The DNA sequence of human chromosome 22 (original) (raw)
Main
Two alternative approaches have been proposed to determine the human genome sequence. In the clone by clone approach, a map of the genome is constructed using clones of a suitable size (for example, 100–200 kilobases (kb)), and then the sequence is determined for each of a representative set of clones that completely covers the map1. Alternatively, a whole genome shotgun2 requires the sequencing of unmapped genomic clones, typically in a size range of 2–10 kb, followed by a monolithic assembly to produce the entire sequence. Although the merits of these two strategies continue to be debated3, the public domain human genome sequencing project is following the clone by clone approach4 because it is modular, allows efficient organization of distributed resources and sequencing capacities, avoids problems arising from distant repeats and results in early completion of significant units of the genome. Here we report the first sequencing landmark of the human genome project, the operationally complete sequence of the euchromatic portion of a human chromosome.
Chromosome 22 is the second smallest of the human autosomes, comprising 1.6–1.8% of the genomic DNA5. It is one of five human acrocentric chromosomes, each of which shares substantial sequence similarity in the short arm, which encodes the tandemly repeated ribosomal RNA genes and a series of other tandem repeat sequence arrays. There is no evidence to indicate the presence of any protein coding genes on the short arm of chromosome 22 (22p). In contrast, direct6 and indirect7,8 mapping methods suggest that the long arm of the chromosome (22q) is rich in genes compared with other chromosomes. The relatively small size and the existence of a high-resolution framework map of the chromosome9 suggested to us that sequencing human chromosome 22 would provide an excellent opportunity to show the feasibility of completing the sequence of a substantial unit of the human genome. In addition, alteration of gene dosage on part of 22q is responsible for the aetiology of a number of human congenital anomaly disorders including cat eye syndrome (CES, Mendelian Inheritance in Man (MIM) 115470, http://www.ncbi.nlm.nih.gov/omim/) and velocardiofacial/DiGeorge syndrome (VCFS, MIM 192430; DGS, MIM 188400). Other regions associated with human disease are the schizophrenia susceptibility locus10,11, and the sequences involved in spinocerebellar ataxia 10 (SCA10)12. Making the sequence of human chromosome 22 freely available to the community early in the data collection phase has benefited studies of disease-related and other genes associated with this human chromosome13,14,15,16,17,18,19.
Genomic sequencing
To identify genomic clones as the substrate for sequencing chromosome 22, extensive clone maps of the chromosome were constructed using cosmids, fosmids, bacterial artificial chromosomes (BACs) and P1-derived artificial chromosomes (PACs). Clones representing parts of chromosome 22 were identified by screening BAC and PAC libraries representing more than 20 genome equivalents using sequence tagged site (STS) markers known to be derived from the chromosome, or by using cosmid and fosmid libraries derived from flow-sorted DNA from chromosome 22. Overlapping clone contigs were assembled on the basis of restriction enzyme fingerprints and STS-content data, and ordered relative to each other using the established framework map of the chromosome9. The resulting nascent contigs were extended and joined by iterative cycles of chromosome walking using sequences from the end of each contig. In two places, yeast artificial chromosome (YAC) clones were used to join or extend contigs (AL049708, AL049760). The sequence-ready map covers 22q in 11 clone contigs with 10 gaps and stretches from sequences containing known chromosome 22 centromeric tandem repeats to the 22q telomere20.
In the final sequence, one additional gap that was intractable to sequencing is found 234 kb from the centromere (see below). The gaps between the clone contigs are located at the two ends of the map, in the 4.3 Mb adjacent to the centromere and in 7.3 Mb at the telomeric end. These regions are separated by a central contig of 23 Mb. We have concluded that the gaps contain sequences that are unclonable with the available host-vector systems, as we were unable to detect clones containing the sequences in these gaps by screening more than 20 genome equivalents of bacterial clones using sequences adjacent to the contig ends.
The size of the seven gaps in the telomeric region has been estimated by DNA fibre fluorescence in situ hybridization (FISH). No gap in this region is judged to be larger than ∼150 kb. For three of these gaps, a number of BAC and PAC clones that contain STSs on either side of the gap were shown to be deleted for at least a minimal core region by DNA fibre FISH. As these clones come from multiple donor DNA sources, these results are unlikely to be due to deletion in the DNA used to make the libraries. Furthermore, the same result was observed for the gap at 32,600 kb from the centromeric end of the sequence, when the DNA fibre FISH experiments were performed on DNA from two different lymphoblastoid cell lines. One possible explanation for this observation is that DNA fragments containing the gap sequences are initially cloned in the BAC library but clones that delete these sequences have a significant selective advantage as the library is propagated. As the observed size range of the cloned inserts in the BAC libraries ranges from 100 kb to more than 230 kb (http://bacpac.med.buffalo.edu/), such deletion events are not distinguishable on the basis of size from undeleted BACs. Additional analysis of the distribution of BAC end sequences from dbGSS (http://www.ncbi.nlm.nih.gov/dbGSS/index.html) suggests that the BAC coverage is sparser closer to the gaps and that this analysis did not identify any BACs spanning the gaps. The three remaining clone-map gaps in the proximal region of the long arm are in regions that may contain segments of previously characterized low-copy repeats21. These gaps could not be sized by DNA fibre FISH because of the extensive intra- and interchromosomal repeat sequences (see below) but were amenable to long-range restriction mapping. The gap between AP000529 and AP000530 was estimated to be shorter than 150 kb by comparison with a previously established long-range restriction map22. The gap closest to the centromere, which is less than 2 kb in size, could not be sequenced despite BAC clone coverage as it was unrepresented in plasmid or M13 libraries, and was intractable to all sequencing strategies applied. Detailed descriptions of several of the clone contigs have been published21,23,24 or will be published elsewhere.
Each sequencing group took responsibility for completion of adjacent areas of the sequence as illustrated in Figure 1 (PDF file: 752kb). A set of minimally overlapping clones (the ‘tile path’) was chosen from the physical map and sequenced using a combination of a random shotgun assembly, followed by directed sequencing to close gaps and resolve ambiguities (‘finishing’). The major problems encountered during completion of the sequence in the directed sequencing phase were CpG islands, tandem repeats and apparent cloning biases. Directed sequencing using oligonucleotide primers, very short insert plasmid libraries, or identification of bridging clones by screening high complexity plasmid or M13 libraries solved these problems.
The completed sequence covers 33.4 Mb of 22q with 11 gaps and has been estimated to be accurate to less than 1 error in 50,000 bases, by internal and external checking exercises25. The order and size of each of the contiguous pieces of sequence is detailed in Table 1. The largest contiguous segment stretches over 23 Mb. From our gap-size estimates, we calculate that we have completed 33,464 kb of a total region spanning 34,491 kb and that therefore the sequence is complete to 97% coverage of 22q. The complete sequence and analysis is available on the internet (http://www.sanger.ac.uk/HGP/Chr22 and http://www.genome.ou.edu/Chr22.html).
Table 1 Sequence contigs on chromosome 22
Sequence analysis and gene content
Analysis of the genomic sequence of the model organisms has made extensive use of predictive computational analysis to identify genes26,27,28. In human DNA, identification of genes by these methods is more difficult because of extensive splicing, lower density of exons and the high proportion of interspersed repetitive sequences. The accuracy of ab initio gene prediction on vertebrate genomic sequence has been difficult to determine because of the lack of sequence that has been completely annotated by experiment. To determine the degree of overprediction made by such algorithms, all genes within a region need to be experimentally identified and annotated, however it is virtually impossible to know when this job is complete. A 1.4-Mb region of human genomic sequence around the BRCA2 locus has been subjected to extensive experimental investigation, and it is believed that the 170 exons identified is close to the total number expressed in the region.
The most recent calibration of ab initio methods against this region (R.B.S.K. and T.H., manuscript in preparation) shows that with the best methods29,30 more than 30% of exon predictions do not overlap any experimental exons, in other words, they are overpredictions. Furthermore, having now applied this analysis to larger amounts of data (more than 15 Mb from the Sanger Annotated Genome Sequence Repository which can be obtained as part of the Genesafe collection (http://www.hgmp.mrc.ac.uk/Genesafe/)), it is confirmed that prediction accuracy also varies considerably between different regions of sequence. It was hoped that these calibration efforts would lead to rules for reliable gene prediction based on ab initio methods alone, perhaps on the basis of combining several different methods, GC content and so on. However, so far this has not been possible. The same analysis also shows that although ∼95% of genes are at least partially predicted by ab initio methods, few gene structures are completely correct (none in BRCA2) and more than 20% of experimental exons are not predicted at all. The comparison of ab initio predictions and the annotated gene structures (see below) in the chromosome 22 sequence is consistent with this, with 94% of annotated genes at least partially detected by a Genscan gene prediction, but only 20% of annotated genes having all exons predicted exactly. Sixteen per cent of all the exons in annotated genes were not predicted at all, although this is only 10% for internal exons (that is, not 5′ and 3′ ends). As a result, we do not consider that ab initio gene prediction software can currently be used directly to reliably annotate genes in human sequence, although it is useful when combined with other evidence (see below), for example, to define splice-site boundaries, and as a starting point for experimental studies.
