Fit-for-Purpose Method Development and Validation for Successful Biomarker Measurement (original) (raw)
Abstract
Despite major advances in modern drug discovery and development, the number of new drug approvals has not kept pace with the increased cost of their development. Increasingly, innovative uses of biomarkers are employed in an attempt to speed new drugs to market. Still, widespread adoption of biomarkers is impeded by limited experience interpreting biomarker data and an unclear regulatory climate. Key differences preclude the direct application of existing validation paradigms for drug analysis to biomarker research. Following the AAPS 2003 Biomarker Workshop (J. W. Lee, R. S. Weiner, J. M. Sailstad, et al. Method validation and measurement of biomarkers in nonclinical and clinical samples in drug development. A conference report. Pharm Res 22:499–511, 2005), these and other critical issues were addressed. A practical, iterative, “fit-for-purpose” approach to biomarker method development and validation is proposed, keeping in mind the intended use of the data and the attendant regulatory requirements associated with that use. Sample analysis within this context of fit-for-purpose method development and validation are well suited for successful biomarker implementation, allowing increased use of biomarkers in drug development.
Access this article
Subscribe and save
- Get 10 units per month
- Download Article/Chapter or eBook
- 1 Unit = 1 Article or 1 Chapter
- Cancel anytime Subscribe now
Buy Now
Price excludes VAT (USA)
Tax calculation will be finalised during checkout.
Instant access to the full article PDF.
Similar content being viewed by others
Abbreviations
AAPS:
American Association of Pharmaceutical Sciences
BQL:
below quantifiable limit
CDER:
Center for Drug Evaluation and Research
CMS:
Centers for Medicare and Medicaid Services
CLAS:
Clinical Ligand Assay Society
CLIA:
Clinical Laboratory Improvement Amendments
CLSI:
Clinical and Laboratory Standards Institute
GLP:
Good Laboratory Practices
LBABFG:
Ligand Binding Assay Bioanalytical Focus Group
LOD:
lower limit of detection
LLOQ:
lower limit of quantification
MRD:
minimum required dilution
NCCLS:
National Committee for Clinical Laboratory Standards
PD:
pharmacodynamic
PK:
pharmacokinetic
QC:
Quality Controls
QL:
quantification limits
ULOQ:
upper limit of quantification
VEGF:
vascular endothelial growth factor
VS:
validation sample
References
- J. W. Lee R. S. Weiner J. M. Sailstad_et al._ (2005)ArticleTitleMethod validation and measurement of biomarkers in nonclinical and clinical samples in drug development. A conference report_Pharm. Res._ 22 499–511Occurrence Handle1:CAS:528:DC%2BD2MXjsFahu7c%3DOccurrence Handle15846456
CAS PubMed Google Scholar - J. A. DiMasi R. W. Hansen H. G. Grabowski (2003)ArticleTitleThe price of innovation: new estimates of drug development costs_J. Health Econ._ 22 151–185Occurrence Handle10.1016/S0167-6296(02)00126-1Occurrence Handle12606142
Article PubMed Google Scholar - J. M. Reichert (2003)ArticleTitleTrends in development and approval times for new therapeutics in the United States_Nat. Rev. Drug. Discov._ 2 695–702Occurrence Handle10.1038/nrd1178Occurrence Handle1:CAS:528:DC%2BD3sXmvVWlsb0%3DOccurrence Handle12951576
Article CAS PubMed Google Scholar - FDA Mar 2004 report. Innovation or Stagnation? Challenge and opportunity on the critical path to new medical products. Available at http://www.fda.gov/oc/initiatives/criticalpath/.
- E. Zerhouni (2003)ArticleTitleMedicine. The NIH roadmap_Science_ 302 63–72Occurrence Handle10.1126/science.1091867Occurrence Handle1:CAS:528:DC%2BD3sXnvVOrt78%3DOccurrence Handle14526066
Article CAS PubMed Google Scholar - G. J. Downing (Eds) (2000) Biomarkers and surrogate endpoints: clinical research and applications. Proceedings of the NIH–FDA Conference held on April 15–16, 1999 Elsevier New York
Google Scholar - G. Levy (1994)ArticleTitleMechanism-based pharmacodynamics modeling_Clin. Pharmacol. Ther._ 56 356–358Occurrence Handle1:STN:280:DyaK2M%2Fktlygsg%3D%3DOccurrence Handle7955796Occurrence Handle10.1038/clpt.1994.134
Article CAS PubMed Google Scholar - C. C. Peck W. H. Barr L. Z. Benet_et al._ (1992)ArticleTitleOpportunities for integration of pharmacokinetics, pharmacodynamics, and toxicokinetics in rational drug development_Pharm. Sci._ 81 600–610
Google Scholar - W. A. Colburn (1997)ArticleTitleSelecting and validating biologic markers for drug development_J. Clin. Pharmacol._ 37 355–362Occurrence Handle1:CAS:528:DyaK2sXjs1CnsLk%3DOccurrence Handle9156368
CAS PubMed Google Scholar - E. S. Vesell (2000)ArticleTitleAdvances in pharmacogenetics and pharmacogenomics_J. Clin. Pharmacol._ 40 930–938Occurrence Handle10.1177/00912700022009666Occurrence Handle1:CAS:528:DC%2BD3cXmsF2nsLg%3DOccurrence Handle10975065
Article CAS PubMed Google Scholar - Guidance for industry on bioanalytical method validation: availability. Fed. Regist. 66:28526–28527 (2001).
