Cancer Cells Expressing Toll-like Receptors and the Tumor Microenvironment (original) (raw)
Introduction
Recent studies have revealed that chronic inflammation increases the risk of cancer development and progression [1]. Inflammation is usually a host defense against invading microbial pathogens, tissue destruction/injury or cancer. In this setting, toll-like receptors (TLRs) play a crucial role in the innate immune response and the subsequent induction of adaptive immune responses [2]. TLRs are expressed not only on immune cells but also on cancer cells. [3–12]. Activated TLR signals on cancer cells may promote cancer progression, anti-apoptotic activity and resistance to host immune responses [3–7, 13].
The tumor microenvironment, which includes cancer cells, stressed normal cells, stromal tissue and extracellular matrix, has recently been implicated as a major factor for progression and metastasis of cancer [14]. Stromal tissue consists of fibroblasts, myofibroblasts, vascular and lymphovascular endothelial cells, and infiltrating immune cells such as antigen-presenting macrophages, dendritic cells (DCs) and T-cells. Downregulation of the anti-tumor activity of infiltrating immune cells has been suggested to support cancer progression, angiogenesis and metastasis [15, 16]. Recent studies show that activated TLRs expressed on cancer cells can dampen the anti-tumor functions of infiltrating immune cells, thereby altering the inflammatory response in a manner that promotes cancer progression [5, 6, 13].
This review will examine interactions between the tumor microenvironment, TLRs expressed on immune and cancer cells, and the pathogen-associated molecular patterns (PAMPs) and damage-associated molecular patterns (DAMPs) that are defined as TLR ligands. Understanding how exogenous (PAMPs) or endogenous (DAMPs) danger signals activate TLRs and oncogenesis in the setting of chronic inflammation will facilitate development of more effective therapeutic strategies against a wide variety of cancers.
Toll-like Receptors and Ligands
TLRs are pattern recognition receptors for ligand molecules derived from microbes or host cells; TLR-ligand binding plays a key role in innate immunity and subsequent acquired immunity against microbial infection or tissue injury [17, 18]. TLRs are evolutionary conserved from invertebrates to humans, and the TLR family has at least 13 members [19]. Eleven members (TLR1 to TLR11) have been identified in humans so far. These are expressed by different types of immune cells; they are located on the cell surface or in the cell cytoplasm; and they recognize different DAMPs and PAMPs. PAMPs are conserved molecular products derived from pathogens that include Gram-positive and Gram-negative bacteria, fungi and viruses. DAMPs are endogenous molecules released from injured or dying cells. Both DAMPs and PAMPs initiate immune responses through TLR signals [20]. The list of ligands for TLRs continues to increase, particularly with recent additions of mammalian cell molecules (Table 1).
Table 1 TLRs and ligands
TLR2 and TLR4 have a key role in recognition of various bacteria: TLR2 can recognize lipoprotein, lipoteichoic acid and peptidoglycan molecules derived from Gram-positive bacteria, whereas TLR4 is necessary for recognizing lipopolysaccharide (LPS) from the Gram-negative bacterial cell wall. Both of these TLRs also are crucial for responses to DAMPs [17, 18]. TLR5 recognizes bacterial flagellin. TLR11 recognizes profilin-like molecule from Toxoplasma. TLR3, 7, 8 and 9 are expressed in the cytoplasm and can recognize invading viruses [19]; TLR3 responds to double-strand RNA, whereas TLR7 and TLR8 respond to single-strand RNA. TLR9 recognizes CpG-ODN derived from bacteria and viruses. TLR heterodimers such as TLR1/2 and TLR2/6 interact with a wider range of ligands than monomeric TLRs. Akira et al. [19] have reviewed TLR signaling pathways during pathogen recognition; they describe in detail the induction of immune reactions via extracellular and intracellular pathways mediated by myeloid differentiation factor 88 (MyD88), nuclear factor _kappa_-light–chain-enhancer of activated B cells (NF-κB), and mitogen-associated protein kinase (MAPK).
Toll-like Receptors and Chronic Inflammation
TLRs are expressed not only by immune cells but also by normal epithelial cells in the digestive system, normal keratinocytes in skin, alveolar and bronchial epithelial cells, and epithelial cells of the female reproductive tract. These epithelial cells lining an organ are the first line of defense against invasion of microorganisms, and TLRs expressed in epithelial cells have a crucial role in regulation of proliferation and apoptosis. Recent studies report abnormally upregulated TLR signals in epithelial cells undergoing carcinogenic changes during chronic inflammation [1, 21]. Chronic inflammation caused by autoimmune disease or microbial infections is an important risk factor for colorectal cancer (inflammatory bowel disease), gastric cancer (Helicobacter pylori), cervical cancer (human papilloma virus), liver cancer (hepatitis virus B and C), and hematologic malignancies (cytomegalovirus and Epstein-Barr virus). More than 15% of cancers worldwide have a direct infectious origin [22].
Chronic inflammation appears to be immunologically distinct from acute infection. The acute phase of infection is characterized by CD8+ T-cell priming and activation of NK cells. CD8+ effector T-cells have a central role in tumor-associated antigen (TAA)-specific immunity and thus in elimination of tumors; activated NK cells stimulate the maturation of DCs and facilitate adaptive anti-tumor immunity. The absence or reduction of these functions during chronic inflammation may promote tumor tolerance [23], carcinogenesis and evolution of the tumor microenvironment. Chronic inflammation has been thought to induce malignant transformation by activation of oncogenes, inhibition of tumor suppressors, and induction of immunosuppression.
TLRs are also expressed by cancer cells (Table 2). TLRs expressed on cancer cells can upregulate the NF-κB cascade and produce anti-apoptotic proteins that contribute to carcinogenesis and cancer cell proliferation. They also can mediate cancer cell release of cytokines and chemokines that can recruit immune cells to enhance immunity in the tumor microenvironment. These optimized immune cells release further proinflammatory cytokines, proangiogenic factors and growth factors, which impair the anti-tumor function of antigen-presenting cells (APCs) and effector T-cells.