Fortunately, a vast resource of experimental data on human genes in the form of complementary DNA and protein sequences and expressed sequence tags (ESTs) is available which can be used to identify genes within genomic DNA. Furthermore about 60% of human genes have distinctive CpG island sequences at their 5′ ends31 which can also be used to identify potential genes. Thus, the approach we have taken to annotating genes in the chromosome 22 sequence relies on a combination of similarity searches against all available DNA and protein databases, as well as a series of ab initio predictions. Upon completion of the sequence of each clone in the tile path, the sequence was subjected to extensive computational analysis using a suite of similarity searches and prediction tools. Briefly, the sequences were analysed for repetitive sequence content, and the repeats were masked using RepeatMasker (http://ftp . genome . washington . edu / RM / RepeatMasker . html). Masked sequence was compared to public domain DNA and protein databases by similarity searches using the blast family of programs32. Unmasked sequence was analysed for C + G content and used to predict the presence of CpG islands, tandem repeat sequences, tRNA genes and exons. The completed analysis was assembled into contigs and visualized using implementations of ACEDB (http://www.sanger.ac.uk/Software/Acedb/). In addition, the contiguous masked sequence was analysed using gene prediction software29,30.
Gene features were identified by a combination of human inspection and software procedures. Figure 1 shows the 679 gene sequences annotated across 22q. They were grouped according to the evidence that was used to identify them as follows: genes identical to known human gene or protein sequences, referred to as ‘known genes’ (247); genes homologous, or containing a region of similarity, to gene or protein sequences from human or other species, referred to as ‘related genes’ (150); sequences homologous to only ESTs, referred to as ‘predicted genes’ (148); and sequences homologous to a known gene or protein, but with a disrupted open reading frame, referred to ‘pseudogenes’ (134). (See Supplementary Information, Table 1, for details of these genes.) The ab initio gene prediction program, Genscan, predicted 817 genes (6,684 exons) in the contiguous sequence, of which 325 do not form part of the annotated genes categorized above. Given the calibration of ab initio prediction methods discussed above, we estimate that of the order of 100 of these will represent parts of ‘real’ genes for which there is currently no supporting evidence in any sequence database, and that the remainder are likely to be false positives.
The total length of the sequence occupied by the annotated genes, including their introns, is 13.0 Mb (39% of the total sequence). Of this, only 204 kb contain pseudogenes. About 3% of the total sequence is occupied by the exons of these annotated genes. This contrasts sharply with the 41.9% of the sequence that represents tandem and interspersed repeat sequences. There is no significant bias towards genes encoded on one strand at the 5% level (χ2 = 3.83).
A striking feature of the genes detected is their variety in terms of both identity and structure. There are several gene families that appear to have arisen by tandem duplication. The immunoglobulin λ locus is a well-known example, but there also are other immunoglobulin-related genes on the chromosome outside the immunoglobulin λ region. These include the three genes of the immunoglobulin λ-like (IGLL) family plus a fourth possible member of the family (AC007050.7). There are five clustered immunoglobulin κ variable region pseudogenes in AC006548, and an immunoglobulin variable-related sequence (VpreB3) in AP000348. Much further away from the λ genes is a variable region pseudogene, ∼123 kb telomeric of IGLL3 in sequence AL008721 (coordinates ∼9,420–9,530 kb from the centromeric end of the sequence), and a cluster of two λ constant region pseudogenes and a variable region pseudogene in sequences AL008723/AL021937 (coordinates ∼16,060–16,390 kb from the centromeric end).
Human chromosome 22 also contains other duplicated gene families that encode glutathione _S_-transferases, Ret-finger-like proteins19, phorbolins or APOBECs, apolipoproteins and β-crystallins. In addition, there are families of genes that are interspersed among other genes and distributed over large chromosomal regions. The γ-glutamyl transferase genes represent a family that appears to have been duplicated in tandem along with other gene families, for instance the BCR-like genes, that span the 22q11 region and together form the well-known LCR22 (low-copy repeat 22) repeats (see below).
The size of individual genes encoded on this chromosome varies over a wide range. The analysis is incomplete as not all 5′ ends have been defined. However, the smallest complete genes are only of the order of 1 kb in length (for example, HMG1L10 is 1.13 kb), whereas the largest single gene (LARGE15) stretches over 583 kb. The mean genomic size of the genes is 19.2 kb (median 3.7 kb). Some complete gene structures appear to contain only single exons, whereas the largest number of exons in a gene (PIK4CA) is 54. The mean exon number is 5.4 (median 3). The mean exon size is 266 bp (median 135 bp). The smallest complete exon we have identified is 8 bp in the PITPNB gene. The largest single exon is 7.6 kb in the PKDREJ, which is an intron-less gene with a 6.7-kb open reading frame. In addition, two genes occur within the introns of other expressed genes. The 61-kb TIMP3 gene, which is involved in Sorsby fundus macular degeneration, lies within a 268-kb intron of the large SYN3 gene, and the 8.5 kb HCF2 gene lies within a 27.5-b intron of the PIK4CA gene. In each case, the genes within genes are oriented in the opposite transcriptional orientation to the outer gene. We also observe pseudogenes frequently lying within the introns of other functional genes.
Peptide sequences for the 482 annotated full-length and partial genes with an open reading frame of greater than or equal to 50 amino acids were analysed against the protein family (PFAM)33, Prosite34 and SWISS-PROT35 databases. These data were processed and displayed in an implementation of ACEDB. Overall, 240 (50%) predicted proteins had matching domains in the PFAM database encompassing a total of 164 different PFAM domains. Of the residues making up these 482 proteins, 25% were part of a PFAM domain. This compares with PFAM's residue coverage of SWISS-PROT/TrEMBL, which is more than 45% and indicates that the human genome is enriched in new protein sequences. Sixty-two PFAM domains were found to match more than one protein, including ten predicted proteins containing the eukaryotic protein kinase domain (PF00069), nine matching the Src homology domain 3 (PF00018) and eight matching the RhoGAP domain (PF00620). Fourteen predicted proteins contain zinc-finger domains (See Supplementary Information, Table 2, for details of the PFAM domains identified in the predicted proteins).
Nineteen per cent of the coding sequences identified were designated as pseudogenes because they had significant similarity to known genes or proteins but had disrupted protein coding reading frames. Because 82% of the pseudogenes contained single blocks of homology and lacked the characteristic intron–exon structure of the putative parent gene, they probably are processed pseudogenes. Of the remaining spliced pseudogenes, most represent segments of duplicated gene families such as the immunoglobulin κ variable genes, the β-crystallins, CYP2D7 and CYP2D8, and the GGT and BCR genes. The pseudogenes are distributed over the entire sequence, interspersed with and sometimes occurring within the introns of annotated expressed genes. However, there also is a dense cluster of 26 pseudogenes in the 1.5-Mb region immediately adjacent to the centromere; the significance of this cluster is currently unclear.
Given that the sequence of 33.4 Mb of chromosome 22q represents 1.1% of the genome and encodes 679 genes, then, if the distribution of genes on the other chromosomes is similar, the minimum number of genes in the entire human genome would be at least 61,000. Previous work has suggested that chromosome 22 is gene rich6 by a factor of 1.38 (http://www.ncbi.nlm.nih.gov/genemap/page.cgi?F=GeneDistrib.html), which would reduce this estimate to ∼45,000 genes. It is important, however, to recognize that the analysis described here only provides a minimum estimate for the gene content of chromosome 22q, and that further studies will probably reveal additional coding sequences that could not be identified with the current approaches.
Two lines of evidence point to the existence of additional genes that are not detected in this analysis. First, the 553 predicted CpG islands, which typically lie at the true 5′ ends of about 60% of human genes31, are in excess of 60% of the number of genes identified (60% = 327, excluding pseudogenes); 282 of the genes identified have CpG islands at or close to the 5′ end (within 5-kb upstream of the first exon, or 1-kb downstream). Thus, there could be up to 271 additional genes associated with CpG islands undetected in the sequence. Second, there are 325 putative genes predicted by the ab initio gene prediction program, Genscan, that are not in regions already containing annotated transcripts. We estimate (see above) that roughly 100 of these will represent parts of real genes. Identifying additional genes will require further computational and experimental studies. These studies are continuing and entail testing candidate sequences for possible messenger RNA expression, implementing new gene prediction software able to detect the regions around or near CpG islands that currently have no identified transcript, and further analysis of sequences that are conserved between human and mouse. Furthermore, full-length cDNA sequences that accumulate in the sequence databases of human and other species will be used to refine the gene structures.