Google Scholar - Code of Federal Regulations, Title 21, Vol. 1. Good Laboratory Practice for Nonclinical Laboratory Studies. Revised April 1, 2001.
- Code of Federal Regulations, Title 42, Vol. 3. Clinical Laboratory Improvement Amendment. Revised October 1, 2001.
- National Committee for Clinical Laboratory Standards (CLSI), Document EP5-A: Evaluation of Precision Performance of Clinical Chemistry Devices: Approved Guideline. 1999. Document EP6-P: Evaluation of the Linearity of Quantitative Analytical Method: Proposed Guideline. 1986. Document EP7-P: Interference Testing in Clinical Chemistry: Proposed Guideline. 1986. Document EP9-A: Method Comparison and Bias Estimation Using Patient Samples: Approved Guideline. 1995.
- Biomarkers Definitions Working Group, Biomarkers and surrogate endpoints: preferred definitions and conceptual framework. Clin. Pharmacol. Ther. 69:89–95 (2001).
Google Scholar - J. A. Wagner (2002)ArticleTitleOverview of biomarkers and surrogate endpoints in drug development_Dis. Markers_ 18 41–46Occurrence Handle1:CAS:528:DC%2BD38Xns1CltbY%3DOccurrence Handle12364809
CAS PubMed Google Scholar - J. A. Wagner. Bridging preclinical and clinical development: biomarker validation and qualification, in R. Krishna and D. Howard (eds.), Dose Optimization in Drug Development, Marcel Dekker, New York, in press.
- J. W. Lee W. C. Smith G. D. Nordblom R. R. Bowsher (2003) Validation of assays for the bioanalysis of novel biomarkers C. Bloom R. A. Dean (Eds) Biomarkers in Clinical Drug Development Marcel Dekker New York 119–149
Google Scholar - A. R. Mire-Sluis Y. C. Barrett V. Devanarayan_et al._ (2004)ArticleTitleRecommendations for the design and of immunoassays used in the detection of host antibodies against biotechnology products_J. Immunol. Methods_ 289 1–16Occurrence Handle1:CAS:528:DC%2BD2cXlsFOrtbc%3DOccurrence Handle15251407
CAS PubMed Google Scholar - B. DeSilva W. Smith R. Weiner_et al._ (2003)ArticleTitleRecommendations for the bioanalytical method validation of ligand-binding assays to support pharmacokinetic assessments of macromolecules_Pharm. Res._ 20 1885–1900Occurrence Handle10.1023/B:PHAM.0000003390.51761.3dOccurrence Handle1:CAS:528:DC%2BD3sXptVChu7Y%3DOccurrence Handle14661937
Article CAS PubMed Google Scholar - V. P. Shah K. K. Midha S. Dighe_et al._ (1992)ArticleTitleAnalytical methods validation: bioavailability, bioequivalence, and pharmacokinetic studies_Pharm. Res._ 9 588–592
Google Scholar - ICH Guidelines, Text on validation of analytical procedures, Q2A; International Conference on Harmonization, Geneva, Switzerland, 1994.