Table 2 TLR expression in human cancer cells
Contribution of TLR Signals to Carcinogenesis
The high risk of gastric cancer in patients with _H. pylori_-associated chronic gastritis illustrates the link between chronic inflammation and development of cancer [1]. TLR2, 4, 5 and 9 are expressed by normal gastric epithelial cells, and TLR4 signaling has a key role in regulating the proliferation and apoptosis of these cells. However, overexpression of TLR4 has been demonstrated in _H. pylori-_infected gastric epithelial cells. TLR4, 5 and 9 are strongly expressed not only by gastric cancer cells but also by metaplastic and dysplastic gastric epithelial cells from patients with H. pylori gastritis [9, 24]. Continuous stimulation of these TLRs by the LPS component of H. pylori might upregulate inflammatory factors such as cyclooxygenase-2 (COX-2) and prostaglandin E2 (PGE2) through activation of NF-κB; upregulation eventually leads to progressive carcinogenic changes in normal gastric epithelial cells [25].
A possible link between chronic inflammation, TLR expression and oncogenesis also can be found in colorectal cancer. Nine TLRs (TLR1-9) are expressed in normal epithelial cells of the colon; three of these TLRs (TLR2-4) are elevated in most colorectal cancer cell lines. Elevated expression seems to be regulated by commensal bacteria in the intestinal lumen [26]. TLR4 reportedly is overexpressed in colorectal cancer cells from patients with colitis and in colorectal cancer cells from a murine model of colitis; interestingly, colorectal neoplasia is reduced in TLR4-deficient mice [4]. In the same study, activation of TLR4 by LPS led to neoplastic transformation via enhanced COX-2 expression and increased epidermal growth factor receptor (EGFR) signaling. This suggests that chronic inflammation caused by commensal bacteria in the microenvironment may be responsible for carcinogenesis through TLR signaling.
Epithelial cells of the female reproductive tract may acquire carcinogenic changes through continuous TLR stimulation by PAMPs. Four TLRs (TLR2-5) are expressed by ovarian cancer cell lines [12]. TLR4 activation by LPS promotes survival of ovarian cancer cells by inducing the expression of antiapoptotic proteins, including X-linked inhibitor of apoptosis (XIAP) and phosphorylated Akt [27].
Two TLRs (TLR5 and TLR9) might contribute to cervical carcinogenesis [8, 28]. The expression of TLR5 and TLR9 is absent or weak in normal cervical squamous epithelial cells but gradually increases during progression of low-grade cervical intraepithelial neoplasia (CIN) to high-grade CIN and then to invasive cervical squamous cell carcinoma.
Four TLRs (TLR2-4 and 9) are expressed in lung cancer cell lines. Activation of TLR4 by LPS induces resistance of lung cancer cells to TNFα or TRAIL-induced apoptosis through NF-κB upregulation [6]. Various levels of TLR9 expression are observed in tumor specimens from patients with prostate cancer [7, 29], breast cancer, astrocytoma and glioblastoma [30]. Activation of TLR9 by CpG-ODN or bacterial DNA increases cancer cell invasion.
We recently reported high expression of three TLRs (TLR2-4) in human cutaneous melanoma. Our in vivo and in vitro studies showed that other TLRs were expressed less frequently or at lower levels. All three TLRs were functionally active. Stimulation with ligands specific for each TLR (zymosan for TLR2, polyIMP/polyCMP [PIC] for TLR3, and LPS for TLR4), upregulated TLR expression and activated the adaptor protein MyD88 and NF-κB. After stimulation, TLRs induced several inflammatory cytokines and chemokines, as discussed in the next section, and melanoma cell migration increased [5]. These findings strongly suggest that activation of TLRs by melanoma cells can activate inflammatory-related molecules that would govern the status of tumor microenvironment.
TLRs expressed in normal epithelial cells appear to contribute to carcinogenesis through NF-κB upregulation and subsequent production of antiapoptotic factors such as Bcl-x, c-IAP-1 and c-IAP-2. By contrast, TLRs expressed in cancer cells appear to promote tumor progression by facilitating cell survival and migration in a tumor microenvironment characterized by chronic inflammation and PAMPs [31, 32].
Cytokines and Chemokines Activated Through TLR Signals
In our study of the immune response to stimulation of specific TLRs in melanoma cell lines [5], we demonstrated that exposure of cells to ligands specific for TLR2-4 significantly upregulated proinflammatory cytokines (TNFα, G-CSF, IL-1a, and IL-6), proinflammatory chemokines (CCL2 and CXCL2), an immunsuppressive cytokine (IL-10), and an inflammatory factor (COX-2). Ligation of TLRs expressed in cancer cells reportedly also increases TGFβ, IL-8, CXCR4, ICAM-1 and VEGF [6, 12, 13, 33]. Almost all of these cytokines and chemokines promote tumor progression, and their presence characterizes the tumor microenvironment’s active release of various factors that have multiple effects on tumor cells, immune cells and normal cells.