The long-range chromosome landscape
Critical to the utility of the genomic sequence to genetic studies is the integration of established genetic maps. The positions of the commonly used microsatellite markers from the Genethon genetic map36 are given in Figure 1. The correlation of the order of markers between the genetic map and the sequence is good, within the limitations of genetic mapping. Only a single marker (D22S1175) is discrepant between the two data sets, and this lies in a sequence that is repeated twice on the chromosome (AL021937, see below). In the telomeric region, four of the Genethon markers must lie in our sequence gaps, and we were unable to identify clones from all libraries tested for these. Comparison of genetic distance against physical distance for all the microsatellites whose order is maintained between the datasets shows a mean value of 1.87 cM Mb-1. However, the relationship between genetic and physical distance across the chromosome partitions into two types of region, areas of high and low recombination (Fig. 2). The areas of high recombination may represent recombinational hot spots, although we have not yet been able to identify any specific sequence characteristics common to these areas.
Figure 2: The relationship between physical and genetic distance.
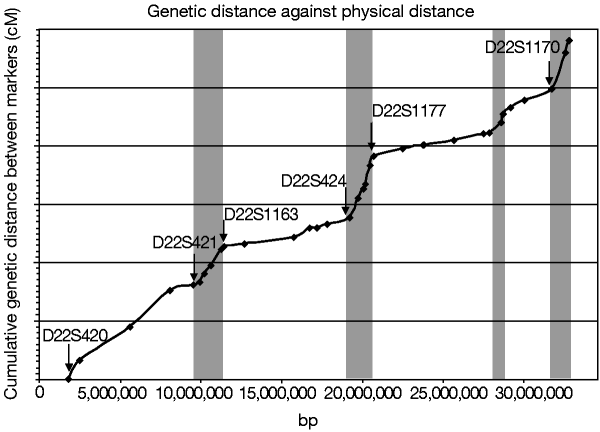
The sex-averaged genetic distances of Dib et al.36 were obtained from ftp://ftp.genethon.fr/pub/Gmap/Nature-1995/ and the cumulative intermarker distances for unambiguously ordered markers (in cM) were plotted against the positions of the microsatellite markers in the genomic sequence. It should be stressed that the y axis does not represent the true genetic distance between distant markers but the sum of the local intermarker distances. The positions of selected genetic markers are labelled. Grey regions are indicative of areas of relatively increased recombination per unit physical distance.
The mean G + C content of the sequence is 47.8%. This is significantly higher than the G + C content calculated for the sum of all human genomic sequence determined so far (42%). Although this result was expected from previous indirect measurements of the G + C content of chromosome 227,8,37, the distribution is not uniform, but regionally segmented as illustrated in Figure 1. There are clear fluctuations in the base content, resulting in areas that are relatively G + C rich and others that are relatively G + C poor. On chromosome 22 these regions stretch over several megabases. For example, the 2 Mb of sequence closest to the centromeric end of the sequence is relatively G + C poor, with the G + C content dropping below 40%. Similarly, the area between 16,000 and 18,800 kb from the centromeric end of the sequence is consistently below 45% G + C. The G + C rich regions often reach more than 55% G + C (for example, at 20,100–23,400 kb from the centromeric end of the sequence). This fluctuation appears to be consistent with previous observations that vertebrate genomes are segmented into ‘isochores’ of distinct G + C content38 and is similar to the structure seen in the human major histocompatibility complex (MHC) sequence39. Isochores correlate with both genes and chromosome structure. The G + C rich isochores are rich in genes and Alu repeats, and are located in the G + C rich chromosomal R-bands, whereas the G + C poor isochores are relatively depleted in genes and Alu repeats, and are located in the G-bands8,37,40. The G + C poor regions of chromosome 22 are depleted in genes and relatively poor in Alu sequences. For example, the region between 16,000 and 18,800 kb from the centromeric end contains just three genes, two of which are greater than 400 kb in length. The G + C poor regions also are depleted in CpG islands, which are clustered in the gene-rich, G + C rich regions. Although it is tempting to correlate the sequence features that we see with the chromosome banding patterns, we believe that high-resolution mapping of the chromosome band boundaries will be required to assign definitively these to genomic sequence.
Over 41.9% of the chromosome 22 sequence comprises interspersed and tandem repeat family sequences (Table 2). The density of repeats across the sequence is plotted in Fig. 1 (PDF file: 752kb). There is variation in the density of Alu repeats and some of the regions with low Alu density correlate with the G + C poor regions, for example, in the region 16,000–18,800 kb from the centromeric end, and these data support the relationship of isochores with Alu distribution. However, in other areas the relationship is less clear. We provide a World-Wide Web interface to the long-range analyses presented here and to further analysis of the many other repeat types and features of the sequence at http://www.sanger.ac.uk/cgi-bin/cwa/22cwa.pl. The 1-Mb region closest to the centromere contains several interesting repeat sequence features that may be typical of other pericentromeric regions. In addition to the density of pseudogenes described above, there is a large 120-kb block of tandemly repeated satellite sequence (D22Z3) centred 500 kb from the centromeric sequence start (not shown in Fig. 1 (PDF file: 752kb), but evident from the absence of Alu and LINE1 sequences at this point). There is also a cluster of satellite II repeats 80-kb telomeric of the D22Z3 sequences. Isolated alphoid satellite repeats are found closer to the centromeric end of the sequence. Furthermore, this pericentromeric 1 Mb closest to the centromere contains many sequences that are shared with a number of different chromosomes, particularly chromosomes 2 and 14. During map construction, 33 out of 37 STSs designed from sequence that was free of high-copy repeats amplified from more than one chromosome in somatic cell hybrid panel analysis.
Table 2 The interspersed repeat content of human chromosome 22
Low-copy repeats on chromosome 22
To detect intra- and interchromosomal repeats, we compared the entire sequence of chromosome 22 to itself, and also to all other existing human genomic DNA sequence using Blastn32 after masking high and medium frequency repeats. The results of the intrachromosomal sequence analysis were plotted as a dot matrix (Fig. 3) and reveal a series of interesting features. Locally duplicated gene families lie close to the diagonal axis of the plot. The most striking is the immunoglobulin λ locus that comprises a cluster of 36 potentially functional V-λ gene segments, 56 V-λ pseudogenes, and 27 partial V-λ pseudogenes (‘relics’), together with 7 each of the J and C λ segments24. Other duplicated gene families that are visible from the dot matrix plot include the clustered genes for glutathione _S_-transferases, β-crystallins, apolipoproteins, phorbolins or APOBECs, the lectins LGALS1 and LGALS2 and the CYP2Ds. A partial inverted duplication of CSF2RB is also observed.
Figure 3: Intrachromosomal repeats on human chromosome 22.
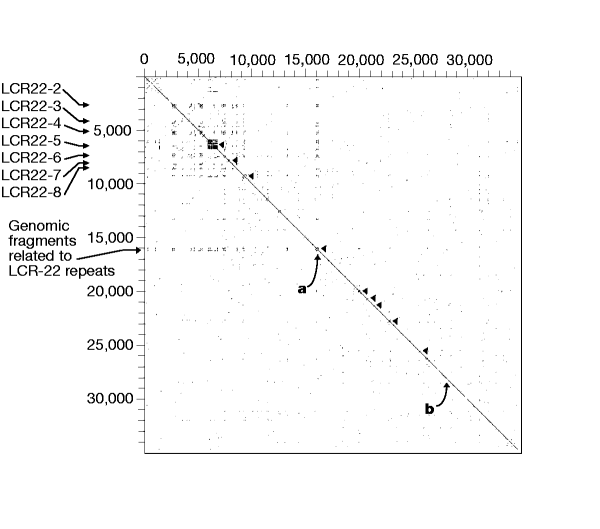
High- and medium-copy repeats and low complexity sequence were masked using RepeatMasker and Dust, and masked sequences were compared using Blastn. The results were filtered to identify regions of more than 50% identity to the query sequence, and were plotted in a 2D matrix with a line proportional to the size of the region of identity. Localized gene family repeats are indicated by arrowheads along the diagonal. From the top, these are the immunoglobulin λ locus, the glutathione _S_-transferase genes, the β-crystallin genes, the Ret-finger-protein-like genes, the apolipoprotein genes, the colony-stimulating factor receptor (CSF2RB) inverted partial duplication, the lectins LGALS1 and LGALS2, the APOBEC genes and the CYP2D genes. Two 60-kb regions of more than 90% homology are labelled ‘a’ (AL008723/AL021937) and ‘b’ (AL031595/AL022339). Seven low-copy repeat regions (LCR22) and a region containing related genomic fragments are indicated at the left margin.
