory domains. J Biol Chem 283(8):5118–5126
Article CAS PubMed Google Scholar - McRobbie AM, Carter LG, Kerou M, Liu H, McMahon SA, Johnson KA, Oke M, Naismith JH, White MF (2009) Structural and functional characterisation of a conserved archaeal RadA paralog with antirecombinase activity. J Mol Biol 389(4):661–673
Article CAS PubMed Google Scholar - Williams GJ, Johnson K, Rudolf J, McMahon SA, Carter L, Oke M, Liu H, Taylor GL, White MF, Naismith JH (2006) Structure of the heterotrimeric PCNA from Sulfolobus solfataricus. Acta Crystallogr Sect F Struct Biol Cryst Commun 62(Pt 10):944–948
Article PubMed Google Scholar - Haring M, Peng X, Brugger K, Rachel R, Stetter KO, Garrett RA, Prangishvili D (2004) Morphology and genome organization of the virus PSV of the hyperthermophilic archaeal genera Pyrobaculum and Thermoproteus: a novel virus family, the Globuloviridae. Virology 323(2):233–242
Article CAS PubMed Google Scholar - Peng X, Blum H, She Q, Mallok S, Brugger K, Garrett RA, Zillig W, Prangishvili D (2001) Sequences and replication of genomes of the archaeal rudiviruses SIRV1 and SIRV2: relationships to the archaeal lipothrixvirus SIFV and some eukaryal viruses. Virology 291(2):226–234
Article CAS PubMed Google Scholar - Oke M, Ching RT, Carter LG, Johnson KA, Liu H, McMahon SA, White MF, Bloch C Jr, Botting CH, Walsh MA, Latiff AA, Kennedy MW, Cooper A, Naismith JH (2008) Unusual chromophore and cross-links in ranasmurfin: a blue protein from the foam nests of a tropical frog. Angew Chem Int Ed Engl 47(41):7853–7856
Article CAS PubMed Google Scholar - Marsden RL, Orengo CA (2008) Target selection for structural genomics: an overview. Methods Mol Biol 426:3–25
Article CAS PubMed Google Scholar - Yard BA, Carter LG, Johnson KA, Overton IM, Dorward M, Liu H, McMahon SA, Oke M, Puech D, Barton GJ, Naismith JH, Campopiano DJ (2007) The structure of serine palmitoyltransferase; gateway to sphingolipid biosynthesis. J Mol Biol 370(5):870–886
Article CAS PubMed Google Scholar - McMahon SA, Roberts GA, Johnson KA, Cooper LP, Liu H, White JH, Carter LG, Sanghvi B, Oke M, Walkinshaw MD, Blakely GW, Naismith JH, Dryden DT (2009) Extensive DNA mimicry by the ArdA anti-restriction protein and its role in the spread of antibiotic resistance. Nucleic Acids Res 37(15):4887–4897
Article CAS PubMed Google Scholar - Sheikh MA, Potter JA, Johnson KA, Sim RB, Boyd EF, Taylor GL (2008) Crystal structure of VC1805, a conserved hypothetical protein from a Vibrio cholerae pathogenicity island, reveals homology to human p32. Proteins 71(3):1563–1571
Article CAS PubMed Google Scholar - Fairhead M, Johnson KA, Kowatz T, McMahon SA, Carter LG, Oke M, Liu H, Naismith JH, van der Walle CF (2008) Crystal structure and silica condensing activities of silicatein alpha-cathepsin L chimeras. Chem Commun (Camb) 15:1765–1767
Article Google Scholar
Acknowledgments
THE SSPF was supported by grants from The Scottish Funding Council (references SSPF and SULSA), The Biotechnology and Biological Sciences Research Council (reference BB/S/B14450), European Union under framework 7 (reference Aeropath).
