Viral exploitation and subversion of the immune system through chemokine mimicry (original) (raw)
To an immunologist, the chemokine signaling system appears primarily to coordinate development and deployment of the immune system and, therefore, to support antimicrobial host defense1,2. However, it is now clear that viruses have thoroughly corrupted this system through molecular mimicry, using it as a toolbox for facilitating diverse steps in pathogenesis. The chemokine system consists of at least 50 structurally related peptide agonists and 20 G protein–coupled receptors. To date, more than 30 distinct virally encoded chemokine and chemokine receptor mimics have been identified, all in the herpesvirus, poxvirus and retrovirus families. These include six medically important viruses: HIV; human cytomegalo-virus; human herpesviruses 6, 7 and 8; and molluscum contagiosum virus. Because immunosuppressed subjects can be coinfected with several of these viruses, it follows that the chemokine mimics produced by each virus might interact in a pathophysiologically significant manner not only with the host chemokine system but also with the chemokine mimics encoded by the other viruses (Fig. 1).
Figure 1: Potential combinatorial effects of chemokine mimicry on viral immunopathogenesis.
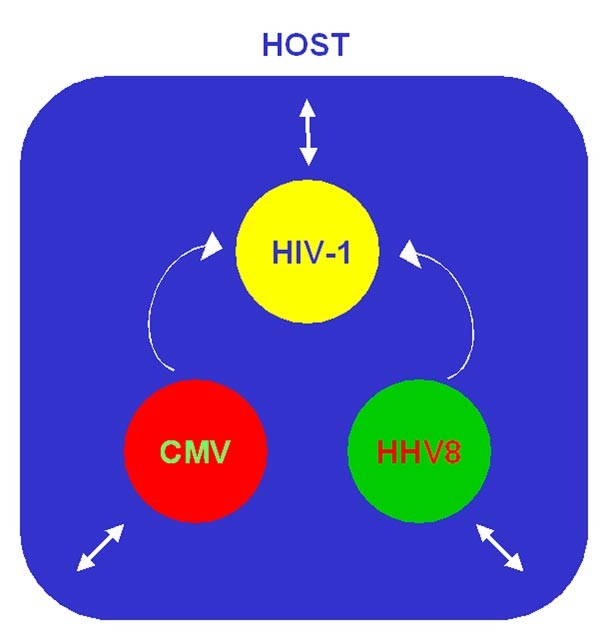
Immunosuppression, such as that caused by HIV-AIDS, may promote productive coinfection of the same host (blue box) with CMV, HHV8 and other herpesviruses as well as the poxvirus molluscum contagiosum virus. Both host and virus may deploy chemokine mimics that, in some cases, have been shown, in vitro, to interact with those of the other viruses and of the host (arrows). Reported interactions include the ability of the CMV-encoded chemokine receptor US28 to support HIV infection by functioning as an HIV coreceptor and the ability of the HHV8 chemokine mimic vMIP-II to block both that activity as well as HIV-1 usage of the host leukocyte receptors CCR5 and CXCR4. Host chemokines can also block gp120 usage of host chemokine receptors for CD4+ target-cell entry. Other viral chemokine mimics can act as agonists at leukocyte chemokine receptors, possibly to promote viral dissemination. Interaction of HIV-1 gp120 with CCR5 is critical for efficient HIV transmission in exposed populations. Whether other potential interactions among viral mimics occur in vivo and affect pathogenesis is not yet known.
One subset of mimics includes actual chemokines and chemokine receptors, which were presumably acquired by the virus in the manner of viral oncogenes, by gene transfer from the host. The captured sequences can be spliced or unspliced3,4 and are clear examples of divergent evolution. A second subset includes molecules with structures that are unrelated to chemokines or chemokine receptors, but which bind to either a chemokine or a chemokine receptor, thus distorting its function. These are examples of convergent evolution. The chemokine and chemokine receptor mimics form the largest known class of viral homologs of host proteins. The majority of the others are mimics either of other types of immunoregulatory molecules or of cell-cycle-control proteins5,6.