TGFβ, VEGF, CCL22 and IL-10 can induce CD4+CD25+Foxp3+ regulatory T cells (Tregs) in the tumor microenvironment and tumor-draining regional lymph nodes of cancer patients [16, 34]. These Tregs secrete additional IL-10 and TGFβ, which suppress anti-tumor functions of non-Treg T cells. Elevated tumor levels of Tregs are linked to poor prognosis in several cancers [35]. IL-10, an immunsuppressive cytokine, upregulates expression of alternatively activated myeloid cells (M2c) in tumor-associated macrophages (TAMs). M2c cells release angiogenic and lymphoangiogenic factors that promote lymphatic metastasis of cancer cells [36]. Inflammatory mediators IL-1b, IL-6 and PGE2 recruit myeloid-derived suppressor cells (MDSCs) in the tumor microenvironment [37]. MDSCs have recently been recognized as critical mediators of cancer progression; they inhibit the anti-tumor immune response by release of arginase, nitric oxide synthase (NOS) and TGFβ [15, 38]. Additionally, mature myeloid DCs induce a strong T helper 1 (Th1)—type immune response and are considered potent inducers of TAA-specific immunity. However, in several cancers the dominant population of DCs in the tumor microenvironment is not functionally mature DCs but dysfunctional DCs. Differentiation and maturation of these myeloid DCs are profoundly suppressed by factors present in the tumor microenvironment, including VEGF, IL-6, IL-1, TGFβ, COX-2 and PGE2 [23]. Cancer cells also induce CXCL12, TNFα and IL-8. CXCL12 recruits plasmacytoid DCs that express CXCR4, the receptor of CXCL12, into the tumor microenvironment. These plasmacytoid DCs induce significant IL-10 production by T cells and therefore act as immunsuppressants. Moreover, CXCL12, TNFα and IL-8 attract vascular DCs to the tumor microenvironment, with a subsequent increase in tumor vascularization and metastasis [39]. This extensive and complex interaction between immune cytokines/chemokines and immune cells is initiated by TLRs and is responsible for an immunsuppressive response in the tumor microenvironment.
Cancer-associated fibroblasts (CAFs) are important components of the tumor microenvironment, and they are the main cellular component of the tumor stroma. Unlike normal fibroblasts, CAFs are perpetually activated [40]. Their origin is not well understood, but they appear to be as important as immune cells in the tumor microenvironment [41]. A recent study proposed that TGFβ has a crucial role in activation of CAFs [42]. Activated CAFs promote the proliferation and progression of cancer through the production of growth factors and metalloproteinases. Therefore, a TLR-related increase in TGFβ might lead to assembly and activation of CAFs in the tumor microenvironment.
In summary, during cancer progression in the setting of chronic inflammation, TLR ligands activate TLRs expressed in cancer cells. Activated cancer cells release cytokines and chemokines that are an important component of the tumor microenvironment. Cytokine-activated infiltrating immune cells subsequently can induce further cytokine release that contributes to activation of CAFs and impairs the function of APCs, effector T-cells and TAA-specific immunity; possibly resulting tumor immunotolerance. The interplay and additive effects of these events facilitate continuous activation of TLR in cancer cells or adjacent normal epithelial cells, thereby maintaining a hostile tumor microenvironment and promoting tumor progression (Fig. 1).
Fig. 1
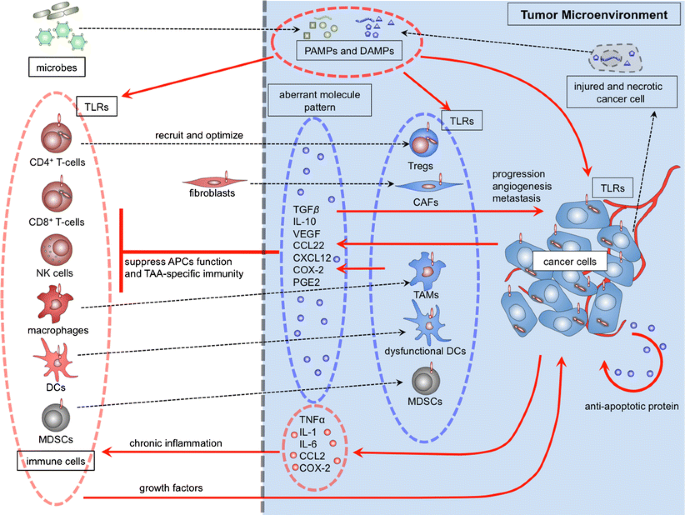
TLR signals contribute to tumor progression in the tumor microenvironment. PAMPs derived from microbes and DAMPs derived from injured and necrotic cancer cells might activate TLRs expressed on immune cells and on cancer cells. These activated cells release cytokines and chemokines; the aberrant molecular pattern of chemokines/cytokines might significantly affect the tumor microenvironment. Tregs: regulatory T cells, TAMs: tumor-associated macrophages, DCs: dendritic cells, CAFs: cancer-associated fibroblasts, MDSCs: myeloid-derived suppressor cells
TLRs and Tumor Angiogenesis
TLRs also seem to have an important role in tumor angiogenesis, i.e., the formation of new capillary blood vessels from existing vessels outside of the tumor. The developing tumor depends on angiogenesis as a source of more oxygen and nutrients for survival and growth. Vascular endothelial growth factor (VEGF) is the main factor involved in tumor angiogenesis and is part of the aberrant molecular pattern associated with TLR signals. VEGF is secreted by cancer cells directly and by immune cells and CAFs. New vessels induced by VEGF are abnormal: they are heterogeneous in distribution, irregular in shape, and not organized into arterioles, venules and capillaries. Their varied permeability leads to high interstitial pressures and further hypoxia, which stimulates additional VEGF production [43]. Hypoxia characterizes solid tumors; it is a stress factor that might cause cells to release DAMPs. These ligands activate TLR signals and contribute to the aberrant molecular pattern in the tumor microenvironment.
