Different molecular patterns in glioblastoma multiforme subtypes upon recurrence (original) (raw)
Abstract
One of the hallmarks of glioblastoma is its inherent tendency to recur. At this point patients with relapsed GBM show a survival time of only few months. The molecular basis of the recurrence process in GBM is still poorly understood. The aim of the present study was to investigate the genetic profile of relapsed GBM compared to their respective primary tumors. We have included 20 paired GBMs. In all tumor samples, we have analyzed p53 and PTEN status by sequencing analysis, EGFR amplification by semiquantitative PCR and a wide-genome fingerprinting was performed by microsatellite analysis. Among primary GBM, we observed twelve type 2 GBM, four type 1 GBM and four further GBM showing neither p53 mutations nor EGFR amplification (non-type 1–non-type 2 GBM). Upon recurrence, we have detected two molecular patterns of tumor progression: GBM initially showing either type 1 or type 2 profiles conserved them at the time of relapse. In contrast, non-type 1–non-type 2 GBM acquired the typical pattern of type 2 GBM and harbor EGFR amplification without p53 mutation. New PTEN mutations upon relapse were only detected in type 2 GBM. Additional LOH were more frequently identified in relapses of type 2 GBM than in those showing the type 1 signature. Taken together, our results strongly suggest that recurrences of GBM may display two distinct pattern of accumulation of molecular alterations depending on the profile of the original tumor.
Similar content being viewed by others
Introduction
Glioblastoma multiforme is the most frequent primary brain tumor in adults. One of its characteristic features is the intrinsic tendency to recur despite aggressive therapy [1]. Upon recurrence, the median survival of patients is only few months [2] whereas the overall median survival time is 15 months. This has not significantly changed in the last 20 years despite of advances in surgery, radio- und chemotherapy [3, 4] A moderate improvement of the 2-year survival rate has been achieved after therapy with the alkylating drug temozolomide in GBM with hypermethylated MGMT compared to those with unmethylated gene (46% vs. 26%) [3, 5, 6].
GBM is one of the most intensively investigated human malignancy but the molecular mechanisms associated with recurrence are still poorly understood. Saxena et al. [7] reported a high rate of homozygous deletion of the genes p16 INK4 and p15 INK4a in GBM relapses. In a further report, this author investigated 10 GBM and their corresponding relapses and observed that recurrences displayed the pattern of genetic alterations frequently observed in the de novo pathway (GBM without a low grade precursor lesion) [8]. Hulsebos et al. [9] analyzed 12 match paired GBM and found in relapse cases new LOH at chromosome regions 1p36, 19q13, 10q23 and 1q25. In contrast to the former study, GBM relapses encompassing new p53 mutation or EGFR amplification were not observed.
The identification of molecular features associated with recurrences of GBM is of major importance, since a better understanding of this process might provide clues for the development of efficient treatments. In the present analysis, we have examined the molecular signatures of forty tumors consisting in relapsed GBM and their corresponding primary neoplasms which had undergone surgery and radio-chemotherapy. This included the status of p53, PTEN and EGFR as well as a wide genome LOH analysis with thirteen highly informative microsatellite markers at chromosome regions 17p13, 10q23, 1p35-36, 19q13, 13q14, 9p21.
Materials and methods
Patient population and tumor samples
All patients had undergone surgery with the goal of maximal possible tumor resection followed by fractionated radiotherapy (mean dose: 58 Gray) and chemotherapy. Forty paired tumors from twenty patients were available. Informed consent for samples and data analysis from each patient or the patient’s caretaker was obtained.
Tumor samples were immediately frozen in liquid nitrogen after surgical resection and stored at −80°C. Tumor tissue was evaluated by experienced pathologists according to the 2000 WHO classification criteria. DNA from tumor specimens was isolated applying the QIAamp® DNA Mini Kit (Qiagen, Hilden, Germany).