Much more striking are the long-range duplications, which are visible away from the diagonal axis. For example, a 60-kb segment of more than 90% similarity is seen between sequences AL008723/AL021937 (at ∼16,060–16,390 kb from the centromeric end) and AL031595/AL022339 (at ∼27,970–28,110 kb from the centromeric end) separated by almost 12 Mb. The 22q11 region is particularly rich in repeated clusters41. Previous work described a low-copy repeat family in 22q11 that might mediate recombination events leading to the chromosomal rearrangements seen in cat eye, velocardiofacial and DiGeorge syndromes21,42. The availability of the entire DNA sequence allows detailed dissection of the molecular structure of these low-copy repeats (LCR22s). Edelmann et al. described eight LCR22 regions21,42. We were unable to find the LCR22 repeat closest to the centromere, but it may lie in the gap at 700 kb from the centromeric end of the sequence. The other LCR22 regions are distributed over 6.5 Mb of 22q11. Analysis of the sequence shows that each LCR22 contains a set of genes or pseudogenes (Fig. 4). For example, five of the LCR22s contain copies of the γ-glutamyl transferase genes and γ-glutamy-transferase-related genes. There is also evidence that a more distant sequence at ∼16,000 kb from the centromeric start of the genomic sequence shares certain sequences with the LCR22 repeats. This similarity involves related genomic fragments including parts of the Ret-finger-protein-like genes, and the IGLC and IGLV genes.
Figure 4: Sequence composition of the LCR22 repeats.
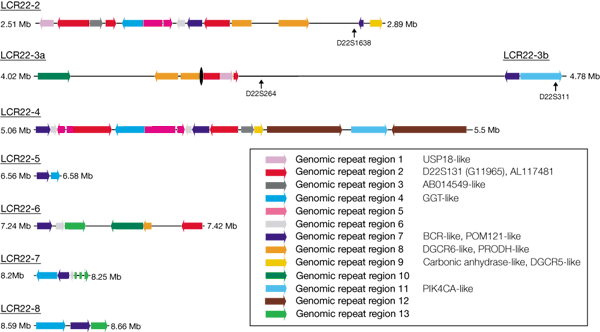
Illustration of the sequence composition of seven LCR22 repeats. The span of each LCR22 region is shown in megabases from the centromere. Coloured arrows indicate the extent of one of the thirteen genomic repeat regions and the orientation of the repeat. The known gene and marker content of these genomic repeat regions is indicated in the key. The black oval indicates the position of the gap in the sequence in LCR22-3.
Regions of conserved synteny with the mouse
The genomic organization of different mammalian species is well known to be conserved43. Comparison of genetic and physical maps across species can aid in predicting gene locations in other species, identifying candidate disease genes13, and revealing various other features relevant to the study of genome organization and evolution. For all the cross-species relationships, that between man and mouse has been most studied. We have examined the relationship of the human chromosome 22 genes to their mouse orthologues.
Of the 160 genes we identified in the human chromosome 22 sequence that have orthologues in mouse, 113 of the murine orthologues have known mouse chromosomal locations (data available at http://www.sanger.ac.uk/HGP/Chr22/Mouse/). Examination of these mouse chromosomal locations mapped onto the human chromosome 22 sequence confirms the conserved linkage groups corresponding to human chromosome 22 on mouse chromosomes 6, 16, 10, 5, 11, 8 and 1518,44,45,46 (Fig. 5). Furthermore, these studies allow placement of the sites of evolutionary rearrangements that have disrupted the conservation of synteny more accurately at the DNA sequence scale. For example, the breakdown of synteny between the mouse 8C1 block and the mouse 15E block occurs between the equivalents of the human HMOX1 and MB genes, which are separated by less than 160 kb that also contains a conspicuous 41-copy 18-nucleotide tandem repeat. A clear prediction from these data is that, for the most part, the unmapped murine orthologues of the human genes lie within these established linkage groups, along with the orthologues of the human genes that currently lack mouse counterparts. Exploitation of the chromosome 22 sequence may hasten the determination of the mouse genomic sequence in these regions.
Figure 5: Regions of conserved synteny between human chromosome 22 and the mouse genome.
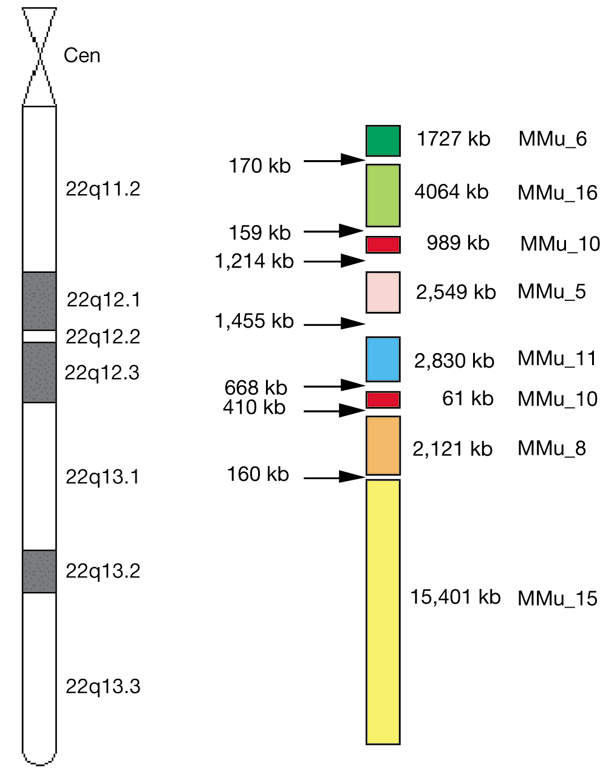
Regions of mouse chromosomes with conserved synteny to human chromosome 22 are shown as adjacent coloured blocks, determined by the mouse map position of mouse orthologues to human chromosome 22 genes. The size of human chromosome 22 corresponding to each mouse chromosomal region is indicated in kb, as well as the size of the gap between the last orthologue in each conserved block. These data are available at http://www.sanger.ac.uk/Chr22/Mouse.
Conclusions
We have shown that the clone by clone strategy is capable of generating long-range continuity sufficient to establish the operationally complete genomic sequence of a chromosome. In doing so, we have generated the largest contiguous segment of DNA sequence to our knowledge to date. The analysis of the sequence gives a foretaste of the information that will be revealed from the remaining chromosomes.
We were unable to obtain sequence over 11 small gaps using the available cloning systems. It may be possible that additional approaches such as using combinations of cloning systems with small insert sizes and low-copy number could reduce the size of these gaps. Direct cloning of restriction fragments that cross these gaps into small insert plasmid or M13 libraries, or direct sequencing approaches might eventually provide access to all the sequence in the gaps. However, closing these gaps is certain to require considerable time and effort, and might be considered as a specialist activity outside the core genome-sequencing efforts. It also is probable that the sequence features responsible for several of these gaps are unlikely to be specific to chromosome 22. In the best case, similar unclonable sequences might be restricted to the centromeric and telomeric regions of the other chromosomes and areas with large tandem repeats, and it will be possible to obtain large contiguous segments for the bulk of the euchromatic genome.
Over the course of the project, the emerging sequence of chromosome 22 has been made available in advance of its final completion through the internet sites of the consortium groups and the public sequence databases47. The benefits of this policy can be seen in both the regular requests received from investigators for materials and information that arise as the result of sequence homology searches, and the publications that have used the data14,15,16,17,18,19. The genome project will continue to pursue this data release policy as we move closer to the anticipated completed sequence of humans, mice and other complex genomes47,48.
Methods
The methods for construction of clone maps have been previously described24,49,50 and can also be found at http://www.sanger.ac.uk/HGP/methods/. Details of sequencing methods and software are available at http://www.sanger.ac.uk/HGP/methods/, http://www.genome.ou.edu/proto.html, http://www-alis.tokyo.jst.go.jp/HGS/team\_KU/team.html and in the literature1,24.
Figure 1: The sequence of human chromosome 22.
