To be EndMT or not to be, that is the question in pulmonary hypertension (original) (raw)
Journal Article
Center for Molecular Medicine, National Heart, Lung and Blood Institute
,
National Institutes of Health
, 20892, Bethesda, MD,
USA
Search for other works by this author on:
 PDF
PDF Navbar Search Filter Mobile Enter search term Search
Pulmonary hypertension (PH) is a progressive and devastating disease of various causes that is associated with structural and functional disorder and inappropriately increased pressure of pulmonary small- to medium-sized vasculature. Extensive pulmonary vascular remodeling with narrowing lumen is well characterized in all forms of PH, which is hemodynamically defined by a mean pulmonary artery pressure exceeding 25 mmHg at rest (Schermuly et al., 2011; Mehari et al., 2014). The morbidity and mortality of PH continues to increase due to no cure (Mehari et al., 2014); however, our understanding of the mechanism and therapeutics underlying PH remains far from complete. There are many competing hypotheses for how PH develops in a genetic or sporadic way (Schermuly et al., 2011). One of them is that the endothelial-to-mesenchymal transition (EndMT) could be implicated in initiation and progression of human PH (Arciniegas et al., 2007). Notably, this point is further emphasized by two recent papers, which provide direct evidence linking EndMT to PH (Good et al., 2015; Ranchoux et al., 2015).
Vascular remodeling of intimal, medial, and adventitial hypertrophy in PH roughly involves endothelium, smooth muscle, and fibroblasts (Schermuly et al., 2011). Although pulmonary vasculature in PH is thought to undergo a series of structure change events with a complex multifactorial etiology, endothelial cells seem to play a central role in this process in view of the following four major reasons. First, the common plexiform lesions in the vessels of patients with PH generally result from excessive endothelial cell proliferation. Second, endothelial cells act with quite widespread autocrine and paracrine effects via secretion of numerous cell signaling effectors including, but certainly not limited to, nitric oxide, endothelin-1, and serotonin. Third, impaired semipermeable barrier of the pulmonary endothelial lining due to endothelial injuries renders the underlying interstitial cells susceptible to diverse blood-borne factors (Budhiraja et al., 2004). Last, germline loss-of-function bone morphogenic protein receptor type 2 (BMPR2) mutations have been significantly linked to the etiology of familial and idiopathic primary pulmonary hypertension (International et al., 2000; Austin and Loyd, 2014). More importantly, BMPR2 is predominantly expressed in endothelium and tightly controls the permeability of the pulmonary artery endothelial wall (Atkinson et al., 2002; Burton et al., 2011).
EndMT is characterized by the acquisition of mesenchymal- and stem-cell-like properties in endothelium subjected to intrinsic or extrinsic cues and functions as a critical source of fibroblasts in various physiological and pathological settings encompassing heart development, tumor progression, and fibrosis (Lin et al., 2012; Yu et al., 2014). One such notorious fibrosis target organ is lung. Reduced fibrinolytic activity and thereby elevated cellular fibronectin concentration have been demonstrated in the lung vessels of monocrotaline-induced rat PH model (Schultze and Roth, 1993; Schultze et al., 1996). Increased collagen deposition is capable of decreasing the distensibility of hypertensive pulmonary arteries (Tozzi et al., 1994). As mentioned earlier, even dysregulation of the intracellular cytoskeleton network causes altered permeability and morphology of pulmonary endothelium (Dudek and Garcia, 2001). Subsequently, Arciniegas and colleagues observed EndMT in pulmonary artery development of chicken embryos in vivo and in vitro (Arciniegas et al., 2005). In addition, myocardin promotes the transdifferentiation of pulmonary arteriolar endothelial cells into smooth muscle-like cells in hypoxia-induced rat PH model and porcine pulmonary artery endothelial cells (Zhu et al., 2006). A prior study showed a spontaneous and transforming growth factor β (TGF-β)-induced EndMT in pulmonary endothelial cells isolated from caveolin-1 knockout mice compared to their wild-type littermates (Li et al., 2013). Moreover, the precisely orchestrated EndMT contributes to bleomycin- and radiation-induced pulmonary fibrosis (Hashimoto et al., 2010; Choi et al., 2015). As progressive pulmonary fibrosis can lead to pulmonary hypertension (Rockey et al., 2015), it will be of considerable interest to ask whether EndMT participates in the regulatory network of human PH and, if so, what is the exact role of EndMT in the development of PH?
