The Emerging Role of p53 in Exercise Metabolism (original) (raw)
Abstract
The major tumour suppressor protein, p53, is one of the most well-studied proteins in cell biology. Often referred to as the Guardian of the Genome, the list of known functions of p53 include regulatory roles in cell cycle arrest, apoptosis, angiogenesis, DNA repair and cell senescence. More recently, p53 has been implicated as a key molecular player regulating substrate metabolism and exercise-induced mitochondrial biogenesis in skeletal muscle. In this context, the study of p53 therefore has obvious implications for both human health and performance, given that impaired mitochondrial content and function is associated with the pathology of many metabolic disorders such as ageing, type 2 diabetes, obesity and cancer, as well as reduced exercise performance. Studies on p53 knockout (KO) mice collectively demonstrate that ablation of p53 content reduces intermyofibrillar (IMF) and subsarcolemmal (SS) mitochondrial yield, reduces cytochrome c oxidase (COX) activity and peroxisome proliferator-activated receptor gamma co-activator 1-α protein content whilst also reducing mitochondrial respiration and increasing reactive oxygen species production during state 3 respiration in IMF mitochondria. Additionally, p53 KO mice exhibit marked reductions in exercise capacity (in the magnitude of 50 %) during fatiguing swimming, treadmill running and electrical stimulation protocols. p53 may regulate contractile-induced increases in mitochondrial content via modulating mitochondrial transcription factor A (Tfam) content and/or activity, given that p53 KO mice display reduced skeletal muscle mitochondrial DNA, Tfam messenger RNA and protein levels. Furthermore, upon muscle contraction, p53 is phosphorylated on serine 15 and subsequently translocates to the mitochondria where it forms a complex with Tfam to modulate expression of mitochondrial-encoded subunits of the COX complex. In human skeletal muscle, the exercise-induced phosphorylation of p53Ser15 is enhanced in conditions of reduced carbohydrate availability in association with enhanced upstream signalling through 5′adenosine monophosphate-activated protein kinase but not p38 mitogen-activated protein kinase. In this way, undertaking regular exercise in carbohydrate restricted states may therefore be a practical approach to achieve the physiological benefits of consistent p53 signalling. Although our knowledge of p53 in exercise metabolism has advanced considerably, much of our current understanding of p53 regulation and associated targets is derived from various non-muscle cells and tissues. As such, many fundamental questions remain unanswered in contracting skeletal muscle. Detailed studies concerning the time-course of p53 activation (including additional post-translational modifications and subsequent subcellular translocation), as well as the effects of exercise modality (endurance versus resistance), intensity, duration, fibre type, age, training status and nutrient availability, must now be performed so that we can optimise exercise prescription guidelines to strategically target p53 signalling. The emerging role of p53 in skeletal muscle metabolism therefore represents a novel and exciting research area for exercise and muscle physiologists.
1 Introduction
Skeletal muscle mitochondrial biogenesis (typically defined as an increase in mitochondrial content per gram of muscle tissue) is one of the hallmark adaptations to regular endurance exercise training [1]. From a functional perspective, the study of the regulation of mitochondrial biogenesis has obvious implications for both human health and performance. Impaired mitochondrial function is associated with the pathology of many metabolic disorders such as insulin resistance and type 2 diabetes [2], obesity [3], ageing [4] and cancer [5]. In relation to exercise performance, the increased mitochondrial content that accompanies exercise training ensures that exercise in the trained state induces less disturbance to metabolic homeostasis for a given exercise intensity. Acute customary exercise is therefore associated with smaller decreases in adenosine triphosphate (ATP), phosphocreatine and muscle glycogen utilisation, and smaller increases in adenosine diphosphate (ADP), adenosine monophosphate (AMP), inorganic phosphate and muscle lactate [6]. Such adaptations are recognised physiologically by the classic rightward shift of the lactate threshold curve [7], increased utilisation of lipid sources to fuel muscle contraction [8] and an improved ability to complete endurance exercise at higher intensities for a longer period [9].
Although the phenomenon of training-induced increases in skeletal muscle mitochondria was first recognised over 40 years ago, the precise molecular mechanisms underpinning mitochondrial biogenesis are only beginning to be understood [10]. It is now generally recognised that acute muscle contraction induces homeostatic perturbations to cellular energy status to activate a number of cell signalling kinases such as the calcium/calmodulin-dependent protein kinase II (CaMKII), the p38 mitogen-activated protein kinase (p38MAPK) and the AMP-dependent protein kinase (AMPK). These kinases converge on the regulation of transcriptional co-activators (e.g. peroxisome proliferator-activated γ receptor coactivator, PGC-1α) and transcription factors (e.g. nuclear respiratory factors 1 and 2, NRF1/2) to co-ordinate regulation of both the nuclear and mitochondrial genomes. In this way, a controlled expression of those proteins involved in the regulation of substrate oxidation, substrate transport and mitochondrial fusion and fission collectively occur in the hours and days following each successive exercise training bout, providing the exercise stimulus is sufficient in terms of intensity and duration [11]. The acute post-translational modifications of cell signalling kinases, the downstream localisation of their associated transcription factors and the subsequent increase in messenger RNA (mRNA) transcripts of their target genes is therefore considered to form the molecular basis of training adaptation [12].