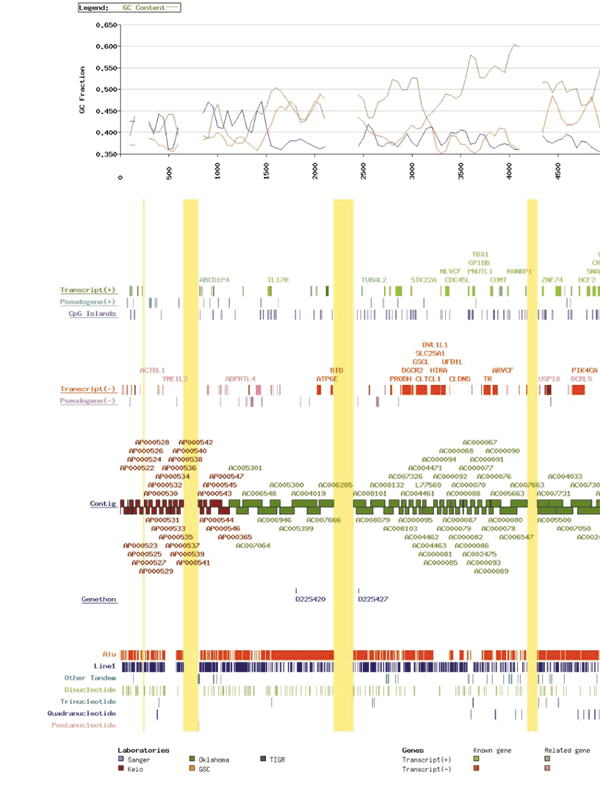
Coloured boxes depict the annotated features of the sequence of human chromosome 22, with the centromere to the left and the telomere to the right. Coordinates are in kilobases. Vertical yellow blocks indicate the positions of the gaps in the sequence and are proportional in size to the estimated size of each gap. From bottom to top the following features are displayed: positions of interspersed repetitive sequences including tandem repeats categorized by nucleotide repeat unit length (at this resolutrion Alu repeats are not visibly separated in some regions); the positions of the microsatellite markers in the genetic map of Dib et al.36; the tiling path of genomic clones used to determine the sequence labelled by their Genbank/EMBL/DDBJ accession number and coloured according to the source of the sequence; and the annotated gene, pseudogene and CpG island content of the sequence. Transcripts and pseudogenes oriented 5′ to 3′ on the DNA strand from centromere to telomere are designated ‘+’, those on the opposite strand ‘-’. In the transcript rows, the annotated genes are subdivided by colour according to the criteria in the text. Annotated genes with approved gene symbols from the HUGO nomenclature committee are labelled. For details of all the genes with their positions in the reference sequence, see Supplementary Information, Table 1. In the case of the immunoglobulin variable region, the entire locus has been drawn as a single block; in reality, this is a complex of variable chain genes (see ref. 27). At the top is a graphical plot of the repeat density for the common interspersed repeats Alu and Line1, and the C + G base frequency across the sequence. Each is calculated as a percentage of the sequence using a sliding 100-kb window moved in 50-kb iterations. Since the production of Fig. 1 (PDF file: 752kb), six accession codes have been updated. The new codes are AL050347 (for Z73987), AL096754 (for Z68686), AL049749 (for Z82197), Z75892 (for Z75891), AL078611 (for Z79997) and AL023733 (for AL023593).
References
- The Sanger Centre & The Genome Sequencing Centre. Toward a complete human genome sequence. Genome Res. 8, 1097–1108 (1998).
Article Google Scholar - Weber,J. L. & Myers,E. W. Human whole-genome shotgun sequencing. Genome Res. 7, 401–409 (1997).
Article CAS Google Scholar - Green,P. Against a whole-genome shotgun. Genome Res. 7, 410 –417 (1997).
Article CAS Google Scholar - Collins,F. S. et al. New goals for the U.S. human genome project: 1998–2003. Science 282, 682–689 (1998).
Article ADS CAS Google Scholar - Morton,N. E. Parameters of the human genome. Proc. Natl Acad. Sci. USA 88, 7474–7476 (1991).
Article ADS CAS Google Scholar - Deloukas,P. et al. A physical map of 30,000 human genes. Science 282, 744–746 (1998).
Article ADS CAS Google Scholar - Craig,J. M. & Bickmore,W. A. The distribution of CpG islands in mammalian chromosomes. Nature Genet. 7, 376–382 (1994).
Article CAS Google Scholar - Saccone,S., Caccio,S., Kusuda,J., Andreozzi,L. & Bernardi, G. Identification of the gene-richest bands in human chromosomes. Gene 174, 85–94 (1996).
Article CAS Google Scholar - Collins,J. E. et al. A high-density YAC contig map of human chromosome 22. Nature 377, 367–379 ( 1995).
CAS PubMed Google Scholar - Pulver,A. E. et al. Psychotic illness in patients diagnosed with velo-cardio-facial syndrome and their relatives. J. Nerv. Ment. Dis. 182 , 476–478 (1994).
Article CAS Google Scholar - Gill,M. et al. A combined analysis of D22S278 marker alleles in affected sib-pairs: support for a susceptibility locus for schizophrenia at chromosome 22q12. Schizophrenia Collaborative Linkage Group (Chromosome 22). Am. J. Med. Genet. 67, 40–45 (1996).
Article CAS Google Scholar - Zu,L., Figueroa,K. P., Grewal,R. & Pulst,S. M. Mapping of a new autosomal dominant spinocerebellar ataxia to chromosome 22. Am. J. Hum. Genet. 64, 594– 599 (1999).
Article CAS Google Scholar - Southard-Smith,E. M. et al. Comparative analyses of the dominant megacolon-SOX10 genomic interval in mouse and human. Mamm. Genome 10, 744–749 (1999).
Article CAS Google Scholar - Nishino,I., Spinazzola,A. & Hirano, M. Thymidine phosphorylase gene mutations in MNGIE, a human mitochondrial disorder. Science 283, 689 –692 (1999).
Article ADS CAS Google Scholar - Peyrard,M. et al. The human LARGE gene from 22q12.3-q13.1 is a new, distinct member of the glycosyltransferase gene family. Proc. natl Acad. Sci. USA 96, 598–603 ( 1999).
Article ADS CAS Google Scholar - Kao,H. T. et al. A third member of the synapsin gene family. Proc. Natl Acad. Sci. USA 95, 4667–4672 (1998).
Article ADS CAS Google Scholar - Mittman,S., Guo,J., Emerick,M. C. & Agnew,W. S. Structure and alternative splicing of the gene encoding alpha1I, a human brain T calcium channel alpha1 subunit. Neurosci. Lett. 269, 121–124 (1999).
Article CAS Google Scholar - Seroussi,E. et al. TOM1 genes map to human chromosome 22q13.1 and mouse chromosome 8C1 and encode proteins similar to the endosomal proteins HGS and STAM. Genomics 57, 380–388 (1999).
Article CAS Google Scholar - Seroussi,E. et al. Duplications on human chromosome 22 reveal a novel ret finger protein-like gene family with sense and endogenous antisense transcripts. Genome Res. 9, 803–814 (1999).
Article CAS Google Scholar - Ning,Y., Rosenberg,M., Biesecker,L. G. & Ledbetter,D. H. Isolation of the human chromosome 22q telomere and its application to detection of cryptic chromosomal abnormalities. Hum. Genet. 97 , 765–769 (1996).
Article CAS Google Scholar - Edelmann,L., Pandita,R. K. & Morrow, B. E. Low-copy repeats mediate the common 3-Mb deletion in patients with velo-cardio-facial syndrome. Am. J. Hum. Genet. 64, 1076–1086 ( 1999).
Article CAS Google Scholar - McDermid,H. E. et al. Long-range mapping and construction of a YAC contig within the cat eye syndrome critical region. Genome Res. 6 , 1149–1159 (1996).
Article CAS Google Scholar - Johnson,A. et al. A 1.5-Mb contig within the cat eye syndrome critical region at human chromosome 22q11.2. Genomics 57, 306–309 (1999).
Article CAS Google Scholar - Kawasaki,K. et al. One-megabase sequence analysis of the human immunoglobulin lambda gene locus. Genome Res. 7, 250– 261 (1997).
Article CAS Google Scholar - Felsenfeld,A., Peterson,J., Schloss,J. & Guyer,M. Assessing the quality of the DNA sequence from the Human Genome Project. Genome Res. 9, 1–4 (1999 ).
CAS PubMed Google Scholar - Mewes,H. W. et al. Overview of the yeast genome. Nature 387, 7–65 (1997).
Article Google Scholar - Blattner,F. R. et al. The complete genome sequence of Escherichia coli K-12. Science 277, 1453–1474 ( 1997).
Article CAS Google Scholar - The C. elegans Sequencing Consortium. Genome sequence of the nematode C. elegans: a platform for investigating biology. Science 282, 2012–2018 (1998).
Article ADS Google Scholar - Solovyev,V. & Salamov,A. The Gene-Finder computer tools for analysis of human and model organisms genome sequences. Ismb 5, 294–302 (1997).
CAS PubMed Google Scholar - Burge,C. & Karlin,S. Prediction of complete gene structures in human genomic DNA. J. Mol. Biol. 268, 78–94 (1997).
Article CAS Google Scholar - Cross,S. H. & Bird,A. P. CpG islands and genes. Curr. Opin. Genet. Dev. 5, 309–314 (1995).
Article CAS Google Scholar - Altschul,S. F., Gish,W., Miller,W., Myers,E. W. & Lipman, D. J. Basic local alignment search tool. J. Mol. Biol. 215, 403–410 (1990).