A key step towards answering these questions has been made by two intriguing studies (Good et al., 2015; Ranchoux et al., 2015) (Fig. 1). As some may recall, Qiao and colleagues took advantage of mice that underwent left pneumonectomy and monocrotaline pyrrole injection to establish a mouse model of PH. Endothelial lineage tracing analyses in this PH mouse model reveals that endothelial cells in the pulmonary neointima have detectable smooth muscle gene expression. Likewise, concurrent expression of endothelial cell and smooth muscle markers occurs in human pulmonary arterial hypertension neointimal lesions (Qiao et al., 2014). Consistent with these observations, Good et al. determined the presence of EndMT by assessing the colocalization of von Willebrand factor and α-smooth muscle actin (α-SMA) in the pulmonary endothelium from the hypoxia/SU5416 preclinical murine pulmonary artery hypertension (PAH) model and systemic sclerosis-associated-PAH (SSc-PAH) patients. Furthermore, a panel of functional assays in vitro lends further support to the notion that EndMT is a bona fide mechanism underlying the pathogenesis of PH (Good et al., 2015). TGF-β has been delineated as a major inducer of EndMT (van Meeteren and ten Dijke, 2012). Nevertheless, utilizing the single reagent TGF-β to galvanize transcription program switching in the EndMT of human pulmonary microvascular endothelial cells seems to require three weeks and several passages (Reynolds et al., 2012). Using a cocktail of TGF-β and inflammatory cytokins tumor necrosis factor α (TNFα) and interleukin 1β (IL-1β) previously described for induction of EndMT in human intestinal microvascular endothelial cells (Rieder et al., 2011), Good et al. obtained the induced EndMT (I-EndMT) cells derived from human pulmonary artery endothelial cells in vitro in a more efficient fashion (Good et al., 2015). These I-EndMT cells secrete high levels of proinflammatory cytokins (such as IL-6, IL-8, and TNFα) and exhibit a similar proinflammatory phenotype in patients with human lung fibroblasts SSc-PAH. Next, it was found that the endothelial barrier integrity is significantly compromised in I-EndMT cells, reminiscent of the features in PH endothelium (Budhiraja et al., 2004; Good et al., 2015).
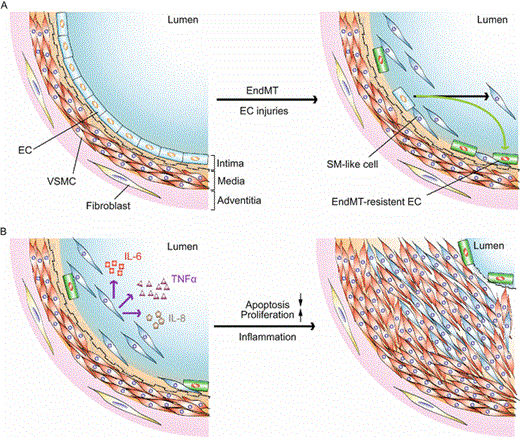
Figure 1
The role of endothelial-to-mesenchymal transition (EndMT) at roughly early (A) and late (B) stages in the pathogenesis of pulmonary hypertension. (A) Genetic and/or microenvironmental insults-induced EC injuries trigger EndMT in pulmonary vasculature. (B) Activated EndMT cells secrete proinflammatory cytokins to potentially stimulate VSMC and/or SM-like cell proliferation and inhibit these cell apoptosis, leading to pulmonary hypertension. Abbreviations: EC, endothelial cell; VSMC, vascular smooth muscle cell; SM-like, smooth muscle-like; IL-6, interleukin-6; IL-8, interleukin-8; TNFα, tumor necrosis factor α
Writing in Circulation at nearly the same time, Ranchoux et al. reported that analysis of endothelial cell-cell junction, and endothelial and subendothelial cell phenotype in intimal and plexiform lesions from PAH lungs relative to control non-tumor lung specimens, was carried out using unambiguous endothelial (CD31, CD34, VE-cadherin) and mesenchymal α-SMA markers. Combined with this analysis result, the protein and mRNA expression patterns consolidate the notion of a key role of EndMT in PH pathology (Ranchoux et al., 2015). Additionally, a unique and refined morphological signature in plexogenic pulmonary arteriopathy has been identified thanks to pioneering research efforts (Smith and Heath, 1979; Weibel, 2012). Remarkably, Ranchoux and co-workers applied transmission electron microscopy, and correlative light and electron microscopy, providing unequivocal ultrastructural-level evidence of ongoing dynamic EndMT in PH samples. Next, the EndMT was examined in the context of conventional monocrotaline and SuHx mouse PH models and the novel BMPR2 deficient rat PH model. Indeed, the in vivo EndMT characteristics in these mouse models are comparable to those in human PH tissues. Excitingly, partial rescue of EndMT-related gene expression and phenotypes has been achieved by rapamycin (Ranchoux et al., 2015).