- D. Borderie C. Roux B. Toussaint_et al._ (2001)ArticleTitleVariability in urinary excretion of bone resorption markers: limitations of a single determination in clinical practice_Clin. Biochem._ 34 571–577Occurrence Handle1:CAS:528:DC%2BD38XptVynsA%3D%3DOccurrence Handle11738394Occurrence Handle10.1016/S0009-9120(01)00269-7
Article CAS PubMed Google Scholar - C. A. Ray R. R. Bowsher W. C. Smith_et al._ (2001)ArticleTitleDevelopment, validation and implementation of a multiplex immunoassay for the simultaneous determination of five cytokines in human serum_J. Pharm. Biomed. Anal._ 36 1037–1044
Google Scholar - J. O. Westgard P. L. Barry M. R. Hunt_et al._ (1981)ArticleTitleA multi-rule Shewhart chart for quality control in clinical chemistry_Clin. Chem._ 27 493–501Occurrence Handle1:CAS:528:DyaL3MXhtlGmt7k%3DOccurrence Handle7471403
CAS PubMed Google Scholar - M. Feinber B. Boulanger W. Dewe_et al._ (2004)ArticleTitleNew advances in method validation and measurement uncertainty aimed at improving the quality of clinical data_Anal. Bioanal. Chem._ 380 502–514
Google Scholar - J. A. Rogers (2005)ArticleTitleStatistical limits for assessment of accuracy and precision in immunoassay validation: a review_J. Clin. Ligand Assay_ 27 256–261
Google Scholar - C. A. Ray, C. Dumaual, and M Willey, et al. Optimization of analytical and pre-analytical variables associated with an ex vivo cytokine secretion assay. J. Pharm. Biomed. Anal., In press.
- R. J. Carroll D. Ruppert (1988) Transformation and Weighting in Regression Chapman & Hall New York
Google Scholar - J. W. A. Findlay W. C. Smith J. W. Lee_et al._ (2000)ArticleTitleValidation of immunoassays for bioanalysis: a pharmaceutical industry perspective_J. Pharm. Biomed. Anal._ 21 1249–1273Occurrence Handle10.1016/S0731-7085(99)00244-7Occurrence Handle1:CAS:528:DC%2BD3cXot1ehOccurrence Handle10708409
Article CAS PubMed Google Scholar
Acknowledgments
The authors are grateful to Ronald R. Bowsher, PhD (Linco Diagnostic Services), for providing encouragement and critical review on the manuscript. We also thank Wesley Tanaka, PhD, and Omar Laterza, PhD (Merck & Co.) for providing input and suggestions.
Author information
Author notes
- Jean W. Lee
Present address: , 1 Amgen Center Drive, Mailstop 30E-3-B, Thousand Oaks, California, 91320, USA
Authors and Affiliations
- Formerly MDS Pharma Services, Lincoln, Nebraska, USA
Jean W. Lee - Merck and Company, Inc., Blue Bell, Pennsylvania, USA
Viswanath Devanarayan - Bristol-Myers Squibb, Princeton, New Jersey, USA
Yu Chen Barrett & Russell Weiner - BAS Analytics Ltd., Kenilworth, UK
John Allinson - Pfizer Global Research and Development, Ann Arbor, Michigan, USA
Scott Fountain - Protein Design Labs, Inc., Fremont, California, USA
Stephen Keller - Wyeth Research, Collegeville, Pennsylvania, USA
Ira Weinryb - Millenium Pharmaceuticals, Cambridge, Massachusetts, USA
Marie Green - Quest Pharmaceutical Services, Newark, Delaware, USA
Larry Duan - Pfizer Global Research and Development, Groton–New London, Connecticut, USA
James A. Rogers & Robert Millham - Therakos, Inc., Exton, Pennsylvania, USA
Peter J. O'Brien - Trimeris Inc., Morrisville, North Carolina, USA
Jeff Sailstad - Covance Laboratories, Inc., Chantilly, Virginia, USA
Masood Khan - Eli Lilly and Company, Indianapolis, Indiana, USA
Chad Ray - Merck and Company, Inc., Rahway, New Jersey, USA
John A. Wagner
Authors
- Jean W. Lee
You can also search for this author inPubMed Google Scholar - Viswanath Devanarayan
You can also search for this author inPubMed Google Scholar - Yu Chen Barrett
You can also search for this author inPubMed Google Scholar - Russell Weiner
You can also search for this author inPubMed Google Scholar - John Allinson
You can also search for this author inPubMed Google Scholar - Scott Fountain
You can also search for this author inPubMed Google Scholar - Stephen Keller
You can also search for this author inPubMed Google Scholar - Ira Weinryb
You can also search for this author inPubMed Google Scholar - Marie Green
You can also search for this author inPubMed Google Scholar - Larry Duan
You can also search for this author inPubMed Google Scholar - James A. Rogers
You can also search for this author inPubMed Google Scholar - Robert Millham
You can also search for this author inPubMed Google Scholar - Peter J. O'Brien
You can also search for this author inPubMed Google Scholar - Jeff Sailstad
You can also search for this author inPubMed Google Scholar - Masood Khan
You can also search for this author inPubMed Google Scholar - Chad Ray
You can also search for this author inPubMed Google Scholar - John A. Wagner
You can also search for this author inPubMed Google Scholar
Corresponding author
Correspondence toJean W. Lee.