In addition to the molecular players alluded to above, the tumour suppressor protein p53 has also recently emerged as a potent regulator of mitochondrial content, function and biogenesis. As the so-called Guardian of the Genome, p53 is one of the most well-studied proteins in cell biology and its list of known functions include regulatory roles in cell cycle arrest, apoptosis, angiogenesis, DNA repair and cell senescence [13]. More recently, studies employing p53 knockout (KO) rodents provide convincing evidence for a regulatory role of p53 in modulating mitochondrial content and exercise performance [14–16]. Additionally, acute exercise also induces post-translational modifications [16, 17] and alterations in subcellular localisation [18], thereby consistent with the notion that p53 contributes to the regulatory network controlling contractile-induced mitochondrial biogenesis.
In this current opinion article, we initially provide a brief overview of some of the acute cell signalling pathways associated with the regulation of exercise-induced mitochondrial biogenesis in skeletal muscle (so as to provide some context for the text to follow) before proceeding to review the evidence supportive of a regulatory role of p53 in modulating mitochondrial content and function. Data from both animal and human studies are subsequently presented to outline a potential signalling pathway through which p53 may regulate contractile-induced increases in mitochondrial content. Finally, we close by offering some perspectives, significance and directions for future research for what is emerging as a novel and exciting area of skeletal muscle biology.
2 Regular Exercise Training Promotes Skeletal Muscle Mitochondrial Biogenesis via Co-Ordinated Regulation of Both the Nuclear and Mitochondrial Genomes
Training-induced increases in mitochondrial biogenesis are thought to be due to the accumulative responses of transient changes in gene expression that occur in the hours during recovery from each training session [11]. During muscle contraction, a number of contractile-induced stressors (e.g. increased AMP/ATP ratio, reactive oxygen species [ROS], Ca2+ flux, lactate, hypoxia, decreased energy availability) are now known to collectively alter the post-translational status of key cell signalling kinases, the most well-studied of which are perhaps the CaMKII, p38MAPK and AMPK. These kinases, acting alone or in combination with each other, can subsequently activate downstream transcription factors and co-activators [19] that exert regulatory roles in co-ordinating the expression of both nuclear and mitochondrial-encoded proteins. Broadly speaking, activation of the aforementioned kinases is dependent on exercise modality [20], intensity [19], duration [21, 22], training history [23], training status [24] and nutrient availability [25]. When taken together, these data provide an insight as to the potential molecular mechanisms underpinning some of the well-known training principles, in that training programs should ensure a continual alteration in workload and manipulation of energy provision in order to repeatedly stimulate the necessary signalling pathways required to support sustained muscle adaptation.
The kinases p38MAPK and AMPK have been identified as two putative signalling proteins, given that they converge on the regulation of PGC-1α, the transcriptional co-activator often cited as the so-called ‘master regulator’ of mitochondrial biogenesis [26]. The importance of PGC-1α in regulating mitochondrial content/function and muscle metabolic health is evident from rodent studies demonstrating that overexpression increases mitochondrial content and oxidative enzyme activity [27], improves insulin sensitivity (albeit when expressed within physiological levels comparable to exercise) [28], protects against sarcopenia [29] and improves exercise capacity [30]. Both p38MAPK [31] and AMPK [32] can directly phosphorylate PGC-1α on different sites, thereby resulting in increased activity and subsequent translocation to both the nucleus [33] and mitochondria [34] during acute exercise. In the nucleus, PGC-1α binds to and activates a host of transcription factors such as NRF1, NRF2, peroxisome proliferator-activated receptor (PPARδ), estrogen-related receptor (EERα) and myocyte enhancer factor 2 (MEF2) to collectively induce upregulation of a variety of proteins involved in the transport and oxidation of glucose and fatty acids. In the mitochondria, PGC-1α modulates the activity of mitochondrial transcription factor A (Tfam), a nuclear-encoded protein that is, itself, regulated by PGC-1α and subsequently incorporated into the mitochondria, so as to increase transcription of mitochondrial-encoded proteins, notably the cytochrome c oxidase (COX) subunits. In this way, PGC-1α is therefore recognised as a major point of control in regulating both nuclear and mitochondrial DNA (mtDNA) expression.