To date, the most important achievement in this area has been to define a central role for the chemokine receptor CCR5 in HIV pathogenesis7,8. This has enabled new antiretroviral drug and vaccine development strategies that have the unusual feature of exploiting a host protein9,10. Additional chemokine mimics have direct potential as anti-inflammatory agents. In this review I will first present a classification system for viral chemokine mimics (Table 1), then focus on recent advances and important unanswered questions with an emphasis on molecules relevant to HIV disease.
Table 1 Viral mimics of chemokines and chemokine receptors
Classification of viral chemokine mimics
Chemokine mimics can be classified into three groups according to their structure: chemokine homologs; chemokine receptor homologs; and unique structures not related in primary amino acid sequence to either chemokines or chemokine receptors (Table 2). Viral chemokine homologs are defined solely by structural similarities. In contrast, because chemokine receptors are related structurally to other types of G protein–coupled receptors, they are defined by functional criteria (chemokine binding and signal transduction).
Table 2 Classification of viral chemokine mimics.
Viral chemokine homologs are found mainly in herpesviruses and include both CC and CXC chemokines, the two main divisions of the chemokine superfamily. Unlike most herpesvirus genes, the genes encoding chemokine and chemokine receptor homologs are often not conserved by closely related herpesviruses. This suggests that they may regulate unique aspects of pathogenesis.
The six known viral chemokine receptors all bind multiple chemokine ligands but differ from each other with respect to specific signaling properties and chemokine specificity. The ligand specificities for these receptors are unusually diverse and, in some cases, even cross the subclass boundaries that govern the mammalian receptors. The receptors have 25–59% amino acid sequence identity to mammalian chemokine receptors as well as shared structural features such as an acidic NH2-terminus and a short basic third-intracellular loop that are characteristic of, but not invariant among or exclusive to, vertebrate chemokine receptors. Additional candidate chemokine receptors have been identified based on high sequence homology with known chemokine receptors. In some cases the candidate open-reading frame (ORF) is syntenic with a known chemokine receptor ORF in a related virus.
To date, five distinct subclasses of mimics have been identified according to in vitro functions: anti-chemokines; cell-entry factors; cell-growth factors; angiogenic factors; and leukocyte chemoattractants. Anti-chemokines subvert the immune system, whereas other types of mimics exploit it. Anti-chemokines can be further subclassified into three groups according to structure and mechanism of action: chemokine homologs that act as chemokine receptor antagonists; chemokine receptor homologs that function as plasma membrane–expressed chemokine scavengers; and chemokine binding proteins with signal sequences, which may have unique and unrelated structures and which function as secreted chemokine scavengers.
Each mimic may carry out more than one function and each function may be carried out by mimics from more than one structural class. Of the 13 known herpesvirus and poxvirus chemokine homologs, five are agonists, two are antagonists and six are still functionally undefined. Six are CC chemokines and seven are CXC chemokines. Viral chemokine agonists act in vitro as leukocyte chemoattractants of narrow specificity, possibly to focus preferred host cells at the site of infection and ensure viral spread.
Chemokine mimicry by HIV-1 gp120
The most notorious example of chemokine mimicry is gp120 of the HIV-1 envelope glycoprotein. gp120 lacks a chemokine structural fold and chemokine sequence motifs and yet can exploit specific chemokine receptors, known as HIV coreceptors, to enable infection of immune system targets7,8. HIV-2, SIV and other lentiviral envelope glycoproteins also do this. Whether this is the only significant activity of gp120 in vivo is not yet clear. In vitro soluble gp120 can also act at both CCR5 and CXCR4 to induce leukocyte signalling11,12,13. It can also act at CXCR4 to directly induce apoptosis of CXCR4+ neurons14 and can indirectly induce apoptosis of CD8+ CXCR4− T cells by stimulating tumor necrosis factor (TNF) release from macrophages15. Together these activities have major implications for how HIV may recruit, infect and deplete immune system cells.