The headline finding of these two studies is that the concept of EndMT in PH has been initially proposed on the basis of compelling experimental proofs (Good et al., 2015; Ranchoux et al., 2015). This discovery opens a new therapeutic window for suppressing or even reversing the pathogenic progression in certain common, but genetically defined, subtype of human PH. Each pulmonary endothelial cell in potential or current patients with PH decides whether its next action is to be EndMT or not to be (with apologies to pre-eminent English dramatist William Shakespeare for scrambling the immortal opening phase in his play Hamlet). In the broad landscape, it will be imperative to determine how the EndMT process is orchestrated, and what its context dependencies may be. Future advances in understanding the distinct stage-specific EndMT events and the druggability of EndMT will require a gene expression profile and more functional analyses to unravel the molecular mechanism in a tempo-spatial manner. Finally, these studies offer the possibility of EndMT as a promising pharmaceutical target in human PH, which warrants further investigations.
Footnotes
The author is grateful for the support from the NIH Intramural Program and the Leducq Foundation. The author thanks Cindy Clark, NIH Library Writing Center, for manuscript editing assistance.
Jianhua Xiong declares that he has no conflict of interest. This article does not contain any studies with human or animal subjects performed by the author.
References
Arciniegas
E
,
Neves
CY
,
Carrillo
LM
,
Zambrano
EA
,
Ramirez
R
Endothelial-mesenchymal transition occurs during embryonic pulmonary artery development
Endothelium
2005
12
193
–
200
Arciniegas
E
,
Frid
MG
,
Douglas
IS
,
Stenmark
KR
Perspectives on endothelial-to-mesenchymal transition: potential contribution to vascular remodeling in chronic pulmonary hypertension
Am J Physiol Lung Cell Mol Physiol
2007
293
L1
–
L8
Atkinson
C
,
Stewart
S
,
Upton
PD
,
Machado
R
,
Thomson
JR
,
Trembath
RC
,
Morrell
NW
Primary pulmonary hypertension is associated with reduced pulmonary vascular expression of type II bone morphogenetic protein receptor
Circulation
2002
105
1672
–
1678
Austin
ED
,
Loyd
JE
The genetics of pulmonary arterial hypertension
Circ Res
2014
115
189
–
202
Budhiraja
R
,
Tuder
RM
,
Hassoun
PM
Endothelial dysfunction in pulmonary hypertension
Circulation
2004
109
159
–
165
Burton
VJ
,
Ciuclan
LI
,
Holmes
AM
,
Rodman
DM
,
Walker
C
,
Budd
DC
Bone morphogenetic protein receptor II regulates pulmonary artery endothelial cell barrier function
Blood
2011
117
333
–
341
Choi
SH
,
Hong
ZY
,
Nam
JK
,
Jang
J
,
Lee
HJ
,
Yoo
RJ
,
Lee
YJ
,
Park
S
,
Ji
YH
,
Lee
YS
et al.
2015
A hypoxia-induced vascular endothelial-to-mesenchymal transition in development of radiation-induced pulmonary fibrosis
Clin Cancer Res
Dudek
SM
,
Garcia
JG
Cytoskeletal regulation of pulmonary vascular permeability
J Appl Physiol
2001
91
1985
1487
–
1500
Good
RB
,
Gilbane
AJ
,
Trinder
SL
,
Denton
CP
,
Coghlan
G
,
Abraham
DJ
,
Holmes
AM
2015
Endothelial to mesenchymal transition contributes to endothelial dysfunction in pulmonary artery hypertension
Am J Pathol
Hashimoto
N
,
Phan
SH
,
Imaizumi
K
,
Matsuo
M
,
Nakashima
H
,
Kawabe
T
,
Shimokata
K
,
Hasegawa
Y
Endothelial-mesenchymal transition in bleomycin-induced pulmonary fibrosis
Am J Respir Cell Mol Biol
2010
43
161
–
172
International
PPHC
,
Lane
KB
,
Machado
RD
,
Pauciulo
MW
,
Thomson
JR
,
Phillips
JA
3rd,
Loyd
JE
,
Nichols
WC
,
Trembath
RC
2000
Heterozygous germline mutations in BMPR2, encoding a TGF-beta receptor, cause familial primary pulmonary hypertension
Nat Genet
26
81
–
84
Li
Z
,
Wermuth
PJ
,
Benn
BS
,
Lisanti
MP
,
Jimenez
SA
Caveolin-1 deficiency induces spontaneous endothelial-to-mesenchymal transition in murine pulmonary endothelial cells in vitro
Am J Pathol
2013
182
325
–
331
Lin
F
,
Wang
N
,
Zhang
TC
The role of endothelial-mesenchymal transition in development and pathological process
IUBMB Life
2012
64
717
–
723
Mehari
A
,
Valle
O
,
Gillum
RF
Trends in pulmonary hypertension mortality and morbidity
Pulm Med
2014
2014
105864
Qiao
L
,
Nishimura
T
,
Shi
L
,
Sessions
D
,
Thrasher
A
,
Trudell
JR
,
Berry
GJ
,
Pearl
RG
,
Kao
PN
Endothelial fate mapping in mice with pulmonary hypertension
Circulation
2014
129
692
–
703
Ranchoux
B
,
Antigny
F
,
Rucker-Martin
C
,
Hautefort
A
,
Pechoux
C
,
Bogaard
HJ
,
Dorfmuller
P
,
Remy
S
,
Lecerf
F
,
Plante
S
et al.