It is noteworthy that many of the acute transcriptional responses to acute exercise are observed prior to an increase in PGC-1α protein [11, 35]. In this regard, it is the alterations in activity of the existing cytoplasmic PGC-1α protein that is considered as the initial regulatory step in controlling the transcriptional network. However, both acute exercise [11, 17, 19] and exercise training [36–38] also increase PGC-1α mRNA and protein content within hours and days after exercise, respectively. Although the changes to existing PGC-1α activity co-ordinate the initial signalling pathways required to stimulate mitochondrial biogenesis, it is the subsequent increase in PGC-1α protein content that occurs with repeated exercise sessions that is therefore considered a necessary requirement to stimulate further mitochondrial adaptations during the course of an endurance training program [35]. For this reason, an understanding of the control of PGC-1α transcription is also important and, with this in mind, the reader is directed to several contemporary and excellent reviews [10, [39](/article/10.1007/s40279-013-0127-9#ref-CR39 "Uguccioni G, D’souza D, Hood DA. Regulation of PPARγ coactivator-1α function and expression in muscle: effect of exercise. PPAR Res. Epub 19 Aug 2010. doi: 10.1155/2010/937123
."), [40](/article/10.1007/s40279-013-0127-9#ref-CR40 "Ljubicic V, Joseph A-M, Saleem A, et al. Transcriptional and post-transcriptional regulation of mitochondrial biogenesis in skeletal muscle: effects of exercise and aging. Biochim Biophys Acta. 2010;1800:223–34.")\].3 p53 Modulates Skeletal Muscle Mitochondrial Content, Function and Exercise Capacity
Evidence indicative of a role of p53 in modulating mitochondrial function (albeit in mouse liver mitochondria and human HCT116 cells) was initially provided by Matoba et al. [14]. In both cell types, these authors observed that loss of p53 reduced oxygen consumption and increased lactate production whilst maintaining rates of ATP production, thus demonstrating that p53 deficiency favours glycolysis as an energy-producing pathway. The reduction in cellular oxygen consumption was also accompanied by parallel decreases in COX activity, which was suggested to be due in part to reduced synthesis of cytochrome c oxidase 2 (SCO2) protein content in both cell types. Indeed, SCO2 is critical for the assembly of COX subunit II into the COX complex, and transactivation of cells with p53 confirmed that SCO2 is a direct transcriptional target of p53. In relation to exercise performance, exercise capacity was reduced in p53 KO mice by approximately 50 % during a fatiguing swimming protocol, thus confirming the functional relevance of p53 activity in vivo.
In relation to muscle tissue, Saleem et al. [16] provided the first report to demonstrate that p53 modulates skeletal muscle mitochondrial content and function. These workers observed that p53 KO mice exhibit reduced subsarcolemmal (SS) and intermyofibrillar (IMF) mitochondrial content as well as reduced COX activity and PGC-1α expression when compared with wild-type animals. Additionally, ablation of p53 also reduced state 3 respiration and increased ROS production in IMF mitochondria. The reductions in mitochondrial content and function also impaired exercise performance, as evidenced by an increased rate of fatigue during acute contractile activity in the form of electrical stimulation.
In a subsequent study, Park et al. [15] confirmed that loss of p53 reduces exercise capacity as they observed that maximal running time to exhaustion was reduced in p53 KO animals. During submaximal running exercise, blood lactate was also threefold higher in p53 KO mice compared with wild-type animals, thus demonstrating that energy production in vivo favours glycolytic metabolism. p53 KO mice were also relatively unresponsive to 5 weeks of treadmill exercise training, as recognised by no significant changes in peak oxygen uptake (_V_O2peak), work capacity or respiratory exchange ratio (as a marker of substrate oxidation). However, unlike findings from mouse liver mitochondria, SCO2 mRNA and protein content in skeletal muscle was not affected by loss of p53. The baseline protein content of PGC-1α (in contrast to Saleem et al. [16]) and exercise-induced mRNA expression of key regulators of mitochondrial biogenesis (e.g. PGC-1α, PGC-1β, NRF1, NRF2) and nuclear (COXIV) and mitochondrial encoded (COXII) members of the electron transport chain (ETC) were also not affected by loss of p53. However, both Tfam mRNA and protein, as well as mtDNA, were significantly lower in p53 KO animals compared with wild-type mice.
When taken together, the above data unequivocally demonstrate that p53 is a potent regulator of mitochondrial content, function and exercise performance. Whilst the mechanisms underpinning how p53 regulates exercise-induced mitochondrial biogenesis are not well understood, it is currently thought that p53 may directly or indirectly modulate Tfam expression and activity, thereby strategically targeting regulation of the mitochondrial genome. Potential signalling mechanisms regulating this pathway in acutely exercised skeletal muscle are discussed in greater detail in the subsequent section.