The generally accepted model of HIV infection holds that gp120 forms a trimolecular complex with CD4 and either CCR5 or CXCR4. This results in deployment of a cryptic fusogenic peptide of gp41, which induces fusion of the viral envelope with the plasma membrane of CD4+ target cells7,16. According to a widely used nomenclature system17, viruses that can use either of these coreceptors are designated R5X4, whereas monotropic strains are designated R5 (CCR5-using) or X4 (CXCR4-using). Because homozygous inheritance of the defective allele CCR5Δ32—which encodes a truncated form of CCR5 not expressed on the plasma membrane—is strongly associated with HIV resistance in populations at risk for infection, normal CCR5 is, by implication, most likely required for efficient person-to-person HIV-1 transmission18,19,20. This, coupled with the lack of apparent health problems in CCR5Δ32 homozygotes, has identified CCR5 as an attractive drug-development target. Several methods to block CCR5 have been published and potent small-molecule antagonists are already in early stages of clinical testing. The therapeutic efficacy of CCR5 blockade may differ for preventing infection as compared to slowing or preventing progression of established infection and could be limited by the ability of virus to evolve usage of other coreceptors, particularly CXCR421. The importance of CXCR4 in vivo is supported by the presence of pure X4 strains in approximately one-third of AIDS patients and rare CCR5Δ32 homozygotes that are HIV+ (ref. 22). However, it is also possible that the 13 or so other “minor” coreceptors identified so far using model systems also operate in disease and might threaten treatment strategies targeting CCR523. Biological evidence for this is limited to a small number of studies of blocking agents or naturally occurring genetic polymorphisms, which are so far inconclusive (CCR2, CCR3, CCR8) or conflicting (CX3CR1)24,25,26,27,28.
The structural basis of chemokine mimicry by gp120 is extremely complex and, despite the availability of a crystal structure29, not yet defined. Like chemokines, gp120 appears to bind to multiple low affinity sites on coreceptors, which together create a high binding energy. Some binding sites—for example, a sulfated tyrosine in the NH2-terminal segment30—also appear to bind chemokines, which explains why the two ligands cross-compete in binding studies. Nevertheless, different gp120s that recognize the same coreceptor can discriminate among other coreceptors and can even bind to different sets of extracellular domains of the same coreceptor31. In addition, X4 and R5 tropism can be switched by changing the specificity of just a single basic amino acid within the V3 loop of gp12032.
Efficient formation of the fusion complex requires sequential interaction of HIV-1 gp120 first with CD4, then with coreceptor33, which indicates that chemokine mimicry by gp120 is CD4-dependent. Several exceptions to this have been noted however7. Recent evidence from immunoprecipitation studies in primary CD4+ T cells, monocytes and macrophages indicates that CD4 binds constitutively to coreceptor monomers, possibly via the second extracellular loop, in the absence of gp12034,35. However, there is, as yet, no hard evidence that this interaction modulates the normal signaling function of either CD4 or coreceptors. Chemokine ligands have been reported to induce dimerization of CCR2, CCR5 and CXCR436,37,38 and studies using an agonistic monoclonal antibody suggest that the CCR2 dimer is the signaling form of the receptor. However, coreceptor dimerization may actually inhibit rather than facilitate HIV fusion37. Still, if, as appears to be the case, the HIV envelope glycoprotein is a multimeric structure, it follows that multiple CD4-coreceptor complexes are likely to be recruited into the fusion complex.
Although when tested in reporter cell lines the specificity of HIV strains for CCR5 and CXCR4 is relatively clear, it does not always correlate with tropism for natural targets in primary cells and tissues. Whether this is due to tissue factors that modulate the structure of gp120 or its targets or both is still unknown. One example is that viruses that by reporter cell-line criteria are dual-tropic paradoxically may infect primary lymphoid tissue in a manner that is consistent with monotropism. This was originally suggested in studies that employed coreceptor-selective blocking agents38 but has now been verified using primary lymphoid tissue from a donor homozygous for CCR5Δ3239. A second example is the paradoxical resistance of primary macrophages—which express CXCR4, CCR5 and CD4—to some X4 HIV strains. This may be due, in part, to competition between CCR5 and CXCR4 for limited amounts of CD4, a competition won by CCR5, at least in macrophages40. Alternatively, a reduced capacity of X4 gp120, relative to R5 gp120, for inducing transmembrane signals in macrophages may be responsible because signaling has been shown to facilitate early post-fusion steps in viral replication41. The signaling pathway involved has not been clearly identified. However, it may not include Gi-type G proteins, which normally couple to CCR5 and CXCR4, because pertussis toxin–treatment of target cells, which efficiently blocks Gi signaling, has been reported not to affect HIV replication.