LOH analysis
LOH of chromosome 10q was studied with markers covering reported deletions on 10q23-24: D10S215, D10S541 and the intragenic PTEN marker PTENCA. Analysis of LOH at 17p13 was performed with a p53 intragenic localized marker. Allelic losses at 9p21 (p16 INK4a, p14 ARF and p15) and 13q14 (RB1) were studied with primers D9S171, D9S1748, D9S1749, D13S153 and D13S267. Loss of heterozygosity at 1p35-p36 and 19q13 was assessed with the markers D1S468, D1S482, D19S112 and D19S412. Amplified PCR products were analyzed on a denaturing 6.5% Long Ranger polyacrylamide gel on an Automated Laser Fluorescence sequencing device and analyzed using the ALLELELINKS® 1.00 software (Amersham Pharmacia Biotech, Freiburg, Germany). Evaluation of LOH was performed as described [10].
Semiquantitative PCR-analysis
For studying amplification of EGFR a differential duplex-PCR with the CFTR gene marker was carried out, as described [11]. Briefly, we calculated the EGFR/CFTR ratios [_x_] from peripheral blood DNA of 20 healthy Caucasian adults. A value [x + 3SD] was considered as the cut-off level for the normal gene copy number. Ratios higher than [x + 3SD] were regarded as evidence of more than 2 copies of the EGFR gene.
Sequencing analysis of p53 and PTEN
Mutational analysis of p53 and PTEN were performed in the DNA from both tumors and corresponding leukocytes. After amplifying all exons and intron-exon boundaries, we analyzed the PCR products on 1% agarose gel and excised and eluted them in sterile H2O. Subsequently, they were subjected to cycle sequencing reactions using the Thermo Sequenase® Fluorescent Cycle Sequencing kit (Amersham Pharmacia Biotech). The cycle sequencing products were resolved using a denaturing 6.5% Long Ranger polyacrylamide gel on a sequencing device. PTEN primer sequences and PCR conditions were previously described [12]. P53 primer sequences and PCR conditions are based on genome database entries (http://www.ncbi.nlm.nih.gov). Briefly, PCR reactions were carried out using a mixture containing 150 ng DNA, 10 mM Tris, 2 mM MgCl2, HCl (pH 8.3), 0.2 mM dNTP, 10 mM concentrations of each primer and 0.5 unit of Taq DNA Polymerase. Temperature cycles and times for PCR reactions were: denaturation at 94°C for 30 s, annealing at 65°C for 30 s, and extension at 72°C for 30 s. Each PCR reaction was preceded by a 3-min denaturation at 94°C, and the final cycle was followed by a 3-min extension at 72°C. The total number of cycles for PCR amplification was 25–30, depending on the sample DNA.
Statistical analysis
Student _t_-test, Chi-square test, Fisher’s exact test and Mann–Whitney U-test, were performed to compare differences between groups depending on the analyzed variables. Spearman correlation test was used to evaluate the correlation between two parameters. Confidence interval (CI) was obtained through logistic regression. Survival studies were performed with the Kaplan Meier analysis and the log-rank test. A value of P < 0.05 was considered to be significant. Analyses were performed with the SPSS software (version 10, SPSS Inc., Chicago, IL, USA).
Results
Clinico-pathological characteristics of the patients
The male/female ratio was 1:1. The median age at diagnosis of primary tumor was 59.5 years (range: 27–69; SD: 12.25). The median time to tumor recurrence was 7.5 months (range: 3–24 months; SD: 5.1; 95%-CI: 5.3–7.4 months). With one only exception, all patients displayed at diagnosis a Karnofsky performance score (KPS) of >70 points. An association between age or KPS and TTR was not evidenced (P = 0.35, ρ = 0.30, Spearman correlation test). There were no significant associations between mutation of p53 or EGFR amplification and either survival time (P = 0.250 and 0.127, respectively, log-rank test and Kaplan–Meier) or time to tumor relapse (P = 0.210 and 0.287, respectively, log-rank test and Kaplan–Meier).
Genetic analyses
In primary GBM we have observed EGFR amplification without mutations of p53 in 12 cases, thus indicating a type II GBM profile. Four further cases showed p53 mutations (Table 1, Fig. 1) in the absence of EGFR amplification, which was indicative of a type 1 GBM signature. In four GBM neither p53 mutations nor EGFR amplification were identified (non-type 1–non-type 2 GBM).