Article CAS Google Scholar - Bateman,A. et al. Pfam 3.1: 1313 multiple alignments and profile HMMs match the majority of proteins. Nucleic Acids Res. 27, 260–262 (1999).
Article CAS Google Scholar - Hofmann,K., Bucher,P., Falquet,L. & Bairoch,A. The PROSITE database, its status in 1999. Nucleic Acis Res. 27, 215–219 (1999).
Article CAS Google Scholar - Bairoch,A. & Apweiler,R. The SWISS-PROT protein sequence data bank and its supplement TrEMBL in 1999. Nucleic Acids Res. 27, 49–54 ( 1999).
Article CAS Google Scholar - Dib,C. et al. A comprehensive genetic map of the human genome based on 5,264 microsatellites. Nature 380, 152– 154 (1996).
Article ADS CAS Google Scholar - Holmquist,G. P. Chromosome bands, their chromatin flavors, and their functional features. Am. J. Hum. Genet. 51, 17– 37 (1992).
CAS PubMed PubMed Central Google Scholar - Bernardi,G. et al. The mosaic genome of warm-blooded vertebrates. Science 228, 953–958 ( 1985).
Article ADS CAS Google Scholar - The MHC sequencing consortium. Complete sequence and gene map of a human major histocompatibility complex. Nature 401, 921–923 (1999).
Article ADS Google Scholar - Bernardi,G. The isochore organization of the human genome. Annu. Rev. Genet. 23, 637–661 ( 1989).
Article CAS Google Scholar - Collins,J. E., Mungall,A. J., Badcock,K. L., Fay,J. M. & Dunham,I. The organization of the gamma-glutamyl transferase genes and other low copy repeats in human chromosome 22q11. Genome Res. 7, 522–531 ( 1997).
Article CAS Google Scholar - Edelman,L. et al. A common molecular basis for rearrangement disorders on chromosome 22q11. Hum. Mol. Genet. 8, 1157– 1167 (1999).
Article Google Scholar - Eppig,J. T. & Nadeau,J. H. Comparative maps: the mammalian jigsaw puzzle. Curr. Opin. Genet. Dev. 5, 709–716 (1995).
Article CAS Google Scholar - Bucan,M. et al. Comparative mapping of 9 human chromosome 22q loci in the laboratory mouse. Hum. Mol. Genet. 2, 1245– 1252 (1993).
Article CAS Google Scholar - Carver,E. A. & Stubbs,L. Zooming in on the human-mouse comparative map: genome conservation re-examined on a high-resolution scale. Genome Res. 7, 1123–1137 (1997).
Article CAS Google Scholar - Puech,A. et al. Comparative mapping of the human 22q11 chromosomal region and the orthologous region in mice reveals complex changes in gene organization. Proc. Natl Acad. Sci. USA 94, 14608– 14613 (1997).
Article ADS CAS Google Scholar - Bentley,D. R. Genomic sequence information should be released immediately and freely in the public domain. Science 274, 533– 534 (1996).
Article ADS CAS Google Scholar - Guyer,M. Statement on the rapid release of genomic DNA sequence. Genome Res. 8, 413 (1998).
Article CAS Google Scholar - Dunham,I., Dewar,K., Kim,U.-J. & Ross,M. T. in Genome Analysis: A Laboratory Manual Series, Volume 3: Cloning Systems (eds Birren, B. et al.) 1–86 (Cold Spring Harbor Laboratory Press, Cold Spring Harbor, New York, 1999).
Google Scholar - Asakawa,S. et al. Human BAC library: construction and rapid screening. Gene 191, 69–79 ( 1997).
Article CAS Google Scholar
Acknowledgements
We thank S. Povey, J. White and H. Wain for the help with gene nomenclature, and M. Adams for making available the sequence trace files of U62317. We thank M. Elharam, H. Jia, L. Lane, R. Morales-Diaz, F. Najar, P. Pham, R. Rahhal, M. Rao, Y. Tilahun, R. Wayt, H. Wright, E. Nakato, J. L. Schmeits, K. Schooler, J. Wang, M. Asahina, M. Takahashi, H. Harigai, Y. G. Xie, F. Y. Han, S. Swahn, B. Funke, R. K. Pandita, C. Chieffo, D. Michaud and all members of the Sanger Centre past and present for their assistance. This work was supported by grants from the Wellcome Trust, the NIH National Human Genome Research Institute to B.A.R. and to B.S.E., the NSF to B.A.R., the University of Oklahoma, the Fund for the Human Genome Sequencing Project of Japan Science and Technology Corporation, the Fund for the ‘Research for the Future’ Program from the Japan Society for the Promotion of Science, the UK Medical Research Council, the Medical Research Council of Canada to H.E.M., the Swedish Cancer Foundation, the Swedish Medical Research Council, and a Senior/Established Investigator Award from the Swedish Cancer Foundation to J.P.D.
Author information
Author notes
- A. M. Bridgeman
Present address: Division of Virology, Department of Pathology, Tennis Court Road, Cambridge, CB12 1QP, UK - D. Buck, R. E. Connor & S. Ho
Present address: Lark Technologies, Radwinter Road, Saffron Walden Essex, CB11 3HY, UK - J. Burgess & J. Davis
Present address: Australian Genome Research Facility, Gehrmann Laboratories, University of Queensland, St Lucia, QLD 4072, Australia - D. Conroy
Present address: Incyte Europe Ltd, 214 Cambridge Science Park, Cambridge, CB4 0WA, UK - S. J. Dodsworth
Present address: Tepnel Life Sciences, Innovation Centre, Scotscroft Building, Wilmslow Road, Didsbury, Manchester, M20 8RY, UK - C. Hall
Present address: Department of Brassica & Oilseeds Research, Cambridge Laboratory, John Innes Centre, Norwich, Norfolk, UK - R. W. Heathcott
Present address: Cancer Genetics Lab, Department of Biochemistry, University of Otago, P.O. Box 56, Dunedin, New Zealand - M. N. Whiteley
Present address: Dept. of Zoology, University of Cambridge, Downing Street, CB2 3EJ, UK - M. Vaudin
Present address: Monsanto, Genomic Team, Mail Zone N3SA, 800 North Lindbergh Boulevard, St. Louis, Missouri, 63167, USA - H. Williamson
Present address: Wellcome Trust Centre for Human Genetics, Roosevelt Drive, Oxford, OX3 7BN, UK
Authors and Affiliations
- The Sanger Centre, Wellcome Trust Genome Campus, Hinxton , Cambridge, CB10 1SA, UK
I. Dunham, A. R. Hunt, J. E. Collins, R. Bruskiewich, D. M. Beare, M. Clamp, L. J. Smink, R. Ainscough, J. P. Almeida, A. Babbage, C. Bagguley, J. Bailey, K. Barlow, K. N. Bates, O. Beasley, C. P. Bird, S. Blakey, W. D. Burrill, J. Burton, C. Carder, N. P. Carter, Y. Chen, G. Clark, S. M. Clegg, V. Cobley, C. G. Cole, R. E. Collier, N. Corby, G. J. Coville, A. V. Cox, E. Dawson, P. D. Dhami, C. Dockree, R. M. Durbin, A. Ellington, K. L. Evans, J. M. Fey, K. Fleming, L. French, A. A. Garner, J. G. R. Gilbert, D. Grafham, M. N. Griffiths, R. Hall, G. Hall-Tamlyn, S. Holmes, S. E. Hunt, M. C. Jones, J. Kershaw, A. Kimberley, A. King, G. K. Laird, C. F. Langford, M. A. Leversha, C. Lloyd, D. M. Lloyd, I. D. Martyn, M. Mashreghi-Mohammadi, L. Matthews, O. T. McCann, J. McClay, S. McLaren, A. A. McMurray, S. A. Milne, B. J. Mortimore, C. N. Odell, R. Pavitt, A. V. Pearce, D. Pearson, B. J. Phillimore, S. H. Phillips, R. W. Plumb, H. Ramsay, Y. Ramsey, L. Rogers, M. T. Ross, C. E. Scott, H. K. Sehra, C. D. Skuce, S. Smalley, M. L. Smith, C. Soderlund, L. Spragon, C. A. Steward, J. E. Sulston, R. M. Swann, M. Wall, J. M. Wallis, D. Willey, L. Williams, S. Williams, T. E. Wilmer, L. Wilming, C. L. Wright, T. Hubbard, D. R. Bentley, S. Beck, J. Rogers & Y. Fu - Department of Molecular Biology, Keio University School of Medicine, 35 Shinanomachi, Shinjuku-ku, Tokyo , 160-8582, Japan
N. Shimizu, S. Minoshima, K. Kawasaki, T. Sasaki, S. Asakawa, J. Kudoh, A. Shintani, K. Shibuya, Y. Yoshizaki, N. Aoki & S. Mitsuyama - Department of Chemistry and Biochemistry, The University of Oklahoma, 620 Parrington Oval, Room 311, Norman , 73019, Oklahoma, USA
B. A. Roe, F. Chen, L. Chu, J. Crabtree, S. Deschamps, A. Do, T. Do, A. Dorman, F. Fang, P. Hu, A. Hua, S. Kenton, H. Lai, H. I. Lao, J. Lewis, S. Lewis, S.-P. Lin, P. Loh, E. Malaj, T. Nguyen, H. Pan, S. Phan, S. Qi, Y. Qian, L. Ray, Q. Ren, S. Shaull, D. Sloan, L. Song, Q. Wang, Y. Wang, Z. Wang, J. White, D. Willingham, H. Wu, Z. Yao, M. Zhan & G. Zhang - Genome Sequencing Center, Washington University School of Medicine, 4444 Forest Park Blvd, St. Louis , 63108, Missouri, USA
S. Chissoe, J. Murray, N. Miller, P. Minx, R. Fulton, D. Johnson, G. Bemis, D. Bentley, H. Bradshaw, S. Bourne, M. Cordes, Z. Du, L. Fulton, D. Goela, T. Graves, J. Hawkins, K. Hinds, K. Kemp, P. Latreille, D. Layman, P. Ozersky, T. Rohlfing, P. Scheet, C. Walker, A. Wamsley, P. Wohldmann, K. Pepin, J. Nelson, I. Korf, J. A. Bedell, L. Hillier, E. Mardis, R. Waterston & R. Wilson - Division of Human Genetics and Molecular Biology, The Children's Hospital of Philadelphia and the Department of Pediatrics, University of Pennsylvania School of Medicine, Philadelphia, 19104, Pennsylvania , USA
B. S. Emanuel, T. Shaikh, H. Kurahashi & S. Saitta - Department of Biological Sciences, University of Alberta, Edmonton, T6G 2E9, Alberta, Canada