Chemokine mimicry by HIV-1 Tat
Biological restrictions on chemokine mimicry by gp120 are also shown by the fact that X4 strains can be isolated from only about one-third of HIV+ subjects and almost never until the late stages of disease are reached42. This is despite the fact that CXCR4+CCR5− cells represent a much larger subset of CD4+ T cells in blood and lymph node than either CXCR4−CCR5+ or CXCR4+CCR5+ cells38. The implication of this subset analysis is that X4 viral infection may accelerate CD4+ T cell depletion and disease onset. This has been borne out in vitro using peripheral blood mononuclear cells (PBMCs) and primary lymphoid tissue, as well as by epidemiological association studies38,42. X4 restriction factors could confer a selective advantage to the virus by increasing the chance of transmission to a new host by prolonging the life of the first host. Such factors may include the relatively low prevalence of X4 strains in infected populations; the differential distribution of CCR5+ versus CXCR4+ target cells in mucosal tissue43; and, apropos of the subject of this review, a second HIV-encoded chemokine mimic, the Tat protein44,45.
Tat is a transcription factor that is specific for the HIV TAR sequence46 but it can also be released from HIV-infected cells and has been detected in serum from a subset of HIV+ patients45. Among other activities, extracellular Tat can function as a chemokine mimic. It acts as a chemotactic agonist for neutrophils, basophils (via CCR3), mast cells (via CCR3) and monocytes (via CCR2) but as an antagonist at CXCR4; importantly Tat has no activity at CCR545,47,46,47,48,49. Consistent with this, Tat can induce evolution of R5 strains from an X4 inoculum in vitro45. Thus, Tat could conceivably contribute to in vivo restriction of X4 strains by selectively blocking use of CXCR4. Balanced against this are reports that Tat can induce CXCR4 expression50 and that a Tat-based vaccine can protect monkeys from SHIV challenge51. This suggests that the overriding effect of Tat in vivo may be harmful, not beneficial, perhaps due to its transactivation activity. The SHIV experiment may not test this hypothesis adequately because this virus probably uses CCR5 for infection of target cells. A more significant challenge to the hypothesis is the question of how Tat blockade of CXCR4 might be lifted in late stages of infection to allow emergence of X4 strains.
Compared to chemokines, Tat is similar in length and charge, contains both CXC and CC motifs but lacks other sequence similarities and a chemokine fold. Of note, highly basic small-molecules have been identified that act as specific high-potency antagonists at CXCR4. These include ALX40-4C52, a simple nine-residue arginine polymer that was designed to mimic the highly basic ARM (arginine-rich motif) domain of Tat. This may explain the differential specificity of Tat for coreceptors because the extracellular surface of CXCR4 is much more acidic than that of CCR5.
Chemokine mimicry by human herpesvirus 8
The most common tumor associated with HIV-AIDS is Kaposi's sarcoma (KS), which is widely considered to be an infectious disease caused by HHV8 (also known as Kaposi's sarcoma–associated herpesvirus, or KSHV). Several epidemiologic forms of KS have been recognized, all of which are associated with HHV8 infection. Unlike cancer, KS appears to result from multicentric hyperplasia not metastatic neoplasia53. Lesions appear to be growth factor–dependent and are composed of characteristic spindle cells, blood vessels and infiltrating leukocytes. Although molecular mechanisms are still undefined, HHV8 encodes cell cycle–control proteins and multiple cytokines, including three chemokine mimics that are plausible links between the prevailing growth factor and infectious theories of KS pathogenesis. They may also regulate pathogenesis of primary effusion lymphoma and multicentric Castleman's disease, which have both been linked to HIV-AIDS and HHV8.