4 Exercise Induces Post-Translational Modification of p53 and Promotes Subcellular Mitochondrial and Nuclear Translocation
As noted by Saleem et al. [41], if p53 does indeed play a regulatory role in contractile-induced mitochondrial biogenesis, it follows that it should exhibit post-translational modification and/or alter its subcellular location upon contraction. Indeed, Saleem et al. [16] initially observed that acute muscle contraction (in the form of a fatiguing electrical stimulation protocol) induces a twofold change in phosphorylation of p53 on serine 15 (a modification that is typically associated with increased stability and activity) immediately post-exercise. This change in phosphorylation status also occurred in concert with the classical exercise-induced phosphorylation of both AMPK and p38MAPK, thus suggesting that these kinases may serve as upstream kinases modifying p53 activity. In addition, we also observed that exercise (both high-intensity interval and moderate-intensity continuous running) induces p53Ser15 phosphorylation in human skeletal muscle (vastus lateralis) at 3 h post-exercise in a time-course that appeared related to upstream signalling through either AMPK and/or p38MAPK [17].
In a further study, we subsequently demonstrated that the exercise-induced activation of p53 appears dependent on CHO availability, given that completing the exercise bout in accordance with conventional sports nutrition guidelines (i.e. glycogen loaded, pre-exercise meal and both CHO provision during exercise and in the recovery period) attenuated p53 signalling [42]. In contrast, commencing and recovering from the exercise bout with low CHO availability induced threefold increases in p53 phosphorylation at 3 h post-exercise. The enhanced p53 response with low carbohydrate (CHO) availability was also associated with enhanced acetyl CoA carboxylase (ACC) phosphorylation on serine 79 immediately post-exercise (but not p38MAPK phosphorylation), thus suggesting that AMPK may be the dominant upstream signalling kinase regulating contractile-induced p53 phosphorylation. Interestingly, we also observed that SCO2 mRNA levels were not affected by exercise or CHO restriction (despite enhanced p53 activation), thus extending previous data from rodent muscle [15] that p53 may not regulate the SCO2 protein in skeletal muscle. It is worth noting, however, that our chosen biopsy sampling points were limited to immediately post-exercise and 3 h post-exercise and hence it is possible that more detailed time-course studies need to be performed to detect any significant effects of exercise on SCO2 expression.
The observation that CHO restriction enhances exercise-induced p53 signalling is consistent with cell culture data demonstrating a potent effect of glucose deprivation on p53 activity [43]. There is also a growing body of literature demonstrating that training in conditions of reduced CHO availability enhances oxidative adaptations of human skeletal muscle [37, 44, 45] and, in this regard, the emergence of an AMPK-p53 signalling axis provides an additional pathway that may potentially contribute to this enhanced training effect. Such an hypothesis is consistent with the well-known observation that p53 promotes a shift from glycolysis towards lipid metabolism in cell lines [5, 46, 47], and the fact that training with low CHO availability also enhances whole body [44, 48] and intra-muscular lipid oxidation [45] during customary submaximal exercise.
In addition to post-translational modifications, Saleem and Hood [18] also recently observed that acute treadmill exercise of moderate intensity (90 min at approximately 70 % _V_O2peak) in rodents induces p53 translocation to both the SS and IMF mitochondria (but not the nucleus) at 3 h post-exercise. Although the authors observed no apparent exercise-induced increase in mitochondrial Tfam content, they did demonstrate that p53 forms a complex with Tfam and mtDNA (within the D-loop region) at 3 h post-exercise. When taken together, these findings suggest that p53 may preferentially function to co-ordinate regulation of the mitochondrial genome either directly or indirectly through modulation of Tfam activity. Supporting evidence for this hypothesis was also provided by the observation that the exercise-induced expression of COX-I mRNA (an mtDNA-encoded protein) was abolished in p53 KO mice compared with wild-type animals. The authors also present an intriguing hypothesis that the apparent exodus of p53 from the nucleus upon muscle contraction may be a controlled regulatory molecular mechanism to offset any negative regulation on PGC-1α, thereby allowing the master regulator to continue its work in regulating the nuclear genome. It is worth noting, however, that other researchers have indeed observed the nuclear translocation of p53 upon muscle contraction [49]. As such, it is possible that p53 may dually function to upregulate Tfam expression in the nucleus and then further modify existing mitochondrial Tfam activity and/or newly expressed Tfam that has been subsequently incorporated into the mitochondria in the hours following each successive exercise bout. Furthermore, p53 has also been shown in other cell types to regulate additional proteins with key roles in mitochondrial biogenesis such as heat shock protein 70 (HSP70, a molecular chaperone), apoptosis-inducing factor (AIF, necessary for correct assembly of complex 1 of the respiratory chain) and the mitochondrial fission (dynamin related protein 1, Drp1) and fusion proteins (mitofusion 2, Mfn2) [50–52], although further work is needed to verify these pathways in exercising skeletal muscle. Nevertheless, with this in mind we present a schematic overview of how acute exercise may regulate p53 activity in skeletal muscle so as to modulate transcription of both nuclear and mtDNA-encoded genes in the hours following each acute exercise bout that are likely translated into protein with successive days of repeated exercise (see Fig. 1).