The TLR contribution to tumor angiogenesis has been investigated in _H. pylori_-associated gastric cancer [44]. This study reported that _H. pylori_-induced COX-2 expression and PGE2 release enhanced tumor angiogenesis via TLR2 and 9. Another in vitro study found a direct endothelial stimulatory role for LPS in initiating angiogenesis through activation of TLR signaling pathways [45]. HMGB1 has been recently recognized as a pro-angiogenic factor [46]. HMGB1 upregulation induces the production of VEGF and endothelial cell proliferation. Moreover, HMGB1 acts on endothelial progenitor cells and hematopoietic stem cells to improve neovascularization of injured or malignant tissue [46]. However, other studies show an anti-angiogenic effect for TLRs. In a colorectal cancer xenograft model, a TLR9 agonist reportedly interfered with EGFR signaling and tumor angiogenesis and had a synergistic effect with other EGFR inhibitors [47]. Imiquimod, a TLR7 agonist used as a topical immune-response modifier in patients with skin cancers, can inhibit tumor angiogenesis [48] by inducing anti-angiogenic cytokines such as IFNs, IL-10 and IL-12; down-regulating pro-angiogenic factors such as fibroblast growth factor β (FGFβ) and metalloproteinase-9 (MMP9); and promoting endothelial cell apoptosis [49]. Although the TLR contribution to tumor angiogenesis remains unclear, interaction with ligands and TLRs seems to have a major role in tumor angiogenesis and hypoxia in tumor microenvironment, which supports tumor growth.
DAMPs Released from Injured or Necrotic Cancer Cells
Under normal conditions, scheduled cell death is regulated by adenosine triphosphate (ATP) and related apoptotic pathway factors; this regulation drives fragmentation of cellular macromolecules and the speedy subsequent phagocytosis and clearance of apoptotic debris. However, in cancerous conditions, cells dying by non-apoptotic pathways, principally necrosis, release DAMPs into the extracellular space. DAMPs are nuclear or cytosolic proteins with defined intracellular functions but different extracellular actions after cytolysis. DAMPs released from injured or dying cells are recognized by TLRs on immune cells; subsequent TLR signals disrupt the anti-tumor immune response and lead to cancer progression [18].
Candidate DAMPs include heat shock proteins (HSP 60, 70), ATP and uric acid, the S100 family of calcium modulated proteins, nuclear protein high-mobility group box 1 (HMGB1), and nucleic acids. HMGB1, a DNA binding protein, is one of the best-characterized DAMP. HMGB1 regulates intracellular transcription and mediates extracellular proinflammatory processes. HMGB1 released during unscheduled cell death activates an immune response via TLR signals. Cell line studies show that HMGB1 is strongly up-regulated in breast cancer, colon cancer, melanoma, pancreatic cancer and prostate cancer; upregulated HMGB1 activates TLR2 and TLR4 expressed on immune cells and induces cancer progression and metastasis [20].
We previously reported elevated expression of S100 proteins in melanoma cell lines relative to normal melanocyte lines. S100 proteins released by melanoma cells stimulated melanoma cells as well as PBLs and acted as an autocrine tumor growth factor [50]. S100A4 is responsible for metastasis and is an indicator of poor prognosis for patients with breast cancer [51]. However, although this inflammatory protein is associated with metastatic cancer cells, in the tumor microenvironment it is also expressed by macrophages, lymphocytes and fibroblasts. Elevated interstitial fluid levels of S100A4 in tumors [52] suggest that stromal cells in the tumor microenvironment externalize S100A4, which then activates TLR signals. Recent studies reveal that S100A8 and S100A9 produced by primary tumors can activate serum amyloid A (SAA) 3 in lung tissue prior to pulmonary metastasis. SAA3 has a role in the accumulation of myeloid cells and acts as a positive-feedback regulator for secretion of S100 proteins. SAA3 is a ligand for TLR4 in lung endothelial cells and macrophages. The activation of TLR4 facilitates migration of cancer cells from the primary tumor to lung tissue by creating a tumor microenvironment [53]. Blocking the S100-TLR4 cascade therefore might be an effective strategy for the prevention of pulmonary metastasis.
Nucleic Acid Fragments Act as DAMPs
During tumor expansion, nucleic acids released from necrotic cancer cells or adjacent injured normal epithelial cells act as DAMPs. Kariko et al. [54] demonstrated that TLR3 expressed in DCs was activated by mRNA released from necrotic cells; subsequent TLR signals upregulated DC maturation, leading to IFN-α secretion. This upregulation could be abolished by pretreatment of necrotic cells with RNase. The mRNA released by cancer cells circulates in the blood [55] and its serum levels have been correlated with disease outcome [56]. In our studies, TLR3 expression was upregulated (24.6–121.3% in mean value) in melanoma cells incubated 12 h with purified total RNA from normal PBL or allogeneic melanoma cells (Fig. 2), and TLR activation promoted melanoma cell migration [5]. Thus, RNA derived from melanoma cells can act as a TLR3 ligand and facilitate migration of melanoma cells, without support from immune cells.
Fig. 2
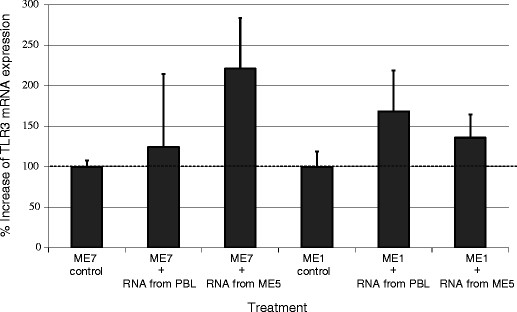
TLR3 ligation and subsequent TLR3 mRNA expression in melanoma cells incubated with purified total RNA from normal donor PBLs or allogeneic melanoma cells. When ME7 and ME1 human melanoma cells were incubated 12 h with total RNA from normal PBL and ME5 melanoma cells, mean TLR3 mRNA expression increased 24.6–121.3% as compared with expression in control cells without total RNA
Likewise, absolute levels of circulating DNA in serum and plasma appear to have diagnostic and prognostic significance for various cancers [57]. The integrity of circulating DNA, measured as the ratio of longer to shorter DNA fragments, is higher in cancer patients than in normal individuals [58]. Apoptotic cells release DNA fragments that are usually 185 to 200 base pairs in length. Uniformly truncated fragments of DNA (and RNA) are produced by a programmed enzymatic cleavage process during apoptosis [59]. As we and other groups have reported, methylation of tumor suppression genes detected in circulating DNA is associated with prognosis [60].