Most compelling, however, is HHV8 ORF 74 (also referred to as KSHV G protein–coupled receptor, or GPCR, and vGPCR). This constitutively active chemokine receptor54 has been shown to induce lesions that are histologically similar to KS when expressed in immunocompetent transgenic mice55. The tumors can be reconstituted in wild-type recipients by adoptive transfer of bone marrow cells from ORF 74 transgenic donors. However, receptor transcripts can be detected in only a small subset of tumor cells. This is also the case for primary KS, in which ORF 74 transcripts colocalize with lytic transcripts56. This aspect of ORF 74 expression in the context of HHV8 infection is not modeled in the transgenic mouse experiment. However, in both cases, the expression data imply an indirect mechanism of tumorigenesis. In this regard, ORF 74 has been shown, in vitro, to activate host immunoregulatory transcription factors (NF-κB and AP-1). ORF 74 has also been shown to induce expression of: the pro-inflammatory cytokines interleukin 1 (IL-1) and TNF; the chemokines IL-8 (also called CXCL8 according to a standard nomenclature1) and monocyte-chemoattracting protein 1 (MCP-1, or CCL2); and the growth factors basic fibroblast growth factor and vascular endothelial cell growth factor54,57. All of these factors have been detected in KS tumors and, together, could coordinate the histological features of KS. Constitutive chemokine induction could even feedback at ORF 74 to modulate its signaling activity. ORF 74 also has oncogenic activity when tested in NIH 3T3 cells54 but, for the reasons cited above, this activity may not play a role in tumorigenesis.
HHV8 ORF 74 was apparently copied from the host chemokine receptor CXCR2 but its functional properties are quite different. CXCR2 is not constitutively active, has not been shown to induce cytokine and growth factor expression and binds only the ELR+ CXC chemokine subset of ORF 74 ligands2. CXCR2 is primarily a leukocyte chemotactic receptor but a possible role in tumorigenesis has been suggested by its expression on some tumor cell lines58. In addition, CXCR2 has been shown to mediate CXC chemokine induction of angiogenesis through its expression on endothelial cells59. Thus, both CXCR2 and ORF 74 may regulate tumorigenesis and angiogenesis but by different mechanisms.
An ORF 74 homolog is found in the same chromosomal region in the related γ2 herpesviruses Herpesvirus saimiri(ORF ECRF3) and γHV68 (ORF 74)60, which infect nonhuman primates and mice, respectively. However, ORF 74 is not conserved in the human γ1 herpesvirus Epstein-Barr virus. CXC chemokines can induce calcium flux in frog oocytes expressing ECRF361 but its properties in mammalian cells and its role in viral pathogenesis are unknown. The function of γHV68 ORF 74 has not yet been reported.
HHV8 also encodes three multifunctional CC chemokines, vMIP-I, vMIP-II and vMIP-III, that act in vitro as leukocyte chemotaxis modulators and HIV-suppressive factors and as angiogenic factors in the chick chorioallantoic membrane assay4,62,63,64. vMIP-I and vMIP-III are chemo-kine receptor agonists, whereas vMIP-II is a broad-spectrum chemokine receptor antagonist. vMIP-III acts selectively at CCR4 and chemoattracts TH2-polarized T cells. However, the significance of this is unclear because these cells have not been linked yet to KS. vMIP-II blocks signaling of CCR1, CCR2, CCR3, CCR5 and CXCR4 and blocks HIV usage of CCR3, CCR5 and CXCR4. vMIP-I also blocks HIV usage of CCR5. vMIP-II can inhibit signaling by HHV8 ORF 7465.
Interestingly, of the four ligands known for the TH2 T cell–associated receptor CCR8, three are viral in origin. These include vMIP-I and vMIP-II, which are agonists, and MC148R of molluscum contagiosum virus (MCV), which is an antagonist66,67,68. This suggests that this receptor may play quite different roles in HHV8 versus MCV infection, MCV also being an opportunistic pathogen in HIV-AIDS. Of note, MCV lesions are virtually devoid of leukocyte infiltration suggesting that it deploys powerful anti-inflammatory factors, including—but perhaps not limited to—MC148R, whereas KS has prominent leukocyte infiltration.