Table 1 Sequence variations of p53 in type 1 glioblastomas
Fig. 1
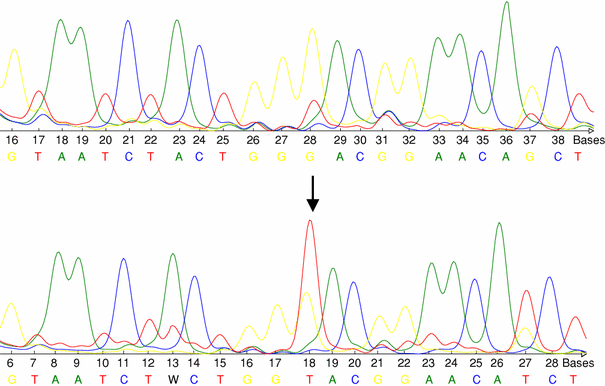
Upper row: wild type sequence of p53. Lower row: base exchange mutation (GGA→GTA) of p53 (exon 8, codon 266), predicting the aminoacid substitution Gly→Val. The arrow indicates the base exchange
At the time of recurrence, every type 1 GBM and type 2 GBM conserved their genetic pattern concerning p53 mutational status and EGFR amplification. On the other hand, relapses of non-type 1–non-type 2 GBM showed in all cases a new EGFR amplification, thus acquiring the signature of type 2 GBM. Additional PTEN mutations in relapse cases were observed only among type 2 GBM (Table 2). Overall, additional LOH were observed more frequently in relapses of type 2 GBM (83.3%), than in those of type 1 (50%) or non-type 1–non-type 2 tumors (25%). Further results are shown in Table 3.
Table 2 Sequence variations of PTEN in first and relapse glioblastomas
Table 3 Review of genetic changes of primary and relapsed glioblastomas
Among p53 mutations there were one C:G to A:T transition mutation at CpG site (codon 273), one C>T transition at non-CpG site (codon 278), two transversion mutations at non-CpG sites (G>C at codon 14 and G>T at codon 266, Fig. 1) as well as one validated polymorphism in exon 4 (G>C at codon 72). Moreover, we have observed two validated intronic polymorphisms in intron 3 (IVS3+21_37dup) and intron 2 (IVS2+38G>C). One of the mutations (c.14G>C) has not been previously described in p53 data bases (http://www.p53.iarc.fr, http://www.p53.free.fr). A detailed characterization of p53 sequence variations is shown in Table 1.
Concerning PTEN mutations, we have identified four transition mutations in exons 2, 5, 6 and 7, in all cases predicting aminoacid substitution. Furthermore, there was an acceptor site splice mutation at the conserved junction nucleotides (IVS8-2A>G). Finally, we have observed two transversion mutations in exon 5 (c.105G>T) and exon 4 (c.75C>A, Fig. 2) as well as four intronic polymorphisms (Table 2). The transition in exon 6 (c.177A>G), the splicing mutation and two of the polymorphisms have not been previously reported (Table 2).
Fig. 2
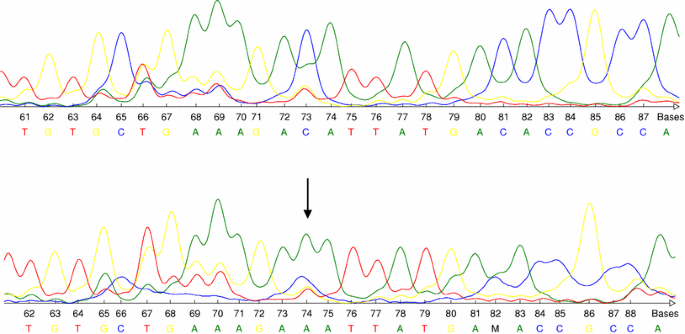
Upper row: wild type sequence of PTEN. Lower row: base exchange mutation (CAT→AAT) of PTEN (exon 4, codon 75) predicting the aminoacid substitution His→Asn. The arrow indicates the base exchange
The results of EGFR amplification analysis and wide-genome LOH are shown in Table 3 and Supplementary Table 1 (EGFR/CFTR ratios). Interestingly, GBM cases harboring mutations of p53 always displayed LOH at 17p13 as well. Similarly, tumors featuring PTEN mutations showed LOH at 10q23, at least in one of the microsatellites used to analyze this region.