Appendices
Appendix A
Precision Profiles of Calibration Curves
The Precision Profile is a plot of the coefficient of variation of the calibrated concentration vs. the true concentration in log scale. The standard error of the calibrated concentration should include the variability of both the assay response and the calibration curve. Complex computation programs can be implemented with the help of a statistician (29) or simple calculations can be performed in an Excel spreadsheet.
A Precision Profile of an ELISA standard curve based on a four-parameter logistic (4PL) model with weighting is shown in Fig. 2a. In this example, the quantitative limits at 10 and 2,000 pg/mL do not approximate the bounds of “linear” portion of the calibration curve that is typically symmetric around EC50. This is attributable to the relatively lower variability (standard deviation) of the low response values and the relatively higher variability of the higher response values. Thus the quantitative range of the curve/assay may not be at the apparently “linear” region of the curve/assay (see Fig. 2b). This is usually true for immunoassays and most other assay formats where the response error variability is not constant across the entire range of the response.
Appendix B
Calibration Curve Model Selection
Using data from a validation experiment, we describe a computation procedure for selecting the appropriate calibration curve model. Some of the calculations below, such as bias, precision and total error, can be easily implemented in Microsoft Excel using available formulae (20), in consultation with a statistician.
- (1)
Fit the calibration curve data from each run using various statistical models and use these models to calibrate the concentrations of the VS (Fig. 3). - (2)
For each VS level, compute the Total Error = Absolute value of Bias + Intermediate Precision from the calibrated results, corresponding to each calibration curve model, as described in DeSilva et al. (20). - (3)
Plot the “Total Error profile” of total error vs. the validation sample concentrations, for each calibration curve model (Fig. 4). - (4)
Select the calibration curve model that has the lowest total error overall across the concentration range of interest.
Fig. 3
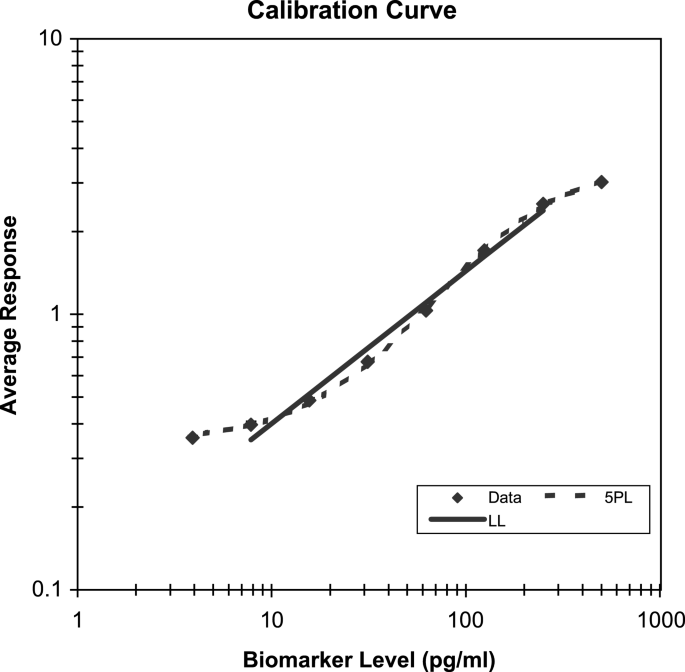
Calibration model selection based on % bias and intermediate precision. The solid line represents the linear model in the log scale of both the concentration and response (LL) that is fit to the linear part of the calibrator range (i.e., 6 out of 8 points were used). The dashed line is the weighted five-parameter logistic model (5PL) that is fit to all the data.