CCR8 is expressed in thymus and on TH2 T lymphocytes, which suggests the potential therapeutic utility of MC148R in TH2-polarized immunologically mediated inflammation. Because vMIP-II has broad target specificity it also has potential as an anti-inflammatory agent. So far it has been shown to be safe and effective in a rat model of glomerulonephritis69.
Although HHV8 chemokine mimics may have some role in KS tumorigenesis and HIV disease, they almost certainly serve another more fundamental role in the HHV8 life cycle because this virus almost certainly evolved outside the context of immunosuppression and HIV. With respect to the chemokine receptor agonists, one such role may be to recruit targets to facilitate viral dissemination. Most likely the HIV suppressive activity is accidental, resulting from independent evolution of opposing exploitation and subversion strategies at the same leukocyte targets by HIV and HHV8. It will be important to test the effects of these proteins on viral replication and latency.
Chemokine mimicry in cytomegalovirus
Human cytomegalovirus (HCMV) can cause severe opportunistic infections that affect multiple organs in immunocompromised hosts, including patients with AIDS. HCMV encodes three chemokine mimics: two CXC chemokine agonists called vCXC-1 and vCXC-270 and a G protein–coupled receptor called US28 (which is specific for multiple CC chemokines and the CX3C chemokine fractalkine)71,72.
In vitro assays have suggested roles for US28 in both immune evasion and viral dissemination. It can scavenge the chemokine RANTES (regulated upon activation, normal T cell–expressed and secreted) from culture media, which suggests a mechanism to prevent recruitment of leukocytes73,74. However, it can also induce chemokine-dependent chemokinesis in vascular smooth muscle cells75, which has suggested a role in atherosclerosis consistent with the epidemiologic and physical association of HCMV with that disease. US28 can also enhance cell-cell fusion mediated by different proteins76 and could conceivably mediate cell-cell or virus-cell adhesion directly by binding fractalkine72, an unusual membrane-bound pro-adhesive chemokine. The latter function would require the presence of US28 in virion membrane, which is not only conceivable but was anticipated by studies with a related viral protein UL33 (see below)77. US28 also functions as an HIV coreceptor in some transfected cell lines78. Finally, US28 has been shown to be constitutively active in transfected COS cells and able to induce NF-κB activation79. Which of these activities occur in actual human infection by HCMV is not yet established.
vCXC-1 induces neutrophil chemotaxis70, with a potency that is on par with IL-8, and acts selectively at CXCR2, one of the two known IL-8 receptor subtypes2. This may explain the association of neutrophils (and mononuclear cells, which also express CXCR2) with HCMV infection. vCXC-2 function has not yet been reported. vCXC-1 and vCXC-2 were detected in low-passage HCMV clinical isolates but not in the attenuated, tissue culture–adapted strain AD169, which suggests that they encode virulence factors.
Neither HCMV chemokine mimic is found in mouse cytomegalovirus (MCMV), which instead encodes a distinct agonistic CC chemokine mimic with a unique structure named m131/129 (also called MCK-1) that is not found in HCMV80. Direct biological analysis of this chemokine using a loss-of-function genetic test has shown its central role in viral dissemination. In particular, normal spread of MCMV to salivary gland—which occurs late after infection via mononuclear leukocytes, probably monocytes and/or macrophages—is abolished in m131/129 mutants81,82. Consistent with this, the chemokine domain of m131/129, induces calcium flux and adherence in murine peritoneal macrophages, although the specific receptors involved have not yet been delineated82. m131/129 may actually play a complex dual role in pathogenesis, functioning also as an immune evasion factor early in infection because viral seeding of liver and spleen is cleared more rapidly in m131/129 mutants. NK cells mediate early defense against MCMV pathogenesis and this is regulated by the host chemokine MIP-1α, however, direct studies of m131/129 action on these cells have not been reported83.