Discussion
GBM mostly exceed in its occurrence and mortality beyond any other primary brain tumor in adults [13]. On molecular level, two subsets of GBM have been recognized on the basis of genetic make-up and clinical features [14, 15]. Type 2 (de novo) GBM occurs commonly in elderly patients and exhibit overexpression and/or amplification of EGFR. Type 1 (secondary) GBM shows typically histological progression from a previous low grade astrocytoma and affects young patients. Paradigmatically, mutations of p53 will be observed (for review see [15]). Most of GBM cases belong to the type 2 group and genetic events are mutually exclusive for EGFR and p53. Moreover, there are GBM without either EGFR amplification or p53 mutations (non-type 1–non-type 2 GBM) [16, 17]. During the past decade, accumulated evidence pointed to that type 1 and type 2 GBM constitute distinct disease entities developing through different genetic pathways [13, 14], showing different RNA and protein patterns [18, 19] and also probably differing in response to treatment [15].
After multimodal therapy, GBM almost always recur. Although this is a capital feature of GBM, only few investigations with series of match paired patients are available from the literature and available data is partially contradictory [7–9, 20, 21]. We aimed to investigate whether molecular subtypes of GBM also show different profiles at the time of relapse.
In our genetic analysis, we have observed two patterns of tumor recurrence (Fig. 3): GBM type 1 and type 2 retained upon recurrence their genetic alterations affecting p53 and EGFR. In contrast, all relapses of non-type 1–non-type 2 GBM showed a new EGFR amplification, thus they acquired a type 2 GBM profile. Accumulation of additional genetic alterations upon relapse included PTEN mutations in four type 2 GBM (but not in the other subtypes). Additional LOH were more frequently observed in relapses of type 2 GBM (in 10/12 cases) compared to relapses of type 1 GBM and non-type 1–non-type 2 GBM (Table 3). The accumulation of alterations during the clonal expansion of transformed glial cells is well known in GBM, confer tumor cells a growth advantage and underlies the genetic heterogeneity of GBM [22, 23]. This phenomenon contributes to explain the molecular differences between primary tumors and relapses [9, 24, 25].
Fig. 3
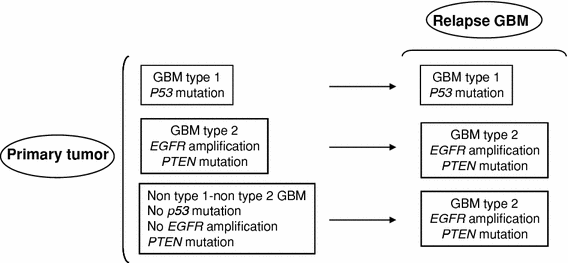
Graphic showing two molecular patterns of GBM recurrence
As a part of one previous investigation we had characterized the promoter methylation status of the DNA repair gene MGMT in all type 1 GBM (Table 3, cases G3, G7, G14 and G16), non-type 1–non-type 2 GBM (Table 3, cases G2, G6, G8 and G9) as well as eight type 2 GBM here described [26]. This analysis together with the present results of p53 and EGFR have evidenced similar rates of MGMT methylation for the three GBM subtypes, and they are in accordance with the overall methylation rates reported in glioblastoma [5, 27]. Typically, MGMT was methylated in the first GBM and remained always methylated upon relapse. Thus, our results also confirmed the observation that, in the pathogenesis and progression of GBM, MGMT becomes hypermethylated at an early stage [28, 29] and remains methylated through progression, a situation which was has been described in small adenomas and carcinomas of colorectal origin as well [30]. The p53 mutational analysis in those type 1 GBM with methylated MGMT showed G:C to A:T transition within CpG site (c.273 CGT→CAT) in two cases. Further mutations of p53 in tumors with methylated MGMT (1 case) occurred in non-CpG sites. From p53 data bases entries, approximately 52% of mutational events are transition mutations and, of this subset, about 72% are G:C to A:T transitions (http://www.p53.iarc.fr, http://www.p53.free.fr) [31], although the signature of the mutational spectrum varies according to tumor type. It was previously suggested a link [29] between MGMT methylation and the presence of G:C to A:T transition mutations in p53 in gliomas. In the present study we could confirm the former in two patients (cases G14p, G14r, G16p and G16r, Table 1); in these cases, G:C to A:T transitions could be attributable to a inactivation in MGMT that allows O _6_-methylguanine to persist and be read as an adenine. Our observations contrast with a recent investigation about GBM reporting only 4% of mutations of p53 in tumors with methylated MGMT within CpG sites [32] although we should take in mind the size of our type 1 GBM collective, clearly too short to state a definite tendency.