We speculate that the high rate of unscheduled cell death in the tumor microenvironment elevates nucleic acid DAMPs. Elevated levels of nucleic acid DAMPs and other DAMPs might foster chronic inflammation, a hallmark of the tumor microenvironment. Figure 3 shows how interactions between TLRs and DAMPs could create and maintain a self-perpetuating tumor microenvironment. In this microenvironment, cancer cell death might stimulate cancer progression if nucleic acid fragments released by the dead tumor cells are transfected into normal cells, thereby changing the normal cell’s properties. Normal cells in the tumor microenvironment might also be transfected by microRNA released from tumor cells, because these small RNA molecules (20–22 base pairs) are easily taken up by cells. Horizontal mediated transfection of microRNA and mRNA in mammalian cells is an intriguing possibility but has yet to be demonstrated in vivo. This phenomenon could explain the expression of tumor-related proteins by normal cells in the tumor microenvironment.
Fig. 3
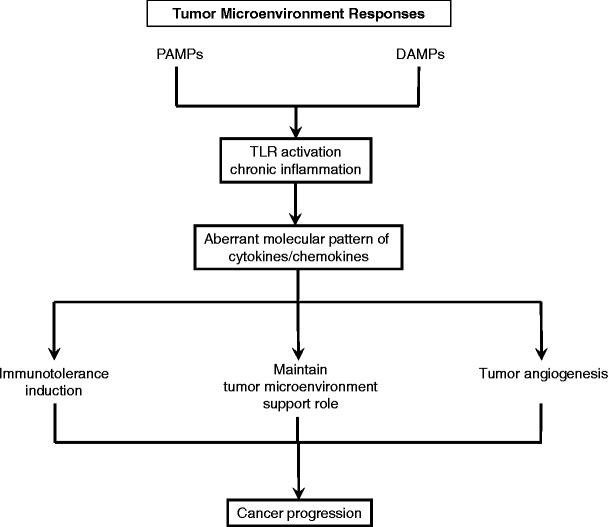
During cancer growth and unscheduled cell death, DAMPs derived from necrotic cancer cells might continuously activate TLRs and create a chronic inflammatory condition as well as PAMPs. TLR ligation activates NF-κB and MAPK signaling, causing the production of proinflammatory cytokines and chemokines. The resulting aberrant molecular pattern of cytokines/chemokines might have a crucial role in immunotolerance, maintain tumor microenvironment, tumor angiogenesis that supports tumor progression
TLR-targeted Therapies
Because several TLRs can induce strong anti-tumor activity by regulating the functions of immune cells that infiltrate the tumor microenvironment, clinical trials are investigating novel anticancer therapies based on TLR ligand delivery. A successful example is imiquimod. This TLR7 agonist is used extensively to treat actinic keratosis and basal cell carcinoma, and it is being studied as an adjuvant therapy for melanoma. A study of imiquimod 5% cream in 90 patients with basal cell carcinoma reported a 96% clearance rate, and only two recurrences during application a mean follow-up period of 36 months. Cutaneous side effects were minimal; there were no systemic side effects [61]. Imiquimod induces IFNα, IFNγ and IL-12, which activate APC function and TAA-specific immunity, thereby correcting the aberrant conditions of the tumor microenvironment [62]. As mentioned above, imiquimod’s ability to inhibit tumor angiogenesis and cause tumor regression suggests a link between TLR7 and tumor angiogenesis.
Another imidazoquinoline agonist for TLR7 is 852A {_N_-[4-(4-amino-2-ethyl-1_H_-imidazo[4,5-_c_]quinolin-1-yl) butyl] methanesulfonamide, 3M-001}. This systemically administered agent has 40 times greater aqueous solubility than imiquimod. It is under clinical investigation for chronic lymphatic leukemia and other solid tumors [63, 64]. CpG-ODN agonists for TLR9 directly induce activation and maturation of DCs, enhance differentiation of B cells into antibody-secreting plasma cells, and promote development of anti-tumor T-cell responses [65]. In a murine model of human ovarian cancer, intraperitoneal administration of CpG-ODN produced a stronger anti-tumor effect than intravenous administration [66]. Early clinical trials are investigating the safety and efficacy of TLR9 agonists for treatment of breast cancer, colorectal cancer, lung cancer, melanoma, glioblastoma and some lymphomas and leukemias [67]. Macrophage activating lipopeptide-2 (MALP2) is a TLR2/6 agonist that has demonstrated encouraging results for treatment of pancreatic cancer: intratumoral injection of MALP2 plus gemcitabine during laparotomy significantly prolonged survival of patients with incompletely resectable disease, from 9 to 17 months [68]. These agents affect the tumor microenvironment and the tumor cells directly and indirectly.
Another therapeutic approach is to target DAMPs, especially HMGB1, in inflammatory diseases and cancers. HMGB1-targeted therapies are grouped according to their ability to sequester HMGB1, target extracellular HMGB1, target receptors, or inhibit HMGB1 release [20]. Targeting DAMPs may neutralize tumor supporting events occurring in the tumor microenvironment.
However, not all TLR agonists and not all TLRs signaling pathways lead to clinically relevant anti-tumor activity. As described in this review, the complicated interactions between cancer cells, immune cells, and PAMPs/DAMPs in the tumor microenvironment can promote the progression of cancer and support inappropriate immune enhancement or anti-tumor immune tolerance through TLR signaling pathways. TLR-targeted therapeutics may also directly affect TLR-expressing tumor cells. Further investigation and better understanding of the relationship between TLRs and the tumor microenvironment are required to clarify mechanisms of tumor progression/metastasis and develop more effective therapeutic approaches to many human cancers.