Fig. 1
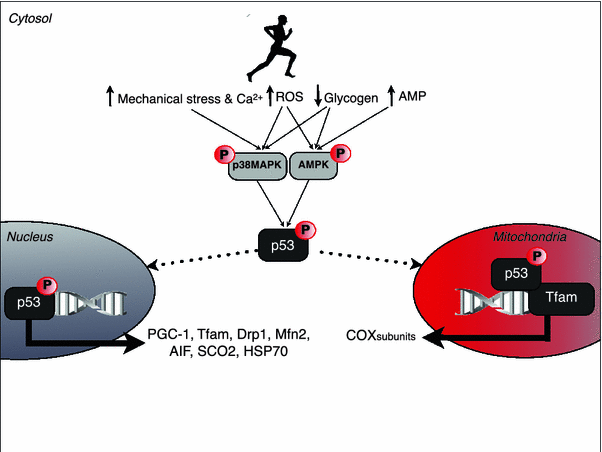
Schematic illustration of the potential signalling pathway through which acute exercise may modulate p53 signalling. During muscle contraction, disruptions to cellular energy homeostasis (e.g. increased mechanical stress, Ca2+, AMP/ATP ratio, ROS and decreased glycogen, etc.) phosphorylate AMPK and/or p38MAPK to converge on the activation of p53 through phosphorylation on serine p53. Upon activation, p53 translocates to the mitochondria where it co-ordinates expression of COX subunits through modulation of Tfam activity. Additionally, p53 may also translocate to the nucleus to induce expression of Tfam (that is subsequently incorporated into the mitochondria) as well as other key proteins involved in mitochondrial biogenesis such as PGC-1α, Drp1, Mfn2, SCO2, AIF and HSP70. Note that many of the potential downstream targets of p53 are based on data from a variety of non-muscle cells and tissues and hence further research is required to verify the existence of such pathways in contracting skeletal muscle as well as additional downstream targets. Any additional exercise-induced post-translational modifications of p53 that are required in order to gain full activity must also be identified. P phosphorylation, AIF apoptosis-inducing factor, AMP adenosine monophosphate, AMPK 5′AMP-activated protein kinase, ATP adenosine triphosphate, COX cytochrome c oxidase, Drp1 dynamin-related protein 1, HSP70 heat shock protein 70, Mfn2 mitofusion 2, p38MAPK p38 mitogen-activated protein kinase, PGC-1α peroxisome proliferator-activated receptor gamma co-activator 1-α, ROS reactive oxygen species, SCO2 synthesis of cytochrome c oxidase 2, Tfam mitochondrial transcription factor A; s_olid lines_ represent activation and dashed lines represent translocation
5 Perspectives and Future Directions
The identification of p53 as a molecular regulator of the mitochondrial and/or nuclear genome represents a significant development in our understanding of p53 function in skeletal muscle, one that has obvious implications for both human health and physical performance. Indeed, reduced p53 function is associated with tumour development, insulin resistance, reduced longevity and impaired exercise performance [41, 46]. Furthermore, the nutritional modulation of contractile-induced p53 signalling (i.e. low CHO availability) also represents an additional approach to potentially achieve the physiological and therapeutic benefits of p53 signalling, especially in relation to reducing the risk of associated metabolic disorders such as cancer, type 2 diabetes, obesity and ageing. The strategic periodization of training with low CHO availability also represents a practical approach for sports scientists to modulate mitochondrial-related training adaptations and, hence, potentially improve endurance performance in the elite athlete.