Strikingly, we have identified in cases G7p and G7r the exonic p53 polymorphism c.72C>G (Arg72Pro) together with the intronic polymorphism IVS3+21_37dup (_P53_PIN3). Several investigations have pointed the association between p53 polymorphisms and an increased risk for different cancers: for instance, in the case of the exonic c.72 variant such association was suggested since the p53 protein with Arg72 is more efficient in inducing apoptosis than the one with the Pro72 variant [33]. In clinical studies, the variants c.72 Pro/Pro and c.72 Arg/Pro have been associated with an elevated risk of lung cancer [34, 35]. Recently, intron 3 duplication (IVS3+21_37dup) has been found to be associated with increased risk of colorectal cancer [36]. However, the authors could not determine whether this intron 3 duplication alone influences mRNA stability or if this effect requires the codon Pro72 variant. To our knowledge, the occurrence of both polymorphisms in GBM patients has not been reported before and pointed to a possible role in brain cancer as well.
With respect to PTEN, we have identified mutations only in type 2 GBM. In four cases, PTEN mutations occurred in both the first GBM and the relapse. In four further cases, new mutations were observed in the recurrences but not in the first tumor. The rate and pattern of mutations are in accordance with previous reported data [37]. All tumors with PTEN mutations also harbored LOH at the chromosome region 10q23, which pointed to the inactivation of this tumor suppressor gene and make patent its contribution to the tumorigenesis and progression in GBM.
Taken together, our results strongly suggest that GBM relapses, compared to the corresponding first tumor, accumulate additional molecular alterations and may develop along two distinct pathways, which contribute to delineate the different profiles of type 1, type 2 and non-type 1–non-type 2 GBM.
Abbreviations
GBM:
Glioblastoma multiforme
TTR:
Time to tumor relapse
MGMT:
O _6_-methyl-guanine-methyl-transferase
CFTR:
Cystic fibrosis transmembrane regulator
KPS:
Karnofsky performance score
References
- Salcman M, Kaplan R (1991) Intracranial tumors in adults. In: Salcman M (ed) Neurology of brain tumors. Williams & Wilkins, Baltimore, pp 1339–1352
Google Scholar - Brandes AA, Ermani M, Basso U, Amistà P, Berti F, Scienza R, Rotilio A, Pinna G, Gardiman M, Monfardini S (2001) Temozolomide as a second-line systemic regimen in recurrent high-grade glioma: a phase II study. Ann Oncol 12:255–257
Article CAS PubMed Google Scholar - Stupp R, Dietrich PY, Ostermann-Kraljevic S, Pica A, Maillard I, Maeder P, Meuli R, Janzer R, Pizzolato G, Miralbell R, Porchet F, Regli L, de Tribolet N, Mirimanoff RO, Leyvraz S (2002) Promising survival for patients with newly diagnosed glioblastoma multiforme treated with concomitant radiation plus temozolomide followed by adjuvant temozolomide. J Clin Oncol 20:1375–1382
Article CAS PubMed Google Scholar - Walker MD, Green SB, Byar DP, Alexander E Jr, Batzdorf U, Brooks WH, Hunt WE, MacCarty CS, Mahaley MS Jr, Mealey J Jr, Owens G, Ransohoff J 2nd, Robertson JT, Shapiro WR, Smith KR Jr, Wilson CB, Strike TA (1980) Randomized comparisons of radiotherapy and nitrosoureas for the treatment of malignant glioma after surgery. N Engl J Med 303:1323–1329