Conclusion
TLRs are expressed on many types of cancer cells, tumor stromal cells and infiltrating immune cells. TLR activation during inflammation and injury plays an active role in the surrounding microenvironment. Similarly, in carcinogenesis and tumor progression TLRs play an active role in the tumor microenvironment. During chronic inflammation, abnormal activation of TLRs in normal fibroblasts and epithelial cells might facilitate neoplastic transformation and carcinogenesis. Cancer cells activated by TLR signals can release cytokines and chemokines that recruit and optimize immune cells to release further cytokines and chemokines. The result is an aberrant cytokine profile associated with immune tolerance, cancer progression and propagation of the tumor microenvironment. DAMPs derived from injured normal epithelial cells and necrotic cancer cells appear to be present at significant levels in the tumor microenvironment, and their stimulation of specific TLRs might foster chronic inflammation. This mechanism is complex and thus far not well understood; however, it is clear that carcinogenesis, cancer progression, and site-specific metastasis are related to interactions between cancer cells, immune cells, DAMPs and PAMPs through TLR signals in the tumor microenvironment. Better understanding of these signals and pathways will lead to development of novel therapeutic approaches to a wide variety of cancers.
References
- Mantovani A, Allavena P, Sica A et al (2008) Cancer-related inflammation. Nature 454:436–444
Article PubMed CAS Google Scholar - O’Neill LA (2008) When signaling pathways collide: positive and negative regulation of toll-like receptor signal transduction. Immunity 29:12–20
Article PubMed CAS Google Scholar - Curtin JF, Liu N, Candolfi M et al (2009) HMGB1 mediates endogenous TLR2 activation and brain tumor regression. PLoS Med 6:e10
Article PubMed CAS Google Scholar - Fukata M, Chen A, Vamadevan AS et al (2007) Toll-like receptor-4 promotes the development of colitis-associated colorectal tumors. Gastroenterology 133:1869–1881
Article PubMed CAS Google Scholar - Goto Y, Arigami T, Kitago M et al (2008) Activation of Toll-like receptors 2, 3, and 4 on human melanoma cells induces inflammatory factors. Mol Cancer Ther 7:3642–3653
Article PubMed CAS Google Scholar - He W, Liu Q, Wang L et al (2007) TLR4 signaling promotes immune escape of human lung cancer cells by inducing immunosuppressive cytokines and apoptosis resistance. Mol Immunol 44:2850–2859
Article PubMed CAS Google Scholar - Ilvesaro JM, Merrell MA, Swain TM et al (2007) Toll like receptor-9 agonists stimulate prostate cancer invasion in vitro. Prostate 67:774–781
Article PubMed CAS Google Scholar - Kim WY, Lee JW, Choi JJ et al (2008) Increased expression of Toll-like receptor 5 during progression of cervical neoplasia. Int J Gynecol Cancer 18:300–305
Article PubMed CAS Google Scholar - Schmausser B, Andrulis M, Endrich S et al (2005) Toll-like receptors TLR4, TLR5 and TLR9 on gastric carcinoma cells: an implication for interaction with Helicobacter pylori. Int J Med Microbiol 295:179–185
Article PubMed CAS Google Scholar - Xie W, Wang Y, Huang Y et al (2009) Toll-like receptor 2 mediates invasion via activating NF-kappaB in MDA-MB-231 breast cancer cells. Biochem Biophys Res Commun 379:1027–1032
Article PubMed CAS Google Scholar - Yoneda K, Sugimoto K, Shiraki K et al (2008) Dual topology of functional Toll-like receptor 3 expression in human hepatocellular carcinoma: differential signaling mechanisms of TLR3-induced NF-kappaB activation and apoptosis. Int J Oncol 33:929–936
PubMed CAS Google Scholar - Zhou M, McFarland-Mancini MM, Funk HM et al (2009) Toll-like receptor expression in normal ovary and ovarian tumors. Cancer Immunol Immunother. Jan 31 [Epub ahead of print]
- Kelly MG, Alvero AB, Chen R et al (2006) TLR-4 signaling promotes tumor growth and paclitaxel chemoresistance in ovarian cancer. Cancer Res 66:3859–3868
Article PubMed CAS Google Scholar - Whiteside TL (2008) The tumor microenvironment and its role in promoting tumor growth. Oncogene 27:5904–5912
Article PubMed CAS Google Scholar - Li H, Han Y, Guo Q et al (2009) Cancer-expanded myeloid-derived suppressor cells induce anergy of NK cells through membrane-bound TGF-beta 1. J Immunol 182:240–249
PubMed CAS Google Scholar - Strauss L, Bergmann C, Whiteside TL (2009) Human circulating CD4+ CD25high Foxp3+ regulatory T cells kill autologous CD8+ but not CD4+ responder cells by Fas-mediated apoptosis. J Immunol 182:1469–1480
PubMed CAS Google Scholar - Gribar SC, Richardson WM, Sodhi CP et al (2008) No longer an innocent bystander: epithelial toll-like receptor signaling in the development of mucosal inflammation. Mol Med 14:645–659
Article PubMed CAS Google Scholar - Lotze MT, Zeh HJ, Rubartelli A et al (2007) The grateful dead: damage-associated molecular pattern molecules and reduction/oxidation regulate immunity. Immunol Rev 220:60–81
Article PubMed CAS Google Scholar - Kumagai Y, Takeuchi O, Akira S (2008) Pathogen recognition by innate receptors. J Infect Chemother 14:86–92
Article PubMed CAS Google Scholar - Ellerman JE, Brown CK, de Vera M et al (2007) Masquerader: high mobility group box-1 and cancer. Clin Cancer Res 13:2836–2848