Fig. 4
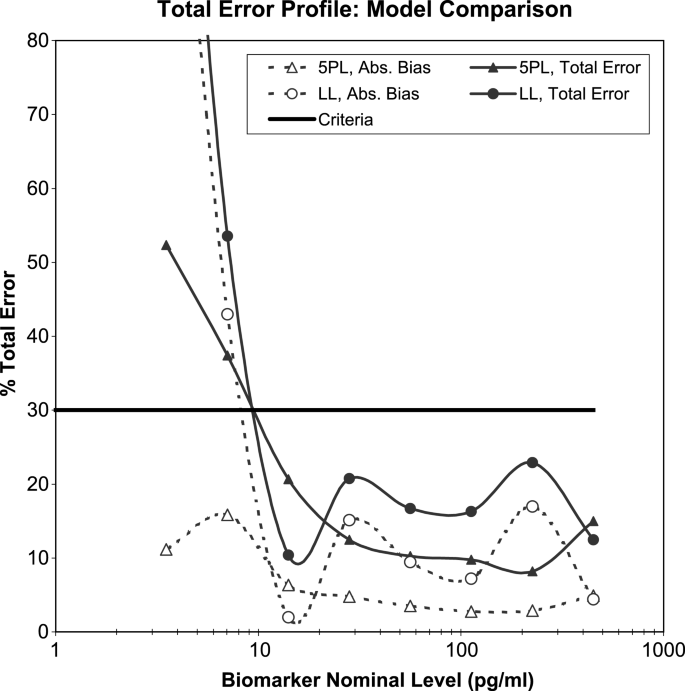
Calibration model selection based on Total Error (TE). The solid and dashed lines with circles represent TE and Absolute Bias, respectively for a linear model in log-scale (LL), the solid and dotted lines with triangles for a five-parameter logistic model (5PL). TE and Absolute Bias are determined using VS from four independent assay runs.
Total Error profiles from the qualification runs should be evaluated as illustrated in the ELISA example in Figs. 3 and 4. The two models considered here for the calibration curve are the linear model in the log scale (LL) and the weighted five-parameter logistic model (5PL). These Total Error profiles are derived from four validation runs. Each run contained eight calibrator standards in triplicate from 500 to 4 pg/mL along with eight VS levels in six replicates. The linear model was fit to only the linear part of the calibrator range (6 out of 8 points were used), resulting in an R 2 of 98.5%. As the clinical test samples from this assay were expected to fall within the range of 14–450 pg/mL, the Total Error profiles were compared for the two calibration curve models within this range. It is evident that the 5PL model is more appropriate, mostly attributable to improved Bias in the lower and higher ends of the curve. The widely used 4PL model, although not plotted here, showed similar performance to the log-linear model.
Some important points regarding this model selection process:
- (1)
If a quick decision on a “working” calibration curve model has to be made based on just one run, independent VS instead of calibrators should be used for the evaluation. The use of calibrator samples alone to select the optimal model will bias a more complex model due to overfitting. In addition, VS levels should not duplicate calibrator concentrations. - (2)
Although widely reported, R 2 is not useful for evaluating the quality of a calibration curve model because it does not penalize model complexity and consequently encourages overfitting. Alternative model selection criteria such as the Akaike's Information Criterion and Schwarz's Bayesian Information Criterion are abstruse, and neither is explicitly designed to choose models with optimal calibration properties. Our proposed method uses an independent set of validation samples to compare the models objectively with respect to the assay performance characteristics such as bias, precision, and total error.
Appendix C
Importance of Weighting for Calibration Curves
Figure 5 shows the bias (% relative error) and precision (error bars) of VS from a prestudy validation of an ELISA, corresponding to the weighted and unweighted 4PL models. The range of the VS covers the range of the calibration curve in Fig. 2b, where the study samples are expected to fall. It is clear from this example that weighting significantly improves the recovery (total error) of the VS across the range of interest, and thus can have a tremendous impact on the performance of an analytical method.
Fig. 5
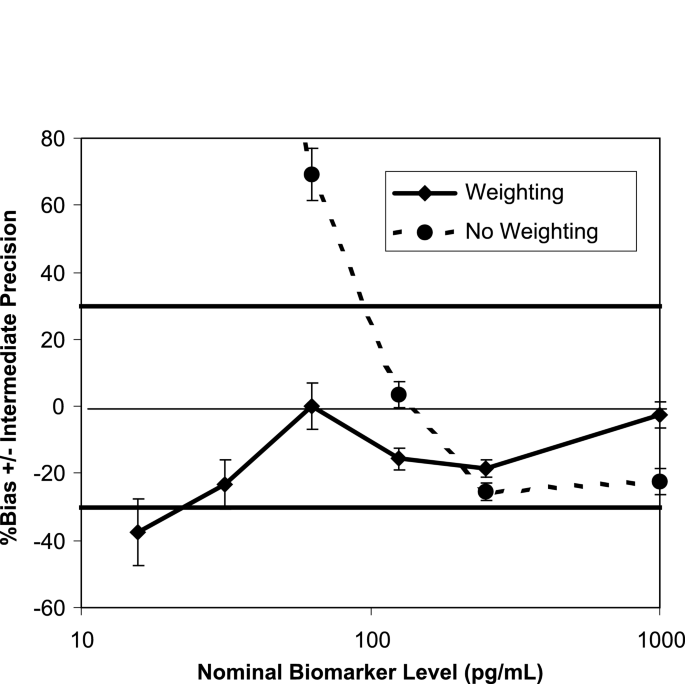
Weighting for calibration curves. The solid line and the dashed line are the % bias corresponding to the weighted and unweighted four-parameter logistic model, respectively. The error bars represent the intermediate precision of the assay. The % relative error and the intermediate precision are determined using VS from four independent assay runs.