CAS PubMed Google Scholar - Gorlia T, van den Bent MJ, Hegi ME, Mirimanoff RO, Weller M, Cairncross JG, Eisenhauer E, Belanger K, Brandes AA, Allgeier A, Lacombe D, Stupp R (2008) Nomograms for predicting survival of patients with newly diagnosed glioblastoma: prognostic factor analysis of EORTC and NCIC trial 26981-22981/CE.3. Lancet Oncol 9:29–38
Article PubMed Google Scholar - Stupp R, Mason WP, van den Bent MJ, Weller M, Fisher B, Taphoorn MJ, Belanger K, Brandes AA, Marosi C, Bogdahn U, Curschmann J, Janzer RC, Ludwin SK, Gorlia T, Allgeier A, Lacombe D, Cairncross JG, Eisenhauer E, Mirimanoff RO, European Organisation for Research and Treatment of Cancer Brain Tumor and Radiotherapy Groups; National Cancer Institute of Canada Clinical Trials Group (2005) Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N Engl J Med 352: 987–996
- Saxena A, Robertson JT, Ali IU (1996) Abnormalities of p16, p15 and CDK4 genes in recurrent malignant astrocytomas. Oncogene 13:661–664
CAS PubMed Google Scholar - Saxena A, Shriml LM, Dean M, Ali IU (1999) Comparative molecular genetic profiles of anaplastic astrocytomas/glioblastomas multiforme and their subsequent recurrences. Oncogene 18:1385–1390
Article CAS PubMed Google Scholar - Hulsebos TJ, Troost D, Leenstra S (2004) Molecular-genetic characterisation of gliomas that recur as same grade or higher grade tumours. J Neurol Neurosurg Psychiatry 75:723–726
Article CAS PubMed Google Scholar - Cawkwell L, Lewis FA, Quirke P (1994) Frequency of allele loss of DCC, p53, RB1, WT1, NF1, NM23 and APC/MCC in colorectal cancer assayed by fluorescent multiplex polymerase chain reaction. Br J Cancer 70:813–818
CAS PubMed Google Scholar - Hunter SB, Abbott K, Varma VA, Olson JJ, Barnett DW, James D (1995) Reliability of differential PCR for the detection of EGFR and MDM2 gene amplification in DNA extracted from FFPE glioma tissue. J Neuropathol Exp Neurol 54:57–64
Article CAS PubMed Google Scholar - Liaw D, Marsh DJ, Li J, Dahia PL, Wang SI, Zheng Z, Bose S, Call KM, Tsou HC, Peacocke M, Eng C, Parsons R (1997) Germline mutations of the PTEN gene in Cowden disease, an inherited breast and thyroid cancer syndrome. Nat Genet 16:64–67
Article CAS PubMed Google Scholar - Ohgaki H, Kleihues P (2005) Population-based studies on incidence, survival rates, and genetic alterations in astrocytic and oligodendroglial gliomas. J Neuropathol Exp Neurol 64:479–489
CAS PubMed Google Scholar - Ohgaki H, Dessen P, Jourde B, Horstmann S, Nishikawa T, Di Patre PL, Burkhard C, Schüler D, Probst-Hensch NM, Maiorka PC, Baeza N, Pisani P, Yonekawa Y, Yasargil MG, Lütolf UM, Kleihues P (2004) Genetic pathways to glioblastoma: a population-based study. Cancer Res 64:6892–6899
Article CAS PubMed Google Scholar - Ohgaki H, Kleihues P (2007) Genetic pathways to primary and secondary glioblastoma. Am J Pathol 170:1445–1453
Article CAS PubMed Google Scholar - Benjamin R, Capparella J, Brown A (2003) Classification of glioblastoma multiforme in adults by molecular genetics. Cancer J 9:82–90
Article CAS PubMed Google Scholar - Lang F, Miller D, Koslow M, Newcomb E (1994) Pathways leading to glioblastoma multiforme: a molecular analysis of genetic alterations in 65 astrocytic tumors. J Neurosurg 81:427–436
Article CAS PubMed Google Scholar - Furuta M, Weil RJ, Vortmeyer AO, Huang S, Lei J, Huang TN, Lee YS, Bhowmick DA, Lubensky IA, Oldfield EH, Zhuang Z (2004) Protein patterns and proteins that identify subtypes of glioblastoma multiforme. Oncogene 23:6806–6814
Article CAS PubMed Google Scholar - Tso CL, Freije WA, Day A, Chen Z, Merriman B, Perlina A, Lee Y, Dia EQ, Yoshimoto K, Mischel PS, Liau LM, Cloughesy TF, Nelson SF (2006) Distinct transcription profiles of primary and secondary glioblastoma subgroups. Cancer Res 66:159–167