Article PubMed CAS Google Scholar - Rakoff-Nahoum S, Medzhitov R (2009) Toll-like receptors and cancer. Nat Rev Cancer 9:57–63
Article PubMed CAS Google Scholar - Kuper H, Adami HO, Trichopoulos D (2000) Infections as a major preventable cause of human cancer. J Intern Med 248:171–183
Article PubMed CAS Google Scholar - Zou W (2005) Immunosuppressive networks in the tumour environment and their therapeutic relevance. Nat Rev Cancer 5:263–274
Article PubMed CAS Google Scholar - Chochi K, Ichikura T, Kinoshita M et al (2008) Helicobacter pylori augments growth of gastric cancers via the lipopolysaccharide-toll-like receptor 4 pathway whereas its lipopolysaccharide attenuates antitumor activities of human mononuclear cells. Clin Cancer Res 14:2909–2917
Article PubMed CAS Google Scholar - Fukata M, Abreu MT (2007) TLR4 signalling in the intestine in health and disease. Biochem Soc Trans 35:1473–1478
Article PubMed CAS Google Scholar - Furrie E, Macfarlane S, Thomson G et al (2005) Toll-like receptors-2, -3 and -4 expression patterns on human colon and their regulation by mucosal-associated bacteria. Immunology 115:565–574
Article PubMed CAS Google Scholar - Dan HC, Sun M, Kaneko S et al (2004) Akt phosphorylation and stabilization of X-linked inhibitor of apoptosis protein (XIAP). J Biol Chem 279:5405–5412
Article PubMed CAS Google Scholar - Lee JW, Choi JJ, Seo ES et al (2007) Increased toll-like receptor 9 expression in cervical neoplasia. Mol Carcinog 46:941–947
Article PubMed CAS Google Scholar - Kundu SD, Lee C, Billips BK et al (2008) The toll-like receptor pathway: a novel mechanism of infection-induced carcinogenesis of prostate epithelial cells. Prostate 68:223–229
Article PubMed CAS Google Scholar - Merrell MA, Ilvesaro JM, Lehtonen N et al (2006) Toll-like receptor 9 agonists promote cellular invasion by increasing matrix metalloproteinase activity. Mol Cancer Res 4:437–447
Article PubMed CAS Google Scholar - Luo JL, Maeda S, Hsu LC et al (2004) Inhibition of NF-kappaB in cancer cells converts inflammation- induced tumor growth mediated by TNFalpha to TRAIL-mediated tumor regression. Cancer Cell 6:297–305
Article PubMed CAS Google Scholar - Pikarsky E, Porat RM, Stein I et al (2004) NF-kappaB functions as a tumour promoter in inflammation-associated cancer. Nature 431:461–466
Article PubMed CAS Google Scholar - Ren T, Wen ZK, Liu ZM et al (2007) Functional expression of TLR9 is associated to the metastatic potential of human lung cancer cell: functional active role of TLR9 on tumor metastasis. Cancer Biol Ther 6:1704–1709
Article PubMed CAS Google Scholar - Linehan DC, Goedegebuure PS (2005) CD25+ CD4+ regulatory T-cells in cancer. Immunol Res 32:155–168
Article PubMed CAS Google Scholar - Perrone G, Ruffini PA, Catalano V et al (2008) Intratumoural FOXP3-positive regulatory T cells are associated with adverse prognosis in radically resected gastric cancer. Eur J Cancer 44:1875–1882
Article PubMed CAS Google Scholar - Martinez FO, Sica A, Mantovani A et al (2008) Macrophage activation and polarization. Front Biosci 13:453–461
Article PubMed CAS Google Scholar - Marigo I, Dolcetti L, Serafini P et al (2008) Tumor-induced tolerance and immune suppression by myeloid derived suppressor cells. Immunol Rev 222:162–179
Article PubMed CAS Google Scholar - Rodriguez PC, Ochoa AC (2008) Arginine regulation by myeloid derived suppressor cells and tolerance in cancer: mechanisms and therapeutic perspectives. Immunol Rev 222:180–191
Article PubMed CAS Google Scholar - Kryczek I, Lange A, Mottram P et al (2005) CXCL12 and vascular endothelial growth factor synergistically induce neoangiogenesis in human ovarian cancers. Cancer Res 65:465–472
PubMed CAS Google Scholar - Li H, Fan X, Houghton J (2007) Tumor microenvironment: the role of the tumor stroma in cancer. J Cell Biochem 101:805–815
Article PubMed CAS Google Scholar - Haviv I, Polyak K, Qiu W et al (2009) Origin of carcinoma associated fibroblasts. Cell Cycle 8:589–595
PubMed CAS Google Scholar - Bhowmick NA, Chytil A, Plieth D et al (2004) TGF-beta signaling in fibroblasts modulates the oncogenic potential of adjacent epithelia. Science 303:848–851
Article PubMed CAS Google Scholar - Carmeliet P (2005) VEGF as a key mediator of angiogenesis in cancer. Oncology 69(Suppl 3):4–10
Article PubMed CAS Google Scholar - Chang YJ, Wu MS, Lin JT et al (2005) Helicobacter pylori-Induced invasion and angiogenesis of gastric cells is mediated by cyclooxygenase-2 induction through TLR2/TLR9 and promoter regulation. J Immunol 175:8242–8252
PubMed CAS Google Scholar - Pollet I, Opina CJ, Zimmerman C et al (2003) Bacterial lipopolysaccharide directly induces angiogenesis through TRAF6-mediated activation of NF-kappaB and c-Jun N-terminal kinase. Blood 102:1740–1742
Article PubMed CAS Google Scholar - van Beijnum JR, Buurman WA, Griffioen AW (2008) Convergence and amplification of toll-like receptor (TLR) and receptor for advanced glycation end products (RAGE) signaling pathways via high mobility group B1 (HMGB1). Angiogenesis 11:91–99
Article PubMed CAS Google Scholar - Damiano V, Caputo R, Bianco R et al (2006) Novel toll-like receptor 9 agonist induces epidermal growth factor receptor (EGFR) inhibition and synergistic antitumor activity with EGFR inhibitors. Clin Cancer Res 12:577–583