In general, the weights should be estimated based on multiple runs of replicate data using one of the methods described by Carroll and Ruppert (29) in consultation with a statistician. We recommend this practice during method development and prestudy validation. The estimated weighting factor/parameter derived from the prestudy validation should then be used to fix the weights for the calibration curves generated during the sample analysis.
Appendix D
Parallelism Experiments
The following are points to consider in constructing parallelism experiments. Some of the following experiments will not be part of exploratory validation, but may be carried out in subsequent stages of validation. The experiments should be performed on a case-by-case and matrix-by-matrix basis with a fit-for-purpose approach.
- (1)
The parallelism sample(s) should be serially diluted to result in a set of samples having analyte concentrations that fall within the quantitative range of the assay. Ideally, the diluent should be the same as the intended sample matrix if an analyte-free matrix is available. In practice, the diluent used should be the same as that used for the calibrators. - (2)
Samples from at least three individual donors should be used. They should not be pooled unless the sample volumes are too small. Pooled samples could result in potential assay interferences (e.g., aggregates) and false nonparallelism. Lot-to-lot consistency of parallelism should be noted. - (3)
When only limited samples are available at a high analyte concentration, the approach of mixing a high- with a low-concentration sample at various ratios and correlating the concentration to the proportion may be considered. - (4)
When no samples of high analyte concentration are available, the endogenous level may be increased by stimulation of transfected cells, or by use of a nontarget population source (e.g., differing in gender or species), if applicable. - (5)
Varying cell counts may be used instead of dilution if the analyte is intracellular.
The measured concentrations of the dilution samples can be plotted against 1/dilution factor using log scales and a linear regression performed. Some individuals use observed results vs. expected where there is confidence in the actual concentration of the analyte in the samples used. Parallelism is proven when the results show a slope of nearly 1. The broken line in Fig. 6a approached parallel with a slope of 0.995, compared to the less parallel, solid line with a slope of 0.877.
Fig. 6
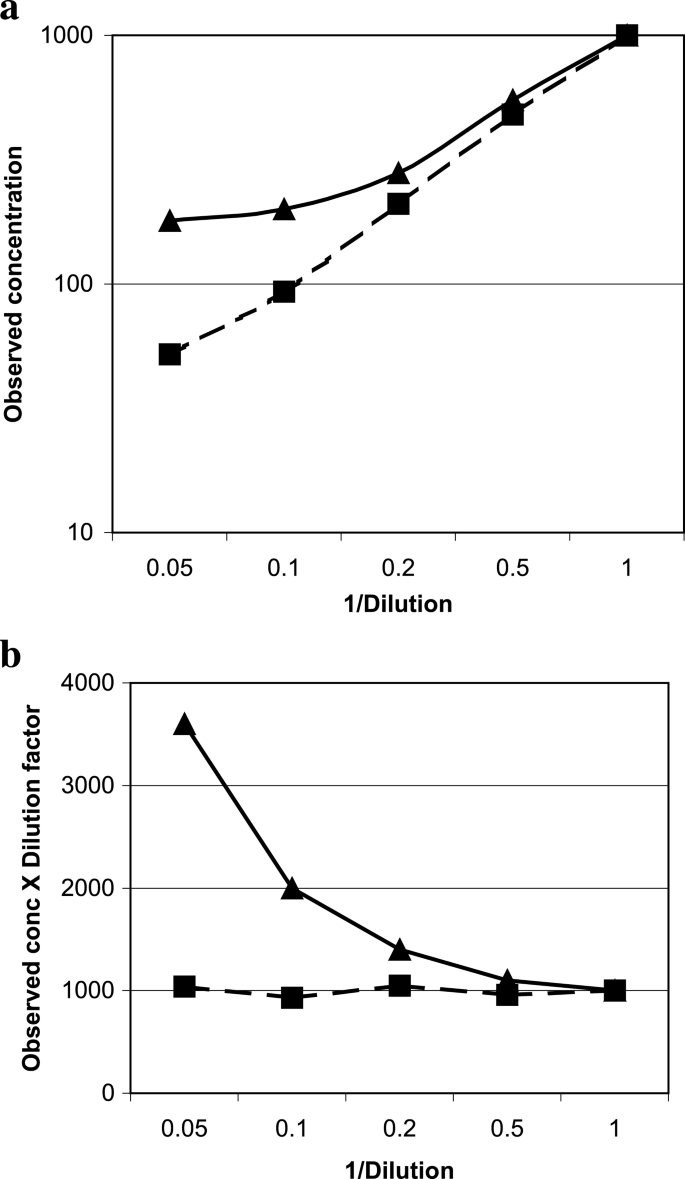
Parallelism. Data from two individual samples to show parallelism (■) or the lack of it (▲). The same dataset was plotted as (a) observed concentration vs. 1/dilution factor, or (b) dilution adjusted concentrations vs. 1/dilution factor.