M. L. Budarf, H. E. McDermid, A. Johnson & A. C. C. Wong - Department of Molecular Genetics, Albert Einstein College of Medicine, 1300 Morris Park Avenue, Bronx, 10461, New York, USA
B. E. Morrow & L. Edelmann - Division of Biology, California Institute of Technology, Pasadena, 91125, California, USA
U. J. Kim, H. Shizuya & M. I. Simon - Department of Molecular Medicine, Clinical Genetics Unit, Karolinska Hospital, CMM bldg. L8:00, Stockholm , 17176, Sweden
A. M. Bridgeman, D. Buck, J. Burgess, R. E. Connor, D. Conroy, J. Davis, S. J. Dodsworth, C. Hall, R. W. Heathcott, S. Ho, M. Vaudin, M. N. Whiteley, H. Williamson, J. P. Dumanski, M. Peyrard, D. Kedra, E. Seroussi, I. Fransson, I. Tapia, C. E. Bruder & K. P. O'Brien
Authors
- I. Dunham
You can also search for this author inPubMed Google Scholar - A. R. Hunt
You can also search for this author inPubMed Google Scholar - J. E. Collins
You can also search for this author inPubMed Google Scholar - R. Bruskiewich
You can also search for this author inPubMed Google Scholar - D. M. Beare
You can also search for this author inPubMed Google Scholar - M. Clamp
You can also search for this author inPubMed Google Scholar - L. J. Smink
You can also search for this author inPubMed Google Scholar - R. Ainscough
You can also search for this author inPubMed Google Scholar - J. P. Almeida
You can also search for this author inPubMed Google Scholar - A. Babbage
You can also search for this author inPubMed Google Scholar - C. Bagguley
You can also search for this author inPubMed Google Scholar - J. Bailey
You can also search for this author inPubMed Google Scholar - K. Barlow
You can also search for this author inPubMed Google Scholar - K. N. Bates
You can also search for this author inPubMed Google Scholar - O. Beasley
You can also search for this author inPubMed Google Scholar - C. P. Bird
You can also search for this author inPubMed Google Scholar - S. Blakey
You can also search for this author inPubMed Google Scholar - A. M. Bridgeman
You can also search for this author inPubMed Google Scholar - D. Buck
You can also search for this author inPubMed Google Scholar - J. Burgess
You can also search for this author inPubMed Google Scholar - W. D. Burrill
You can also search for this author inPubMed Google Scholar - J. Burton
You can also search for this author inPubMed Google Scholar - C. Carder
You can also search for this author inPubMed Google Scholar - N. P. Carter
You can also search for this author inPubMed Google Scholar - Y. Chen
You can also search for this author inPubMed Google Scholar - G. Clark
You can also search for this author inPubMed Google Scholar - S. M. Clegg
You can also search for this author inPubMed Google Scholar - V. Cobley
You can also search for this author inPubMed Google Scholar - C. G. Cole
You can also search for this author inPubMed Google Scholar - R. E. Collier
You can also search for this author inPubMed Google Scholar - R. E. Connor
You can also search for this author inPubMed Google Scholar - D. Conroy
You can also search for this author inPubMed Google Scholar - N. Corby
You can also search for this author inPubMed Google Scholar - G. J. Coville
You can also search for this author inPubMed Google Scholar - A. V. Cox
You can also search for this author inPubMed Google Scholar - J. Davis
You can also search for this author inPubMed Google Scholar - E. Dawson
You can also search for this author inPubMed Google Scholar - P. D. Dhami
You can also search for this author inPubMed Google Scholar - C. Dockree
You can also search for this author inPubMed Google Scholar - S. J. Dodsworth
You can also search for this author inPubMed Google Scholar - R. M. Durbin
You can also search for this author inPubMed Google Scholar - A. Ellington
You can also search for this author inPubMed Google Scholar - K. L. Evans
You can also search for this author inPubMed Google Scholar - J. M. Fey
You can also search for this author inPubMed Google Scholar - K. Fleming
You can also search for this author inPubMed Google Scholar - L. French
You can also search for this author inPubMed Google Scholar - A. A. Garner
You can also search for this author inPubMed Google Scholar - J. G. R. Gilbert
You can also search for this author inPubMed Google Scholar - M. E. Goward
You can also search for this author inPubMed Google Scholar - D. Grafham
You can also search for this author inPubMed Google Scholar - M. N. Griffiths
You can also search for this author inPubMed Google Scholar - C. Hall
You can also search for this author inPubMed Google Scholar - R. Hall
You can also search for this author inPubMed Google Scholar - G. Hall-Tamlyn
You can also search for this author inPubMed Google Scholar - R. W. Heathcott
You can also search for this author inPubMed Google Scholar - S. Ho
You can also search for this author inPubMed Google Scholar - S. Holmes
You can also search for this author inPubMed Google Scholar - S. E. Hunt
You can also search for this author inPubMed Google Scholar - M. C. Jones
You can also search for this author inPubMed Google Scholar - J. Kershaw
You can also search for this author inPubMed Google Scholar - A. Kimberley
You can also search for this author inPubMed Google Scholar - A. King
You can also search for this author inPubMed Google Scholar - G. K. Laird
You can also search for this author inPubMed Google Scholar - C. F. Langford
You can also search for this author inPubMed Google Scholar - M. A. Leversha
You can also search for this author inPubMed Google Scholar - C. Lloyd
You can also search for this author inPubMed Google Scholar - D. M. Lloyd
You can also search for this author inPubMed Google Scholar - I. D. Martyn
You can also search for this author inPubMed Google Scholar - M. Mashreghi-Mohammadi
You can also search for this author inPubMed Google Scholar - L. Matthews
You can also search for this author inPubMed Google Scholar - O. T. McCann
You can also search for this author inPubMed Google Scholar - J. McClay
You can also search for this author inPubMed Google Scholar - S. McLaren
You can also search for this author inPubMed Google Scholar - A. A. McMurray
You can also search for this author inPubMed Google Scholar - S. A. Milne
You can also search for this author inPubMed Google Scholar - B. J. Mortimore
You can also search for this author inPubMed Google Scholar - C. N. Odell
You can also search for this author inPubMed Google Scholar - R. Pavitt
You can also search for this author inPubMed Google Scholar - A. V. Pearce
You can also search for this author inPubMed Google Scholar - D. Pearson
You can also search for this author inPubMed Google Scholar - B. J. Phillimore
You can also search for this author inPubMed Google Scholar - S. H. Phillips
You can also search for this author inPubMed Google Scholar - R. W. Plumb
You can also search for this author inPubMed Google Scholar - H. Ramsay
You can also search for this author inPubMed Google Scholar - Y. Ramsey
You can also search for this author inPubMed Google Scholar - L. Rogers
You can also search for this author inPubMed Google Scholar - M. T. Ross
You can also search for this author inPubMed Google Scholar - C. E. Scott
You can also search for this author inPubMed Google Scholar - H. K. Sehra
You can also search for this author inPubMed Google Scholar - C. D. Skuce
You can also search for this author inPubMed Google Scholar - S. Smalley
You can also search for this author inPubMed Google Scholar - M. L. Smith
You can also search for this author inPubMed Google Scholar - C. Soderlund
You can also search for this author inPubMed Google Scholar - L. Spragon
You can also search for this author inPubMed Google Scholar - C. A. Steward
You can also search for this author inPubMed Google Scholar - J. E. Sulston
You can also search for this author inPubMed Google Scholar - R. M. Swann
You can also search for this author inPubMed Google Scholar - M. Vaudin
You can also search for this author inPubMed Google Scholar - M. Wall
You can also search for this author inPubMed Google Scholar - J. M. Wallis
You can also search for this author inPubMed Google Scholar - M. N. Whiteley
You can also search for this author inPubMed Google Scholar - D. Willey
You can also search for this author inPubMed Google Scholar - L. Williams
You can also search for this author inPubMed Google Scholar - S. Williams
You can also search for this author inPubMed Google Scholar - H. Williamson
You can also search for this author inPubMed Google Scholar - T. E. Wilmer