Article PubMed CAS Google Scholar - Majewski S, Marczak M, Mlynarczyk B et al (2005) Imiquimod is a strong inhibitor of tumor cell-induced angiogenesis. Int J Dermatol 44:14–19
Article PubMed CAS Google Scholar - Li VW, Li WW, Talcott KE et al (2005) Imiquimod as an antiangiogenic agent. J Drugs Dermatol 4:708–717
PubMed Google Scholar - Klein JR, Hoon DS, Nangauyan J et al (1989) S-100 protein stimulates cellular proliferation. Cancer Immunol Immunother 29:133–138
Article PubMed CAS Google Scholar - Helfman DM, Kim EJ, Lukanidin E et al (2005) The metastasis associated protein S100A4: role in tumour progression and metastasis. Br J Cancer 92:1955–1958
Article PubMed CAS Google Scholar - Cabezon T, Celis JE, Skibshoj I et al (2007) Expression of S100A4 by a variety of cell types present in the tumor microenvironment of human breast cancer. Int J Cancer 121:1433–1444
Article PubMed CAS Google Scholar - Hiratsuka S, Watanabe A, Sakurai Y et al (2008) The S100A8-serum amyloid A3-TLR4 paracrine cascade establishes a pre-metastatic phase. Nat Cell Biol 10:1349–1355
Article PubMed CAS Google Scholar - Kariko K, Ni H, Capodici J et al (2004) mRNA is an endogenous ligand for Toll-like receptor 3. J Biol Chem 279:12542–12550
Article PubMed CAS Google Scholar - Tsui NB, Ng EK, Lo YM (2006) Molecular analysis of circulating RNA in plasma. Methods Mol Biol 336:123–134
PubMed CAS Google Scholar - Ng EK, Tsui NB, Lam NY et al (2002) Presence of filterable and nonfilterable mRNA in the plasma of cancer patients and healthy individuals. Clin Chem 48:1212–1217
PubMed CAS Google Scholar - Gal S, Fidler C, Lo YM et al (2004) Quantitation of circulating DNA in the serum of breast cancer patients by real-time PCR. Br J Cancer 90:1211–1215
Article PubMed CAS Google Scholar - Wang BG, Huang HY, Chen YC et al (2003) Increased plasma DNA integrity in cancer patients. Cancer Res 63:3966–3968
PubMed CAS Google Scholar - Giacona MB, Ruben GC, Iczkowski KA et al (1998) Cell-free DNA in human blood plasma: length measurements in patients with pancreatic cancer and healthy controls. Pancreas 17:89–97
Article PubMed CAS Google Scholar - Mori T, O’Day SJ, Umetani N et al (2005) Predictive utility of circulating methylated DNA in serum of melanoma patients receiving biochemotherapy. J Clin Oncol 23:9351–9358
Article PubMed CAS Google Scholar - Tillman DK Jr, Carroll MT (2008) A 36-month clinical experience of the effectiveness of curettage and imiquimod 5% cream in the treatment of basal cell carcinoma. J Drugs Dermatol 7:s7–s14
PubMed Google Scholar - Miller RL, Gerster JF, Owens ML et al (1999) Imiquimod applied topically: a novel immune response modifier and new class of drug. Int J Immunopharmacol 21:1–14
Article PubMed CAS Google Scholar - Dudek AZ, Yunis C, Harrison LI et al (2007) First in human phase I trial of 852A, a novel systemic toll-like receptor 7 agonist, to activate innate immune responses in patients with advanced cancer. Clin Cancer Res 13:7119–7125
Article PubMed CAS Google Scholar - Dummer R, Hauschild A, Becker JC et al (2008) An exploratory study of systemic administration of the toll-like receptor-7 agonist 852A in patients with refractory metastatic melanoma. Clin Cancer Res 14:856–864
Article PubMed CAS Google Scholar - Hanten JA, Vasilakos JP, Riter CL et al (2008) Comparison of human B cell activation by TLR7 and TLR9 agonists. BMC Immunol 9:39
Article PubMed CAS Google Scholar - De Cesare M, Calcaterra C, Pratesi G et al (2008) Eradication of ovarian tumor xenografts by locoregional administration of targeted immunotherapy. Clin Cancer Res 14:5512–5518
Article PubMed Google Scholar - Krieg AM (2008) Toll-like receptor 9 (TLR9) agonists in the treatment of cancer. Oncogene 27:161–167
Article PubMed CAS Google Scholar - Schmidt J, Welsch T, Jager D et al (2007) Intratumoural injection of the toll-like receptor-2/6 agonist ‘macrophage-activating lipopeptide-2’ in patients with pancreatic carcinoma: a phase I/II trial. Br J Cancer 97:598–604
Article PubMed CAS Google Scholar - Rhee SH, Im E, Pothoulakis C (2008) Toll-like receptor 5 engagement modulates tumor development and growth in a mouse xenograft model of human colon cancer. Gastroenterology 135:518–528
Article PubMed CAS Google Scholar - Nishimura M, Naito S (2005) Tissue-specific mRNA expression profiles of human toll-like receptors and related genes. Biol Pharm Bull 28:886–892
Article PubMed CAS Google Scholar - Droemann D, Albrecht D, Gerdes J et al (2005) Human lung cancer cells express functionally active Toll-like receptor 9. Respir Res 6:1
Article PubMed CAS Google Scholar - Salaun B, Lebecque S, Matikainen S et al (2007) Toll-like receptor 3 expressed by melanoma cells as a target for therapy? Clin Cancer Res 13:4565–4574
Article PubMed CAS Google Scholar - Hassan F, Islam S, Tumurkhuu G et al (2006) Intracellular expression of toll-like receptor 4 in neuroblastoma cells and their unresponsiveness to lipopolysaccharide. BMC Cancer 6:281
Article PubMed CAS Google Scholar - Szczepanski M, Stelmachowska M, Stryczynski L et al (2007) Assessment of expression of toll-like receptors 2, 3 and 4 in laryngeal carcinoma. Eur Arch Otorhinolaryngol 264:525–530
Article PubMed Google Scholar