Alternatively, the coefficient of variation (CV) among the recovered concentrations at different dilutions of the test sample can be used to verify parallelism (30). The value of this CV can be adapted on a case-by-case basis based on considerations of other assay performance parameters as well. Figure 6b shows a plot with the “recovered” concentrations replaced by “dilution adjusted” concentrations (observed concentration × dilution factor) because the absolute concentration is usually unknown. CV was 5.1% for the matrix that showed parallelism, and 58.7% for the one that did not.
Glossary
1. Accuracy
Per the FDA Guidance on Bioanalytical Method Validation (May, 2001), Accuracy of an analytical method describes the closeness of mean test results obtained by the method to the true value (concentration) of the analyte. This is sometimes referred to as Trueness or Bias.
2. Advanced Validation
A method validation that requires more rigor and thorough investigation, both in validation tasks and documentation, to support pivotal studies or critical decisions; e.g., differentiating subtle graded drug effects, monitoring drug safety, or for submission to regulatory agencies for drug approval.
3. Biomarker
A characteristic that is objectively measured and evaluated as an indicator of normal biologic processes, pathogenic processes, or pharmacologic response to a therapeutic intervention.
4. Clinical Endpoint
A characteristic or variable that reflects how a patient feels, functions, or survives.
5. Clinical Qualification
The evidentiary and statistical process linking biologic, pathologic and clinical endpoints to the drug effect, or linking a biomarker to biologic and/or clinical endpoints.
6. Definitive Quantitative Assay
An assay with well-characterized reference standards, which represents the endogenous biomarker, and uses a response–concentration standardization function to calculate the absolute quantitative values for unknown samples.
7. Dilution(al) Linearity
A test to demonstrate that the analyte of interest, when present in concentrations above the range of quantification, can be diluted to bring the analyte concentrations into the validated range for analysis by the method. Samples used for this test are, in general, the ones containing high concentrations of spiked analyte, not endogenous analyte.
8. Dynamic Range
The range of the assay that is demonstrated from the prestudy validation experiments to be reliable for quantifying the analyte levels with acceptable levels of bias, precision, and total error.
9. Exploratory Validation
Method validation that is less rigorous but adequate to meet study needs; e.g., looking for big effects in drug candidate screen, mechanism exploration, or internal decision with relatively minor impact to the final product, and not used for submission to regulatory agencies.
10. Interference
(1) Analytical interference: presence of entities in samples that causes a difference in the measured concentration from the true value. (2) Physicochemical interference (matrix interference): A change in measured physical chemical property of the specimen (e.g., excess bilirubin or hemoglobin, ionic strength, and pH) that causes a difference between the population mean and an accepted reference value.
11. Intermediate Precision
Closeness of agreement of results measured under changed operating conditions within a laboratory; e.g., different runs, analysts, equipments, or plates, etc. This is one of the three types of Precision.
12. Limit of Detection
A concentration resulting in a signal that is significantly different (higher or lower) from that of background. Limit of detection is commonly calculated from mean signal at background ± 2 or 3 standard deviations. This is often described as the analytical “sensitivity” of the assay in a diagnostic kit.
13. Limit of Quantification
Highest and lowest concentrations of analyte that have been demonstrated to be measurable with acceptable levels of bias, precision, and total error. The highest concentration is termed the Upper Limit of Quantification, and the lowest concentration is termed the Lower Limit of Quantification.
14. Minimum Required Dilution
The minimum dilution required to dilute out matrix interference in the sample for acceptable analyte recovery.
15. Parallelism
Relative accuracy from recovery tests on the biological matrix, incurred study samples, or diluted matrix against the calibrator calibrators in a substitute matrix. It is commonly assessed with multiple dilutions of actual study samples or samples that represent the same matrix and analyte combination of the study samples.