At a fundamental level, however, there are many basic questions that remain unanswered in relation to exercise-induced p53 signalling. Perhaps most notably, much of our current understanding of p53 regulation and associated targets is derived from various cell lines and tissues with limited data on skeletal muscle. In this regard, and similar to other key molecular players, detailed studies concerning the time-course of p53 activation (including additional post-translational modifications and subsequent subcellular translocation), as well as the effects of exercise mode (i.e. endurance versus resistance), intensity, duration, fibre type, age, training status and nutrient availability, must now be performed so that we can optimise exercise prescription guidelines to strategically target p53 signalling. Additional downstream gene targets of p53 must also be identified so as to provide direct evidence for a crucial regulatory role in modulating mitochondrial biogenesis. The impact of exercise modality on p53 regulation is of particular interest given that endurance and resistance exercise are known to induce very different muscle phenotypes. As such, p53 may represent an additional player that is also involved in influencing concurrent training adaptations and the molecular interference effect that may occur when divergent exercise modalities are performed in close proximity to one another. The emerging role of p53 in skeletal muscle metabolism therefore represents a novel and exciting research area for exercise and muscle physiologists.
References
- Holloszy JO. Biochemical adaptations in muscle: effects of exercise on mitochondrial oxygen uptake and respiratory enzyme activity in skeletal muscle. J Biol Chem. 1967;10(242):2278–82.
Google Scholar - Joseph A-M, Joanisse DR, Baillot RG, et al. Mitochondrial dysregulation in the pathogenesis of diabetes: potential for mitochondrial biogenesis-mediated interventions. Exp Diabetes Res. 2012;2012:1–16.
Article Google Scholar - Jelenik T, Roden M. Mitochondrial plasticity in obesity and diabetes mellitus. Antioxid Redox Signal. 2013;20(19):258–68.
Article Google Scholar - Safdar A, Hamadeh MJ, Kaczor JJ, et al. Aberrant mitochondrial homeostasis in the skeletal muscle of sedentary older adults. PLoS ONE. 2010;5:e10778.
Article PubMed Central PubMed Google Scholar - Lago CU, Sung HJ, Ma W, et al. p53, aerobic metabolism, and cancer. Antioxid Redox Signal. 2011;15(15):1739–48.
Article CAS PubMed Google Scholar - Leblanc PJ, Howarth KR, Gibala MJ, et al. Effects of 7 wk of endurance training on human skeletal muscle metabolism during submaximal exercise. J Appl Physiol. 2004;97:2148–53.
Article CAS PubMed Google Scholar - Bassett DR, Howley ET. Limiting factors for maximum oxygen uptake and determinants of endurance performance. Med Sci Sports Exerc. 2000;32:70–84.
PubMed Google Scholar - Henriksson J. Training induced adaptation of skeletal muscle and metabolism during submaximal exercise. J Physiol. 1977;270:661–75.
CAS PubMed Google Scholar - Hawley JA. Adaptations of skeletal muscle to prolonged, intense endurance training. Clin Exp Pharmacol Physiol. 2002;29:218–22.
Article CAS PubMed Google Scholar - Holloszy JO. Regulation of mitochondrial biogenesis and GLUT4 expression by exercise. Compr Physiol. 2011;1:921–40.
PubMed Google Scholar - Perry CGR, Lally J, Holloway GP, et al. Repeated transient mRNA bursts precede increases in transcriptional and mitochondrial proteins during training in human skeletal muscle. J Physiol. 2010;1(588):4795–810.
Article Google Scholar - Coffey VG, Hawley JA. The molecular bases of training adaptation. Sports Med. 2007;37:737–63.
Article PubMed Google Scholar - Levine AJ, Hu W, Feng Z. The P53 pathway: what questions remain to be explored? Cell Death Differ. 2006;13:1027–36.
Article CAS PubMed Google Scholar - Matoba S, Kang J-G, Patino WD, et al. p53 regulates mitochondrial respiration. Science. 2006;16(312):1650–3.
Article Google Scholar - Park J-Y, Wang P-Y, Matsumoto T, et al. p53 improves aerobic exercise capacity and augments skeletal muscle mitochondrial DNA content. Circ Res. 2009;25(105):705–12.
Article Google Scholar - Saleem A, Adhihetty PJ, Hood DA. Role of p53 in mitochondrial biogenesis and apoptosis in skeletal muscle. Physiol Genom. 2009;3(37):58–66.
Article Google Scholar - Bartlett JD, Hwa Joo C, Jeong T-S, et al. Matched work high-intensity interval and continuous running induce similar increases in PGC-1α mRNA, AMPK, p38, and p53 phosphorylation in human skeletal muscle. J Appl Physiol. 2012;112:1135–43.
Article CAS PubMed Google Scholar - Saleem A, Hood DA. Acute exercise induces p53 translocation to the mitochondria and promotes a p53–Tfam–mtDNA complex in skeletal muscle. J Physiol. 2013;591:3625–36.
CAS PubMed Google Scholar - Egan B, Carson BP, Garcia-Roves PM, et al. Exercise intensity-dependent regulation of peroxisome proliferator-activated receptor coactivator-1 mRNA abundance is associated with differential activation of upstream signalling kinases in human skeletal muscle. J Physiol. 2010;15(588):1779–90.