You can also search for this author inPubMed Google Scholar - L. Wilming
You can also search for this author inPubMed Google Scholar - C. L. Wright
You can also search for this author inPubMed Google Scholar - T. Hubbard
You can also search for this author inPubMed Google Scholar - D. R. Bentley
You can also search for this author inPubMed Google Scholar - S. Beck
You can also search for this author inPubMed Google Scholar - J. Rogers
You can also search for this author inPubMed Google Scholar - N. Shimizu
You can also search for this author inPubMed Google Scholar - S. Minoshima
You can also search for this author inPubMed Google Scholar - K. Kawasaki
You can also search for this author inPubMed Google Scholar - T. Sasaki
You can also search for this author inPubMed Google Scholar - S. Asakawa
You can also search for this author inPubMed Google Scholar - J. Kudoh
You can also search for this author inPubMed Google Scholar - A. Shintani
You can also search for this author inPubMed Google Scholar - K. Shibuya
You can also search for this author inPubMed Google Scholar - Y. Yoshizaki
You can also search for this author inPubMed Google Scholar - N. Aoki
You can also search for this author inPubMed Google Scholar - S. Mitsuyama
You can also search for this author inPubMed Google Scholar - B. A. Roe
You can also search for this author inPubMed Google Scholar - F. Chen
You can also search for this author inPubMed Google Scholar - L. Chu
You can also search for this author inPubMed Google Scholar - J. Crabtree
You can also search for this author inPubMed Google Scholar - S. Deschamps
You can also search for this author inPubMed Google Scholar - A. Do
You can also search for this author inPubMed Google Scholar - T. Do
You can also search for this author inPubMed Google Scholar - A. Dorman
You can also search for this author inPubMed Google Scholar - F. Fang
You can also search for this author inPubMed Google Scholar - Y. Fu
You can also search for this author inPubMed Google Scholar - P. Hu
You can also search for this author inPubMed Google Scholar - A. Hua
You can also search for this author inPubMed Google Scholar - S. Kenton
You can also search for this author inPubMed Google Scholar - H. Lai
You can also search for this author inPubMed Google Scholar - H. I. Lao
You can also search for this author inPubMed Google Scholar - J. Lewis
You can also search for this author inPubMed Google Scholar - S. Lewis
You can also search for this author inPubMed Google Scholar - S.-P. Lin
You can also search for this author inPubMed Google Scholar - P. Loh
You can also search for this author inPubMed Google Scholar - E. Malaj
You can also search for this author inPubMed Google Scholar - T. Nguyen
You can also search for this author inPubMed Google Scholar - H. Pan
You can also search for this author inPubMed Google Scholar - S. Phan
You can also search for this author inPubMed Google Scholar - S. Qi
You can also search for this author inPubMed Google Scholar - Y. Qian
You can also search for this author inPubMed Google Scholar - L. Ray
You can also search for this author inPubMed Google Scholar - Q. Ren
You can also search for this author inPubMed Google Scholar - S. Shaull
You can also search for this author inPubMed Google Scholar - D. Sloan
You can also search for this author inPubMed Google Scholar - L. Song
You can also search for this author inPubMed Google Scholar - Q. Wang
You can also search for this author inPubMed Google Scholar - Y. Wang
You can also search for this author inPubMed Google Scholar - Z. Wang
You can also search for this author inPubMed Google Scholar - J. White
You can also search for this author inPubMed Google Scholar - D. Willingham
You can also search for this author inPubMed Google Scholar - H. Wu
You can also search for this author inPubMed Google Scholar - Z. Yao
You can also search for this author inPubMed Google Scholar - M. Zhan
You can also search for this author inPubMed Google Scholar - G. Zhang
You can also search for this author inPubMed Google Scholar - S. Chissoe
You can also search for this author inPubMed Google Scholar - J. Murray
You can also search for this author inPubMed Google Scholar - N. Miller
You can also search for this author inPubMed Google Scholar - P. Minx
You can also search for this author inPubMed Google Scholar - R. Fulton
You can also search for this author inPubMed Google Scholar - D. Johnson
You can also search for this author inPubMed Google Scholar - G. Bemis
You can also search for this author inPubMed Google Scholar - D. Bentley
You can also search for this author inPubMed Google Scholar - H. Bradshaw
You can also search for this author inPubMed Google Scholar - S. Bourne
You can also search for this author inPubMed Google Scholar - M. Cordes
You can also search for this author inPubMed Google Scholar - Z. Du
You can also search for this author inPubMed Google Scholar - L. Fulton
You can also search for this author inPubMed Google Scholar - D. Goela
You can also search for this author inPubMed Google Scholar - T. Graves
You can also search for this author inPubMed Google Scholar - J. Hawkins
You can also search for this author inPubMed Google Scholar - K. Hinds
You can also search for this author inPubMed Google Scholar - K. Kemp
You can also search for this author inPubMed Google Scholar - P. Latreille
You can also search for this author inPubMed Google Scholar - D. Layman
You can also search for this author inPubMed Google Scholar - P. Ozersky
You can also search for this author inPubMed Google Scholar - T. Rohlfing
You can also search for this author inPubMed Google Scholar - P. Scheet
You can also search for this author inPubMed Google Scholar - C. Walker
You can also search for this author inPubMed Google Scholar - A. Wamsley
You can also search for this author inPubMed Google Scholar - P. Wohldmann
You can also search for this author inPubMed Google Scholar - K. Pepin
You can also search for this author inPubMed Google Scholar - J. Nelson
You can also search for this author inPubMed Google Scholar - I. Korf
You can also search for this author inPubMed Google Scholar - J. A. Bedell
You can also search for this author inPubMed Google Scholar - L. Hillier
You can also search for this author inPubMed Google Scholar - E. Mardis
You can also search for this author inPubMed Google Scholar - R. Waterston
You can also search for this author inPubMed Google Scholar - R. Wilson
You can also search for this author inPubMed Google Scholar - B. S. Emanuel
You can also search for this author inPubMed Google Scholar - T. Shaikh
You can also search for this author inPubMed Google Scholar - H. Kurahashi
You can also search for this author inPubMed Google Scholar - S. Saitta
You can also search for this author inPubMed Google Scholar - M. L. Budarf
You can also search for this author inPubMed Google Scholar - H. E. McDermid
You can also search for this author inPubMed Google Scholar - A. Johnson
You can also search for this author inPubMed Google Scholar - A. C. C. Wong
You can also search for this author inPubMed Google Scholar - B. E. Morrow
You can also search for this author inPubMed Google Scholar - L. Edelmann
You can also search for this author inPubMed Google Scholar - U. J. Kim
You can also search for this author inPubMed Google Scholar - H. Shizuya
You can also search for this author inPubMed Google Scholar - M. I. Simon
You can also search for this author inPubMed Google Scholar - J. P. Dumanski
You can also search for this author inPubMed Google Scholar - M. Peyrard
You can also search for this author inPubMed Google Scholar - D. Kedra
You can also search for this author inPubMed Google Scholar - E. Seroussi
You can also search for this author inPubMed Google Scholar - I. Fransson
You can also search for this author inPubMed Google Scholar - I. Tapia
You can also search for this author inPubMed Google Scholar - C. E. Bruder
You can also search for this author inPubMed Google Scholar - K. P. O'Brien
You can also search for this author inPubMed Google Scholar
Corresponding author
Correspondence toI. Dunham.
Supplementary Information
Supplementary Information
Supplementary Information
Rights and permissions
About this article
Cite this article
Dunham, I., Hunt, A., Collins, J. et al. The DNA sequence of human chromosome 22.Nature 402, 489–495 (1999). https://doi.org/10.1038/990031
- Received: 05 November 1999
- Accepted: 11 November 1999
- Issue Date: 02 December 1999
- DOI: https://doi.org/10.1038/990031