16. Pharmacodynamic
The relationship between drug concentrations and biochemical and physiological effects of drugs and mechanisms of drug action.
17. Precision
Precision is a quantitative measure (usually expressed as standard deviation and coefficient of variation) of the random variation between a series of measurements from multiple sampling of the same homogenous sample under the prescribed conditions. If it is not possible to obtain a homogenous sample, it may be investigated using artificially prepared samples or a sample solution. Precision may be considered at three levels: 1. Repeatability, 2. Intermediate Precision, and 3. Reproducibility.
18. Precision Profile
A plot of the coefficient of variation of the calibrated concentration vs. the concentration in log scale. It provides preliminary estimates of the quantification limits and feasibility assessments on the intended range.
19. Quality Controls
A set of stable pools of analyte, prepared in the intended biological matrix with concentrations that span the range claimed for the test method, used in each sample assay run to monitor assay performance for batch acceptance.
20. Qualitative Assay
The assay readout does not have a continuous proportionality relationship to the amount of analyte in a sample; the data is categorical in nature. Data may be nominal (positive or negative) such as presence or absence of a gene or gene product. Alternatively, data might be ordinal, with discrete scoring scales (1 to 5, −+, +++, etc.), such as immunohistochemistry assays.
21. Quasiquantitative Assay
(Quasi: “possesses certain attributes”) A method that has no calibrator, has a continuous response, and the analytical result is expressed in terms of a characteristic of the test sample. An example would be an antidrug antibody assay that is express as titer or % bound.
22. Relative Quantitative Assay
A method which uses calibrators with a response–concentration calibration function to calculate the values for unknown samples. The quantification is considered relative because the reference standard is either not well characterized, not available in a pure form, or is not fully representative of the endogenous biomarker.
23. Relative Accuracy
For relative quantitative methods, absolute accuracy is not possible to evaluate due to the unknown nature of the endogenous biomarker. Relative accuracy is the recovery (see below) of the reference standard spiked into the study matrix.
24. Recovery
The quantified closeness of an observed result to its theoretical true value, expressed as a percent of the nominal (theoretical) concentration. Recovery is often used as a measure of accuracy.
25. Repeatability
Closeness of agreement between results of successive measurements of the same samples carried out in the same laboratory under the same operating condition within short intervals of time. It is also termed intraassay or intrabatch precision. This is one of the three types of Precision.
26. Reproducibility
Closeness of agreement of results measured under significantly changed conditions; e.g., inter laboratory, alternate vendor of a critical reagent. This is also referred to as cross validation.
27. Robustness of the assay
A measure of the capacity of a method to remain unaffected by small, but deliberate changes in method parameters and provides an indication of its reliability during normal run conditions.
28. Selectivity
The ability of a method to determine the analyte unequivocally in the presence of components that may be expected to be present in the sample.
29. Sensitivity
The lowest concentration of analyte that an analytical method can reliably differentiate from background (limit of detection).
30. Specificity
The ability of assay reagents (e.g., antibody) to distinguish between the analyte, to which the reagents are intended to detect, and other components.
31. Surrogate Endpoint
A biomarker that is intended to substitute for a clinical endpoint. A surrogate endpoint is expected to predict clinical benefit or harm (or lack of benefit or harm) based on epidemiologic, therapeutic, pathophysiologic, or other scientific evidence.
32. Target Range
Range of analyte concentrations where the study samples are expected to fall.
33. Total Error
The sum of all systematic bias and variance components that affect a result; i.e., the sum of the absolute value of the Bias and Intermediate Precision. This reflects the closeness of the test results obtained by the analytical method to the true value (concentration) of the analyte.
34. Validation
It is the confirmation via extensive laboratory investigations that the performance characteristics of an assay are suitable and reliable for its intended analytical use. It describes in mathematical and quantifiable terms the performance characteristics of an assay.
35. Validation samples
Test samples in biological matrix mimicking study samples, endogenous and/or spiked, used in prestudy validation to provide characterizations of assay performances; e.g., intra- and inter-run accuracy and precision, and analyte stability.
Rights and permissions
About this article
Cite this article
Lee, J.W., Devanarayan, V., Barrett, Y.C. et al. Fit-for-Purpose Method Development and Validation for Successful Biomarker Measurement.Pharm Res 23, 312–328 (2006). https://doi.org/10.1007/s11095-005-9045-3
- Received: 28 July 2005
- Accepted: 07 October 2005
- Published: 12 January 2006
- Issue Date: February 2006
- DOI: https://doi.org/10.1007/s11095-005-9045-3