Article Google Scholar - Atherton PJ, Babraj J, Smith K, et al. Selective activation of AMPK-PGC-1alpha or PKB-TSC2-mTOR signaling can explain specific adaptive responses to endurance or resistance training-like electrical muscle stimulation. FASEB J. 2005;19:786–8.
CAS PubMed Google Scholar - Rose AJ, Hargreaves M. Exercise increases Ca2+-calmodulin-dependent protein kinase II activity in human skeletal muscle. J Physiol. 2003;15(553):303–9.
Article Google Scholar - Rose AJ, Kiens B, Richter EA. Ca2+-calmodulin-dependent protein kinase expression and signalling in skeletal muscle during exercise. J Physiol. 2006;1(574):889–903.
Article Google Scholar - Coffey V, Zhong Z, Shield A, et al. Early signaling responses to divergent exercise stimuli in skeletal muscle from well-trained humans. FASEB J. 2006;20:190–2.
CAS PubMed Google Scholar - Benziane B, Burton TJ, Scanlan B, et al. Divergent cell signaling after short-term intensified endurance training in human skeletal muscle. Am J Physiol Endocrinol Metab. 2008;295:E1427–38.
Article CAS PubMed Google Scholar - Yeo WK, McGee SL, et al. Acute signalling responses to intense endurance training commenced with low or normal muscle glycogen. Exp Physiol. 2010;95:351–8.
Article CAS PubMed Google Scholar - Puigserver P, Spiegelman BM. Peroxisome proliferator-activated receptor-gamma coactivator 1 alpha (PGC-1 alpha): transcriptional coactivator and metabolic regulator. Endocr Rev. 2003;24:78–90.
Article CAS PubMed Google Scholar - Lin J, Wu H, Tarr PT, et al. Transcriptional co-activator PGC-1 alpha drives the formation of slow-twitch muscle fibres. Nature. 2002;15(418):797–801.
Article Google Scholar - Benton CR, Wright DC, Bonen A. PGC-1alpha-mediated regulation of gene expression and metabolism: implications for nutrition and exercise prescriptions. Appl Physiol Nutr Metab. 2008;33:843–62.
Article CAS PubMed Google Scholar - Wenz T, Rossi SG, Rotundo RL, et al. Increased muscle PGC-1alpha expression protects from sarcopenia and metabolic disease during aging. Proc Natl Acad Sci USA. 2009;1(106):20405–10.
Article Google Scholar - Calvo JA, Daniels TG, Wang X, et al. Muscle-specific expression of PPARgamma coactivator-1alpha improves exercise performance and increases peak oxygen uptake. J Appl Physiol. 2008;104:1304–12.
Article CAS PubMed Google Scholar - Fan M, Rhee J, St-Pierre J, et al. Suppression of mitochondrial respiration through recruitment of p160 myb binding protein to PGC-1alpha: modulation by p38 MAPK. Genes Dev. 2004;1(18):278–89.
Article Google Scholar - Jäger S, Handschin C, St-Pierre J, et al. AMP-activated protein kinase (AMPK) action in skeletal muscle via direct phosphorylation of PGC-1alpha. Proc Natl Acad Sci USA. 2007;17(104):12017–22.
Article Google Scholar - Little JP, Safdar A, Cermak N, et al. Acute endurance exercise increases the nuclear abundance of PGC-1alpha in trained human skeletal muscle. Am J Physio Regul Integr Comp Physiol. 2010;22(298):R912–7.
Article Google Scholar - Safdar A, Little JP, Stokl AJ, et al. Exercise increases mitochondrial PGC-1alpha content and promotes nuclear-mitochondrial cross-talk to coordinate mitochondrial biogenesis. J Biol Chem. 2011;25(286):10605–17.
Article Google Scholar - Wright D, Geiger P, Han D, et al. Calcium induces increases in peroxisome proliferator-activated receptor γ coactivator-1α and mitochondrial biogenesis by a pathway leading to p38 mitogen-activated protein kinase activation. J Biol Chem. 2007;282(26):18793–9.
Article CAS PubMed Google Scholar - Russell AP, Feilchenfeldt J, Schreiber S, et al. Endurance training in humans leads to fiber type-specific increases in levels of peroxisome proliferator-activated receptor-gamma coactivator-1 and peroxisome proliferator-activated receptor-alpha in skeletal muscle. Diabetes. 2003;52:2874–81.
Article CAS PubMed Google Scholar - Morton JP, Croft L, Bartlett JD, et al. Reduced carbohydrate availability does not modulate training-induced heat shock protein adaptations but does upregulate oxidative enzyme activity in human skeletal muscle. J Appl Physiol. 2009;106:1513–21.