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Author information
Author notes
- Lester G. Carter
Present address: Stanford Synchrotron Radiation Light Source, 2575 Sand Hill Road, MS 69, Menlo Park, CA, 94025, USA - Kenneth A. Johnson
Present address: The Norwegian Structural Biology Centre, University of Tromsø, 9037, Tromsø, Norway - Nadine D. Weikart
Present address: Faculty of Chemistry, Technische Universität Dortmund, Otto-Hahn-Str. 6, 44227, Dortmund, Germany - Nadia Kadi
Present address: Institute of Cancer Research, 15 Cotswold Road, Belmont, Sutton, Surrey, SM2 5NG, UK - Mark Dorward
Present address: Division of Signal Transduction Therapy, College of Life Sciences, University of Dundee, Dundee, DD1 5EH, Scotland, UK - Michal Zawadzki
Present address: Syngenta Ltd, Jealott’s Hill International Research Centre, Bracknell, Berkshire, RG42 6EY, UK - Christopher Cozens
Present address: Medical Research Council Laboratory of Molecular Biology, Hills Road, Cambridge, CB2 0QH, UK - Helen Falconer
Present address: Institute of Structural and Molecular Biology, Edinburgh University, Kings Buildings, Edinburgh, EH9 3JR, UK - Ian M. Overton
Present address: MRC Human Genetics Unit, Crewe Road South, Edinburgh, EH4 2XU, UK
Authors and Affiliations
- Biomedical Sciences Research Complex, University of St Andrews, St Andrews, KY16 9ST, UK
Muse Oke, Lester G. Carter, Kenneth A. Johnson, Huanting Liu, Stephen A. McMahon, Xuan Yan, Melina Kerou, Nadine D. Weikart, Md. Arif Sheikh, Stefan Schmelz, Mark Dorward, Michal Zawadzki, Christopher Cozens, Helen Falconer, Helen Powers, Catherine H. Botting, Peter J. Coote, Ulrich Schwarz-Linek, Garry L. Taylor, Malcolm F. White & James H. Naismith - Department of Chemistry, University of Warwick, Coventry, CV4 7AL, UK
Nadia Kadi, Prakash Patel & Gregory L. Challis - Division of Biological Chemistry and Drug Discovery, College of Life Sciences, University of Dundee, Dundee, DD1 5EH, Scotland, UK
Ian M. Overton, C. A. Johannes van Niekerk & Geoffrey J. Barton - Department of Biology, Archaea Centre, University of Copenhagen, Ole Maaløes Vej 5, 2200, Copenhagen N, Denmark
Xu Peng & Roger A. Garrett - Institut Pasteur, 25 rue Dr. Roux, 75724, Paris cedex 15, France
David Prangishvili - EaStChem School of Chemistry, University of Edinburgh, The King’s Buildings, Edinburgh, EH9 3JJ, UK
David T. F. Dryden
Authors
- Muse Oke
You can also search for this author inPubMed Google Scholar - Lester G. Carter
You can also search for this author inPubMed Google Scholar - Kenneth A. Johnson
You can also search for this author inPubMed Google Scholar - Huanting Liu
You can also search for this author inPubMed Google Scholar - Stephen A. McMahon
You can also search for this author inPubMed Google Scholar - Xuan Yan
You can also search for this author inPubMed Google Scholar - Melina Kerou
You can also search for this author inPubMed Google Scholar - Nadine D. Weikart
You can also search for this author inPubMed Google Scholar - Nadia Kadi
You can also search for this author inPubMed Google Scholar - Md. Arif Sheikh
You can also search for this author inPubMed Google Scholar - Stefan Schmelz
You can also search for this author inPubMed Google Scholar - Mark Dorward
You can also search for this author inPubMed Google Scholar - Michal Zawadzki
You can also search for this author inPubMed Google Scholar - Christopher Cozens
You can also search for this author inPubMed Google Scholar - Helen Falconer
You can also search for this author inPubMed Google Scholar - Helen Powers
You can also search for this author inPubMed Google Scholar - Ian M. Overton
You can also search for this author inPubMed Google Scholar - C. A. Johannes van Niekerk
You can also search for this author inPubMed Google Scholar - Xu Peng
You can also search for this author inPubMed Google Scholar - Prakash Patel
You can also search for this author inPubMed Google Scholar - Roger A. Garrett
You can also search for this author inPubMed Google Scholar - David Prangishvili
You can also search for this author inPubMed Google Scholar - Catherine H. Botting
You can also search for this author inPubMed Google Scholar - Peter J. Coote
You can also search for this author inPubMed Google Scholar - David T. F. Dryden
You can also search for this author inPubMed Google Scholar - Geoffrey J. Barton
You can also search for this author inPubMed Google Scholar - Ulrich Schwarz-Linek
You can also search for this author inPubMed Google Scholar - Gregory L. Challis
You can also search for this author inPubMed Google Scholar - Garry L. Taylor
You can also search for this author inPubMed Google Scholar - Malcolm F. White
You can also search for this author inPubMed Google Scholar - James H. Naismith
You can also search for this author inPubMed Google Scholar
Corresponding author
Correspondence toJames H. Naismith.
Additional information
Muse Oke, Lester G. Carter, Kenneth A. Johnson, Huanting Liu, Stephen A. McMahon—Joint first authors.
Electronic supplementary material
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License (https://creativecommons.org/licenses/by-nc/2.0), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Oke, M., Carter, L.G., Johnson, K.A. et al. The Scottish Structural Proteomics Facility: targets, methods and outputs.J Struct Funct Genomics 11, 167–180 (2010). https://doi.org/10.1007/s10969-010-9090-y
- Received: 09 March 2010
- Accepted: 06 April 2010
- Published: 24 April 2010
- Issue Date: June 2010
- DOI: https://doi.org/10.1007/s10969-010-9090-y