Endothelial-to-mesenchymal transition in pulmonary hypertension
Circulation
2015
131
1006
–
1018
Reynolds
AM
,
Holmes
MD
,
Danilov
SM
,
Reynolds
PN
Targeted gene delivery of BMPR2 attenuates pulmonary hypertension
Eur Respir J
2012
39
329
–
343
Rieder
F
,
Kessler
SP
,
West
GA
,
Bhilocha
S
,
de la Motte
C
,
Sadler
TM
,
Gopalan
B
,
Stylianou
E
,
Fiocchi
C
Inflammation-induced endothelial-to-mesenchymal transition: a novel mechanism of intestinal fibrosis
Am J Pathol
2011
179
2660
–
2673
Rockey
DC
,
Bell
PD
,
Hill
JA
Fibrosis—a common pathway to organ injury and failure
N Engl J Med
2015
372
1138
–
1149
Schermuly
RT
,
Ghofrani
HA
,
Wilkins
MR
,
Grimminger
F
Mechanisms of disease: pulmonary arterial hypertension
Nat Rev Cardiol
2011
8
443
–
455
Schultze
AE
,
Roth
RA
Fibrinolytic activity in blood and lungs of rats treated with monocrotaline pyrrole
Toxicol Appl Pharmacol
1993
121
129
–
137
Schultze
AE
,
Emeis
JJ
,
Roth
RA
Cellular fibronectin and von Willebrand factor concentrations in plasma of rats treated with monocrotaline pyrrole
Biochem Pharmacol
1996
51
187
–
191
Smith
P
,
Heath
D
Electron microscopy of the plexiform lesion
Thorax
1979
34
177
–
186
Tozzi
CA
,
Christiansen
DL
,
Poiani
GJ
,
Riley
DJ
Excess collagen in hypertensive pulmonary arteries decreases vascular distensibility
Am J Respir Crit Care Med
1994
149
1317
–
1326
van Meeteren
LA
,
ten Dijke
P
Regulation of endothelial cell plasticity by TGF-beta
Cell Tissue Res
2012
347
177
–
186
Weibel
ER
Fifty years of Weibel-Palade bodies: the discovery and early history of an enigmatic organelle of endothelial cells
J Thromb Haemost
2012
10
979
–
984
Yu
W
,
Liu
Z
,
An
S
,
Zhao
J
,
Xiao
L
,
Gou
Y
,
Lin
Y
,
Wang
J
The endothelial-mesenchymal transition (EndMT) and tissue regeneration
Curr Stem Cell Res Ther
2014
9
196
–
204
Zhu
P
,
Huang
L
,
Ge
X
,
Yan
F
,
Wu
R
,
Ao
Q
Transdifferentiation of pulmonary arteriolar endothelial cells into smooth muscle-like cells regulated by myocardin involved in hypoxia-induced pulmonary vascular remodelling
Int J Exp Pathol
2006
87
463
–
474
© The Author(s) 2015
This is an Open Access article distributed under the terms of the Creative Commons Attribution Non-Commercial License (https://creativecommons.org/licenses/by/4.0/), which permits non-commercial reuse, distribution, and reproduction in any medium, provided the original work is properly cited. For commercial re-use, please contact [email protected]
Citations
Views
Altmetric
Email alerts
Citing articles via
More from Oxford Academic