Article CAS PubMed Google Scholar - Spiegl-Kreinecker S, Pirker C, Marosi C, Buchroithner J, Pichler J, Silye R, Fischer J, Micksche M, Berger W (2007) Dynamics of chemosensitivity and chromosomal instability in recurrent glioblastoma. Br J Cancer 96:960–969
Article CAS PubMed Google Scholar - Stark AM, Witzel P, Strege RJ, Hugo HH, Mehdorn HM (2003) p53, mdm2, EGFR, and msh2 expression in paired initial and recurrent glioblastoma multiforme. J Neurol Neurosurg Psychiatry 74:779–783
Article CAS PubMed Google Scholar - Berkman RA, Clark WC, Saxena A, Robertson JT, Oldfield EH, Ali IU (1992) Clonal composition of glioblastoma multiforme. J Neurosurg 77:432–437
Article CAS PubMed Google Scholar - Morse RP, Darras BT, Ye Z, Wu JK (1994) Clonal analysis of human astrocytomas. J. Neurooncol 21:151–157
Article CAS PubMed Google Scholar - Grasbon-Frodl EM, Kreth FW, Ruiter M, Schnell O, Bise K, Felsberg J, Reifenberger G, Tonn JC, Kretzschmar HA (2007) Intratumoral homogeneity of MGMT promoter hypermethylation as demonstrated in serial stereotactic specimens from anaplastic astrocytomas and glioblastomas. Int J Cancer 121:2458–2464
Article CAS PubMed Google Scholar - Parkinson JF, Wheeler HR, Clarkson A, McKenzie CA, Biggs MT, Little NS, Cook RJ, Messina M, Robinson BG, McDonald KL (2008) Variation of O(6)-methylguanine-DNA methyltransferase (MGMT) promoter methylation in serial samples in glioblastoma. J Neurooncol 87:71–78
Article CAS PubMed Google Scholar - Martínez R, Setien F, Voelter C, Casado S, Quesada MP, Schackert G, Esteller M (2007) CpG island promoter hypermethylation of the pro-apoptotic gene caspase-8 is a common hallmark of relapsed glioblastoma multiforme. Carcinogenesis 28:1264–1268
Article PubMed CAS Google Scholar - Hegi ME, Diserens AC, Gorlia T, Hamou MF, de Tribolet N, Weller M, Kros JM, Hainfellner JA, Mason W, Mariani L, Bromberg JE, Hau P, Mirimanoff RO, Cairncross JG, Janzer RC, Stupp R (2005) MGMT gene silencing and benefit from temozolomide in glioblastoma. N Engl J Med 352:997–1003
Article CAS PubMed Google Scholar - Esteller M, Hamilton SR, Burger PC, Baylin SB, Herman JG (1999) Inactivation of the DNA repair gene O6-methylguanine-DNA methyltransferase by promoter hypermethylation is a common event in primary human neoplasia. Cancer Res 59:793–797
CAS PubMed Google Scholar - Nakamura M, Watanabe T, Yonekawa Y, Kleihues P, Ohgaki H (2001) Promoter methylation of the DNA repair gene MGMT in astrocytomas is frequently associated with G:C→A:T mutations of the TP53 tumor suppressor gene. Carcinogenesis 22:1715–1719
Article CAS PubMed Google Scholar - Esteller M, Toyota M, Sanchez-Cespedes M, Capella G, Peinado MA, Watkins DN, Issa JP, Sidransky D, Baylin SB, Herman JG (2000) Inactivation of the DNA repair gene O6-methylguanine-DNA methyltransferase by promoter hypermethylation is associated with G to A mutations in K-ras in colorectal tumorigenesis. Cancer Res 60:2368–2371
CAS PubMed Google Scholar - Greenblatt MS, Bennett WP, Hollstein M, Harris CC (1994) Mutations in the p53 tumor suppressor gene: clues to cancer etiology and molecular pathogenesis Cancer Res 54: 4855–4878
Google Scholar - Cancer Genome Atlas Research Network (2008) Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature 455:1061–1068