HCMV ORF UL3384 may also encode a chemokine receptor because it is syntenic with HHV6 ORF U1285, which has been shown to encode a CC chemokine receptor. UL33 is conserved in both mouse and rat CMV. Although their biochemical functions are undefined, targeted disruption of UL33 homologs in both rat and mouse CMV results in reduced viral virulence, which, in part, is due to defective viral spread to salivary gland86,87.
Recently the stealth variant of simian CMV has been reported to encode three CXC chemokine homologs and five US28 homologs88,89. Together these ORFs account for 7% of the recognized ORFs in this virus.
Chemokine mimicry in HHV6
HHV6, which has been implicated as an opportunistic pathogen in HIV disease90, has three chemokine system mimics: two are receptors and one is a chemokine agonist. ORF U51 has exceptionally low sequence relatedness to mammalian and other viral chemokine receptors (<23% amino acid identity) but, like US28, it binds multiple CC chemokines. It may function as an immune evasion molecule but by a unique mechanism: suppression of endogenous RANTES transcription. This has been reported to occur even in the absence of chemokine ligation91 and could be viewed as a mechanism for viral spread from the initial site of infection in epithelia. HHV6 also encodes a CC chemokine agonist (ORF U83)92 and a second CC chemokine receptor (ORF U12)85 but there is limited information available about their functional roles.
Secreted chemokine scavengers
One group of chemokine mimics that are less clearly related to HIV-AIDS but are of great conceptual and potential practical importance is the secreted chemokine scavengers. Soluble chemokine scavengers may be well suited for clinical application in inflammation93,94, although issues of delivery and antigenicity may severely limit the indications.
To date, three structurally unique classes of virally encoded chemokine scavengers have been identified: the 35-kD CC chemokine scavengers of diverse orthopoxviruses and leporipoxviruses; the mixed CC chemokine and interferon γ scavenger of myxoma virus, a rabbit poxvirus; and, the first herpesvirus example, the recently discovered M3 protein of γ herpesvirus 6895,96,97,98. M3 binds a broad spectrum of CC and CXC chemokines and blocks chemokine signaling97,98. Its relevance to CXC chemokines is still unclear because, although it binds IL-8 well, it does not appear to bind MIP-2 or KC, the functional mouse counterparts of IL-8. The M3 sequence is related to another γHV68 protein M1—which functions as a latency factor—and the poxvirus serine protease Spi-2, neither of which has been reported yet to bind chemokines.
These factors have variable effects on virulence. Deletion of the myxoma virus 35-kD protein M-T1 had no significant effects on disease progression in or mortality of infected European rabbits. However, increased infiltration of leukocytes—particularly monocytes and macrophages—in primary tissue sites early after infection is observed, which suggests a role for M-T1 in blocking chemokine-mediated leukocyte mobilization99. Nevertheless, viral clearance was not reduced in this experiment, which suggests the importance of concerted action of other viral proteins.
The structure of the 35-kD protein from cowpox has now been elucidated. It is a unique structure with a negatively charged surface that presumably corresponds to the chemokine-binding domain100. The chemo-kine and chemokine receptor mimics with unique structure raise the question of whether nonchemokine host proteins might exist that can activate chemokine receptors. Recently several examples of this have been reported including the β–defensins, which bind to CCR6 and may provide a link between innate and adaptive immune responses101.
It has been reported that CCR5 (and other chemokine receptors) can facilitate infection by myxoma, vaccinia and other poxviruses102. If this holds true for variola, which is now being tested, then smallpox will be the leading candidate for the selective pressure that is responsible for fixation of the CCR5Δ32 HIV-resistance allele in modern Caucasians, the finding that provided proof of principle for the chemokine hypothesis of HIV pathogenesis.
Conclusions
Why chemokine mimicry is such a common theme in viral pathogenesis is a mystery. The simplest answer, that viruses need to block chemokine action to evade the immune system, clearly applies to only a few of the known examples. The chemokine receptors, in particular, appear to have been adapted to completely new functions. In addition, disease associations such as Kaposi's sarcoma may be misleading with regard to the function of a particular chemokine mimic in the normal viral life cycle. Further study of these diverse molecules holds great promise for improved understanding of viral immunopathogenesis and, in some cases, for clinical application in immunologically mediated diseases.