Article CAS PubMed Google Scholar - Burgomaster KA, Howarth KR, Phillips SM, et al. Similar metabolic adaptations during exercise after low volume sprint interval and traditional endurance training in humans. J Physiol. 2008;1(586):151–60.
Google Scholar - Uguccioni G, D’souza D, Hood DA. Regulation of PPARγ coactivator-1α function and expression in muscle: effect of exercise. PPAR Res. Epub 19 Aug 2010. doi:10.1155/2010/937123.
- Ljubicic V, Joseph A-M, Saleem A, et al. Transcriptional and post-transcriptional regulation of mitochondrial biogenesis in skeletal muscle: effects of exercise and aging. Biochim Biophys Acta. 2010;1800:223–34.
Article CAS PubMed Google Scholar - Saleem A, Carter HN, Iqbal S, et al. Role of p53 within the regulatory network controlling muscle mitochondrial biogenesis. Exerc Sport Sci Rev. 2011;39:199–205.
PubMed Google Scholar - Bartlett JD, Louhelainen J, Iqbal Z, et al. Reduced carbohydrate availability enhances exercise-induced p53 signaling in human skeletal muscle: implications for mitochondrial biogenesis. Am J Physiol Regul Integr Comp Physiol. 2013;304:R450–8.
Article CAS PubMed Google Scholar - Madan E, Gogna R, Bhatt M, et al. Regulation of glucose metabolism by p53: emerging new roles for the tumor suppressor. Oncotarget. 2011;2:948–57.
PubMed Google Scholar - Yeo WK, Paton CD, Garnham AP, et al. Skeletal muscle adaptation and performance responses to once a day versus twice every second day endurance training regimens. J Appl Physiol. 2008;105:1462–70.
Article CAS PubMed Google Scholar - Hulston C, Venables MC, Mann CH, et al. Training with low muscle glycogen enhances fat metabolism in well-trained cyclists. Med Sci Sport Exerc. 2010;42:2046–55.
CAS Google Scholar - Vousden KH, Ryan KM. p53 and metabolism. Nat Rev Cancer. 2009;9:691–700.
Article CAS PubMed Google Scholar - Maddocks ODK, Vousden KH. Metabolic regulation by p53. J Mol Med. 2011;89:237–45.
Article CAS PubMed Central PubMed Google Scholar - Yeo WK, Lessard SJ, Chen Z-P, et al. Fat adaptation followed by carbohydrate restoration increases AMPK activity in skeletal muscle from trained humans. J Appl Physiol. 2008;105:1519–26.
Article CAS PubMed Google Scholar - Philp A, Chen A, Lan D, et al. Sirtuin 1 (SIRT1) deacetylase activity is not required for mitochondrial biogenesis or peroxisome proliferator-activated receptor-gamma coactivator-1alpha (PGC-1alpha) deacetylation following endurance exercise. J Biol Chem. 2011;2(286):30561–70.
Article Google Scholar - Stambolsky P, Weisz L, Shats I, et al. Regulation of AIF expression by p53. Cell Death Differ. 2006;13:2140–9.
Article CAS PubMed Google Scholar - Li J, Donath S, Li Y, et al. miR-30 regulates mitochondrial fission through targeting p53 and the dynamin-related protein-1 pathway. PLoS Genet. 2010;6:e1000795.
Article PubMed Central PubMed Google Scholar - Wang W, Cheng X, Lu J, et al. Mitofusin-2 is a novel direct target of p53. Biochem Biophys Res Commun. 2010;1(400):587–92.
Article Google Scholar
Acknowledgments
The authors declare no conflicts of interest. No form of external funding was received to prepare this manuscript.
Author information
Authors and Affiliations
- Research Institute for Sport and Exercise Sciences, Liverpool John Moores University, Tom Reilly Building, Byrom St Campus, Liverpool, L3 3AF, UK
Jonathan D. Bartlett, Graeme L. Close, Barry Drust & James P. Morton
Authors
- Jonathan D. Bartlett
You can also search for this author inPubMed Google Scholar - Graeme L. Close
You can also search for this author inPubMed Google Scholar - Barry Drust
You can also search for this author inPubMed Google Scholar - James P. Morton
You can also search for this author inPubMed Google Scholar
Corresponding author
Correspondence toJames P. Morton.
Rights and permissions
About this article
Cite this article
Bartlett, J.D., Close, G.L., Drust, B. et al. The Emerging Role of p53 in Exercise Metabolism.Sports Med 44, 303–309 (2014). https://doi.org/10.1007/s40279-013-0127-9
- Published: 22 November 2013
- Issue Date: March 2014
- DOI: https://doi.org/10.1007/s40279-013-0127-9