Article CAS Google Scholar - Dumont P, Leu JI, Della Pietra ACIII, George DL, Murphy M (2003) The codon 72 polymorphic variants of p53 have markedly different apoptotic potential. Nat Genet 33:357–365
Article CAS PubMed Google Scholar - Wu X, Zhao H, Amos CI, Shete S, Makan N, Hong WK, Kadlubar FF, Spitz MR (2002) p53 genotypes and haplotypes associated with lung cancer susceptibility and ethnicity. J Natl Cancer Inst 94:681–690
CAS PubMed Google Scholar - Fan R, Wu MT, Miller D, Wain JC, Kelsey KT, Wiencke JK, Christiani DC (2000) The p53 codon 72 polymorphism and lung cancer risk. Cancer Epidemiol Biomark Prev 9:1037–1042
CAS Google Scholar - Gemignani F, Moreno V, Landi S, Moullan N, Chabrier A, Gutiérrez-Enríquez S, Hall J, Guino E, Peinado MA, Capella G, Canzian F (2004) A TP53 polymorphism is associated with increased risk of colorectal cancer and with reduced levels of TP53 mRNA. Oncogene 23:1954–1956
Article CAS PubMed Google Scholar - Bonneau D, Longy M (2000) Mutations of the human PTEN gene. Hum Mutat 16: 109–122
Google Scholar - Ishii N, Maier D, Merlo A, Tada M, Sawamura Y, Diserens AC, Van Meir EG (1999) Frequent co-alterations of TP53, p16/CDKN2A, p14ARF, PTEN tumor suppressor genes in human glioma cell lines. Brain Pathol 9:469–479
Article CAS PubMed Google Scholar - Liu J, Babaian DC, Liebert M, Steck PA, Kagan J (2000) Inactivation of MMAC1 in bladder transitional-cell carcinoma cell lines and specimens. Mol Carcinog 29:143–150
Article CAS PubMed Google Scholar - Pezzolesi MG, Li Y, Zhou XP, Pilarski R, Shen L, Eng C (2006) Mutation-positive and mutation-negative patients with Cowden and Bannayan-Riley-Ruvalcaba syndromes associated with distinct 10q haplotypes. Am J Hum Genet 79:923–934
Article CAS PubMed Google Scholar - Pleasants LM, Hansen MF (1994) Identification of a polymorphism in intron 2 of the p53 gene. Hum Genet 93:607–608
Article CAS PubMed Google Scholar - Poetsch M, Lorenz G, Kleist B (2002) Detection of new PTEN/MMAC1 mutations in head and neck squamous cell carcinomas with loss of chromosome 10. Cancer Genet Cytogenet 132:20–24
Article CAS PubMed Google Scholar - Zhang SJ, Endo S, Ichikawa T, Yoshimura J, Onda K, Tanaka R, Washiyama K, Kumanishi T (1999) Rare-type mutations of MMAC1 tumor suppressor gene in human glioma cell lines and their tumors of origin. Jpn J Cancer Res 90:934–941
CAS PubMed Google Scholar
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Author information
Authors and Affiliations
- Department of Neurosurgery, University of Göttingen, Robert-Koch-Str. 40, 37075, Göttingen, Germany
Ramon Martinez & Veit Rohde - Department of Neurosurgery, University of Dresden, Fetscherstr. 74, 01307, Dresden, Germany
Gabriele Schackert
Authors
- Ramon Martinez
You can also search for this author inPubMed Google Scholar - Veit Rohde
You can also search for this author inPubMed Google Scholar - Gabriele Schackert
You can also search for this author inPubMed Google Scholar
Corresponding author
Correspondence toRamon Martinez.
Electronic supplementary material
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License (https://creativecommons.org/licenses/by-nc/2.0), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Martinez, R., Rohde, V. & Schackert, G. Different molecular patterns in glioblastoma multiforme subtypes upon recurrence.J Neurooncol 96, 321–329 (2010). https://doi.org/10.1007/s11060-009-9967-4
- Received: 13 April 2009
- Accepted: 06 July 2009
- Published: 31 July 2009
- Issue Date: February 2010
- DOI: https://doi.org/10.1007/s11060-009-9967-4